

Abstract
The conserved protein kinase Chk1 is believed to play an important role in checkpoint responses to aberrant DNA structures; however, genetic analysis of Chk1 functions in metazoans is complicated by lethality of Chk1-deficient embryonic cells. We have used gene targeting to eliminate Chk1 function in somatic DT40 B-lymphoma cells. We find that Chk1-deficient DT40 cells are viable, but fail to arrest in G2/M in response to and are hypersensitive to killing by ionizing radiation. Chk1-deficient cells also fail to maintain viable replication forks or suppress futile origin firing when DNA polymerase is inhibited, leading to incomplete genome duplication and diminished cell survival after release from replication arrest. In contrast to embryonic cells, however, Chk1 is not required to delay mitosis when DNA synthesis is inhibited. Thus, Chk1 is dispensable for normal cell division in somatic DT40 cells but is essential for DNA damage-induced G2/M arrest and a subset of replication checkpoint responses. Furthermore, Chk1-dependent processes promote tumour cell survival after perturbations of DNA structure or metabolism.
Keywords: cell survival/checkpoints/Chk1/DNA damage/DNA replication
Introduction
Chk1 was originally identified in fission yeast by genetic studies as a protein kinase required to delay entry of cells to mitosis after DNA damage (Walworth et al., 1993; al-Khodairy et al., 1994; Walworth and Bernards, 1996). DNA damage-induced mitotic delay in fission yeast is imposed primarily through inhibitory Tyr15 phosphorylation of Cdc2 (O’Connell et al., 2000). In response to DNA damage, Chk1 is activated in a Rad3-dependent manner (the yeast homologue of the mammalian ATM and ATR proteins) (Lopez-Girona et al., 2001), and prevents activation of Cdc2 by inhibiting Cdc25 phosphatase, a Cdc2 activator (Rhind et al., 1997; Furnari et al., 1999; Lopez-Girona et al., 1999), and stimulating the activity of the Mik1 tyrosine kinase, a Cdc2 inhibitor (Baber-Furnari et al., 2000; Christensen et al., 2000).
In fission yeast, mitosis is also delayed by a specific checkpoint mechanism when DNA replication is blocked [the S–M checkpoint (Enoch et al., 1992)]. In contrast to DNA damage, mitotic delay under conditions of replication arrest in wild-type fission yeast cells is not implemented by Chk1 (Walworth et al., 1993; al-Khodairy et al., 1994), but instead by a second checkpoint effector kinase, Cds1 (Murakami and Okayama, 1995; the homologue of Chk2 in mammalian cells). Cds1 phosphorylates Cdc25 in vitro (Zeng et al., 1998), and in vivo evidence suggests that the S–M checkpoint is also mediated via inhibitory tyrosine phosphorylation of Cdc2 (Rhind and Russell, 1998). Mitotic delay in response to DNA damage and replication arrest in fission yeast therefore share a common mechanistic endpoint, even though distinct effector kinases are used to implement the checkpoint response in each case.
Under conditions of replication arrest, additional checkpoint processes operate in S phase to maintain the viability of stalled replication forks and to suppress futile origin firing. In fission and budding yeast, these replication checkpoint functions require the Cds1 and Rad53 protein kinases respectively (Santocanale and Diffley, 1998; Kim and Huberman, 2001); however, the molecular mechanisms and targets involved are currently less well understood.
In mammalian cells, several reports have provided biochemical evidence that Chk1 participates in the G2/M DNA damage checkpoint by phosphorylating and modulating the activity of the Cdc2 regulators Cdc25C phosphatase and Wee1 kinase, relatives of fission yeast Cdc25 and Mik1, respectively (reviewed in Rhind and Russell, 2000). Additional evidence for an in vivo role for Chk1 in the mammalian G2/M DNA damage checkpoint has been obtained from analysis of chk1-deficient mouse blastocysts and embryonic stem (ES) cells (Liu et al., 2000; Takai et al., 2000). However, these studies also revealed that Chk1 is essential for embryonic cell viability, and the acute lethality of the Chk1-deficient cells prevented a detailed evaluation of the extent to which the G2/M checkpoint was impaired in the absence of Chk1, or investigation of the fate of cells undergoing checkpoint failure.
More recently, evidence that Chk1 plays a role in replication checkpoint responses in metazoans has begun to accumulate. Xenopus Chk1 is activated by replication arrest and required to prevent premature mitosis in egg extracts in vitro (Kumagai et al., 1998; Michael et al., 2000). In addition, Chk1-deficient mouse blastocytes were found to undergo premature entry to mitosis when DNA synthesis was inhibited with aphidicolin (Takai et al., 2000), suggesting a role for Chk1 in the S–M checkpoint in vivo. In addition, a selective inhibitor of Chk1 (UCN-01) has been shown to impair replication fork viability and disturb the control of origin firing in mammalian cells when DNA synthesis is inhibited (Feijoo et al., 2001). However, because UCN-01 is known to inhibit other checkpoint kinases (Busby et al., 2000), a potential role for other effectors in these processes could not be formally excluded.
Yeast mutants which lack Chk1 are more sensitive to killing by DNA damage but not replication arrest (Walworth et al., 1993; Walworth and Bernards, 1996; Sanchez et al., 1996). In comparison, little is known about the potential role of Chk1 in determining vertebrate cell survival in response to DNA structure perturbations. This issue is potentially of more than academic interest, since radiation and many other clinically important anti-cancer therapies act by damaging or inhibiting the replication of DNA. Furthermore, Chk1 inhibitors such as UCN-01 are currently under evaluation as potential therapeutic agents (Senderowicz, 2000).
We reasoned that, as in yeast, a systematic dissection of Chk1 functions in vertebrates would be greatly facilitated by analysis of cells which are genetically devoid of this function. To this end, we have used the avian B-lymphoma cell line DT40 to generate Chk1-deficient vertebrate somatic cells by gene targeting. In contrast to embryonic cells, Chk1-deficient DT40 cells are viable but exhibit striking defects in specific checkpoint responses to both DNA damage and replication arrest. Chk1-deficient cells are also markedly more sensitive to killing by DNA damage and DNA synthesis inhibition, demonstrating that Chk1-dependent processes promote tumour cell survival after perturbations of DNA structure or metabolism.
Results
Characterization of avian Chk1 and generation of Chk1-deficient DT40 cells by gene targeting
To investigate the physiological role of Chk1 in vertebrate somatic cells, we generated Chk1-deficient DT40 cells by gene targeting. An avian chk1 cDNA was isolated from a chicken B-cell cDNA library using human chk1 as a probe. The predicted avian Chk1 protein of 476 amino acids encoded by this cDNA shows 85, 81 and 75% amino acid identity to the human, mouse and Xenopus Chk1 homologues, respectively, and shares the conserved N-terminal kinase domain, C-terminal putative regulatory regions and several potential serine–glutamine ATM/ATR phosphorylation site motifs (Chen et al., 2000; Guo et al., 2000; Figure 1A).

Fig. 1. Sequence of avian Chk1 and generation of Chk1-deficient DT40 cells. (A) Predicted amino acid sequence of avian Chk1 (red) and comparison with human, mouse and Xenopus homologues; identical and highly conserved amino acid residues are shown. The conserved kinase domain is grey, C-terminal regulatory regions are yellow, potential ATM/ATR phosphorylation sites are magenta. Ser345 (human sequence) is identified by an asterisk. (B) Southern (left panel) and western (right panel) blotting analysis of Chk1+/+, Chk1+/– and Chk1–/– cells. Diagnostic bands representing the wild-type (6.0 kb) and targeted (1.4 kb) chk1 alleles are indicated. Chk1 protein was detected using an anti-Chk1 monoclonal antibody.
Targeting vectors designed to disrupt the DT40 chk1 gene were constructed by PCR of DT40 genomic DNA using oligonucleotide primers derived from the avian chk1 cDNA sequence (see Materials and methods for details). After two sequential rounds of gene targeting, Chk1–/– clones in which both copies of the chk1 gene had been disrupted were identified by Southern blotting (Figure 1B). Western blotting of cell extracts using several independent anti-Chk1 antibodies confirmed that Chk1–/– cells were devoid of functional Chk1 protein (Figure 1B; data not shown). Thus, in contrast to embryonic cells, Chk1 is not essential for the survival of somatic DT40 cells in culture.
Chk1-deficient cells proliferate more slowly and exhibit increased levels of spontaneous cell death
Although Chk1-deficient DT40 cells were viable, they multiplied more slowly during exponential growth phase and ceased to proliferate at lower saturation densities than wild-type cells (Figure 2A). These growth defects were associated with accumulation of more dead cells in Chk1–/– cultures compared with wild-type as determined by trypan blue staining and optical microscopy (Figure 2B). To investigate the mechanism of cell death in more detail, exponentially growing Chk1+/+ and Chk1–/– cultures were stained with Annexin V and propidium iodide (PI) and analysed by flow cytometry. This analysis (Figure 2C) revealed that Chk1–/– cultures consistently contained more Annexin V-positive cells in both the early (bottom right quadrant) and late (upper right quadrant) stages of apoptosis at the expense of viable cells (bottom left quadrant) than Chk1+/+ cultures.

Fig. 2. Proliferation properties of Chk1+/+ and Chk1–/– cells. (A) Growth curves of Chk1+/+ and Chk1–/– cells. Error bars show the standard deviation of the mean for three experiments. (B) Accumulation of dead cells with time as determined by trypan blue staining. Error bars show the standard deviation of the mean for three experiments. (C) Flow cytometry analysis of cells stained with Annexin V and PI. Viable cells are negative for both Annexin V and PI (lower left quadrant), whilst cells in the early (Annexin V-positive/PI-negative) or late (Annexin V-positive/PI-positive) stages of apoptosis appear in the lower right and upper right quadrants respectively. Numbers indicate the percentage (%) of cells in the respective quadrant.
To determine whether loss of Chk1 disturbed cell cycle progression per se, we pulse-labelled Chk1–/– and Chk1+/+ cells with bromo-deoxyuridine (BrdU) and followed the progress of the labelled cohort through the subsequent cell cycle as described previously (Clark and Gillespie, 1997). Chk1-deficient cultures exhibited a modest increase in the percentage of cells in the G1 and G2/M phases of the cell cycle and a corresponding decrease in S phase compared to Chk1+/+; however, both cell types divided with an overall cell cycle time of ∼10 h (data not shown). Thus, whilst loss of Chk1 may lead to subtle alterations in cell cycle control, the slower multiplication of Chk1-deficient cells under normal growth conditions is primarily due to an increase in the incidence of spontaneous apoptotic cell death.
Chk1 is essential for G2/M arrest in response to ionizing radiation in DT40 cells
Chk1 is activated by γ-ionizing radiation (IR) in wild-type DT40 cells (data not shown) as judged both by decreased electrophoretic mobility (Walworth and Bernards, 1996) and increased phosphorylation of Ser345 (human sequence), a site whose modification by ATM/ATR is required for Chk1 activity (Lopez-Girona et al., 2001; Zhao and Piwnica-Worms, 2001). To determine whether Chk1 is required for DNA damage-induced G2/M arrest, Chk1+/+ and Chk1–/– DT40 cells were exposed to 20 Gy of γ-IR and the effect on cell cycle progression was determined by flow cytometry. Chk1+/+ DT40 cells lack functional p53 (Takao et al., 1999) and 6–8 h after irradiation accumulated predominantly in G2/M (Figure 3A). In comparison, irradiated Chk1–/– cells exhibited no measurable arrest in G2/M and continued to synthesize DNA and cycle actively (Figure 3A; data not shown). This checkpoint failure was solely attributable to the absence of Chk1, since Chk1–/– cells expressing exogenous Chk1 encoded by a transfected transgene (‘revertant’ cells, see Materials and methods for details) arrested normally in G2/M after irradiation (Figure 3A).

Fig. 3. Chk1-deficient cells fail to arrest in G2/M in response to IR. (A) DNA content flow cytometry analysis of Chk1+/+, Chk1–/– and revertant (Rev) cells at indicated times after irradiation (20 Gy γ-IR). (B) Mitotic indices of the indicated cells incubated with nocodazole (Nocod) for 8 h with or without prior irradiation (20 Gy γ-IR); un: untreated cells. A minimum of 200 nuclei was counted for each mitotic index measurement. (C) Cdc2-associated H1 kinase activity and Cdc2 levels (Tyr15 phosphorylated and total) at the indicated times after exposure of Chk1+/+, Chk1–/– and revertant (Rev) cells to 20 Gy γ-IR.
To further confirm that irradiated Chk1-deficient cells entered mitosis, Chk1+/+, Chk1–/– and revertant cells were incubated in medium containing nocodazole for 8 h with or without prior irradiation. The percentage of phospho-Ser10 histone H3-positive (mitotic) cells was then determined by fluorescence microscopy (Ajiro et al., 1996). As shown in Figure 3B, irradiation completely prevented nocodazole-treated Chk1+/+ and revertant cells from accumulating in mitosis, indicating that these cells arrest efficiently in G2. In marked contrast, Chk1–/– cells entered mitosis with similar efficiency regardless of prior irradiation. Additional time course experiments revealed that G2 arrest measured using this method (Xu et al., 2002) was extremely rapid in Chk1+/+ cells (within 1 h); however, Chk1–/– cells exhibited no measurable arrest at any time point tested (data not shown).
We also immunoprecipitated Cdc2 from Chk1+/+, Chk1–/– and revertant cells at 0, 4 and 8 h after 20 Gy of γ-irradiation and monitored changes in Cdc2 catalytic activity and Tyr15 phosphorylation levels in kinase assays using histone 1 (H1) as a substrate and by immunoblotting, respectively (Figure 3C). In Chk1+/+ and revertant cells, accumulation of cells in G2 correlated temporally with inhibition of Cdc2 kinase activity and increased Cdc2–Tyr15 phosphorylation; however, none of these biochemical changes occurred in Chk1–/– cells (Figure 3C). Taken together, these results provide genetic proof that Chk1 is essential for the G2 arrest which delays mitosis in response to IR in somatic DT40 cells.
Chk1-deficient cells are hypersensitive to IR
To determine whether loss of Chk1, and thus of DNA damage-induced G2/M arrest, renders cells more vulnerable to killing by IR, we compared the clonogenic survival of Chk1+/+, Chk1–/– and revertant cells after exposure to increasing doses of IR (Figure 4A). Strikingly, Chk1–/– cells (filled circles) were significantly more sensitive to IR than Chk1+/+ (filled diamonds) at all doses tested. This increase in radiosensitivity was solely due to the absence of Chk1, since revertant cells (open circles) were indistinguishable from wild-type (Figure 4A).

Fig. 4. Chk1-deficient cells are hypersensitive to killing by IR. (A) Clonogenic survival of Chk1+/+, Chk1–/– and revertant (Rev) cells after exposure to the indicated doses of γ-IR. Error bars show the standard deviation of the mean for three experiments. (B) Increase in apoptosis (Annexin V staining) of Chk1+/+ and Chk1–/– cells at various times after exposure to 20 Gy γ-IR. Error bars show the standard deviation of the mean for three experiments.
To determine whether the radiosensitivity of Chk1–/– cells was associated with increased apoptosis, Chk1+/+ and Chk1–/– cells were treated with 12 Gy γ-IR and the proportion of apoptotic cells determined by Annexin V staining and flow cytometry over a time period corresponding to the duration of the clonogenic survival assay (7 days). As shown in Figure 4B, the percentage of apoptotic cells in Chk1+/+ cultures (filled diamonds) increased markedly 3 days after irradiation and then decreased, presumably due to repopulation of the culture by a small fraction of surviving viable cells. In contrast, irradiation of Chk1–/– cultures (filled circles) failed to induce any significant increase in the percentage of apoptotic cells (Figure 4B), even though a greater proportion of the cells were killed as determined by clonogenic survival (Figure 4A). Similar results were obtained when cell cultures were irradiated with 4 or 8 Gy of IR (data not shown). These results indicate that reproductive cell death is likely to account for the decreased clonogenic survival of Chk1-defective cells after irradiation. They also indicate that Chk1 is required for acute radiation-induced apoptosis in DT40 cells.
Chk1 is not essential for the S–M checkpoint but is required for recovery from replication arrest in DT40 cells
Previous analysis of Chk1-deficient mouse blastocytes suggested a role for Chk1 in the S–M checkpoint (Takai et al., 2000). To evaluate the possible role of Chk1 during replication arrest in DT40 cells, we first determined whether aphidicolin, an inhibitor of DNA polymerase α and δ, activated Chk1. As shown in Figure 5A, treatment of wild-type DT40 cells with 20 µM aphidicolin led to activation of Chk1 as evidenced by both altered electrophoretic mobility and increased phosphorylation of Ser345. Chk1 activation was rapid and persistent (Figure 5A; data not shown), suggesting a functional role for Chk1 during replication arrest.
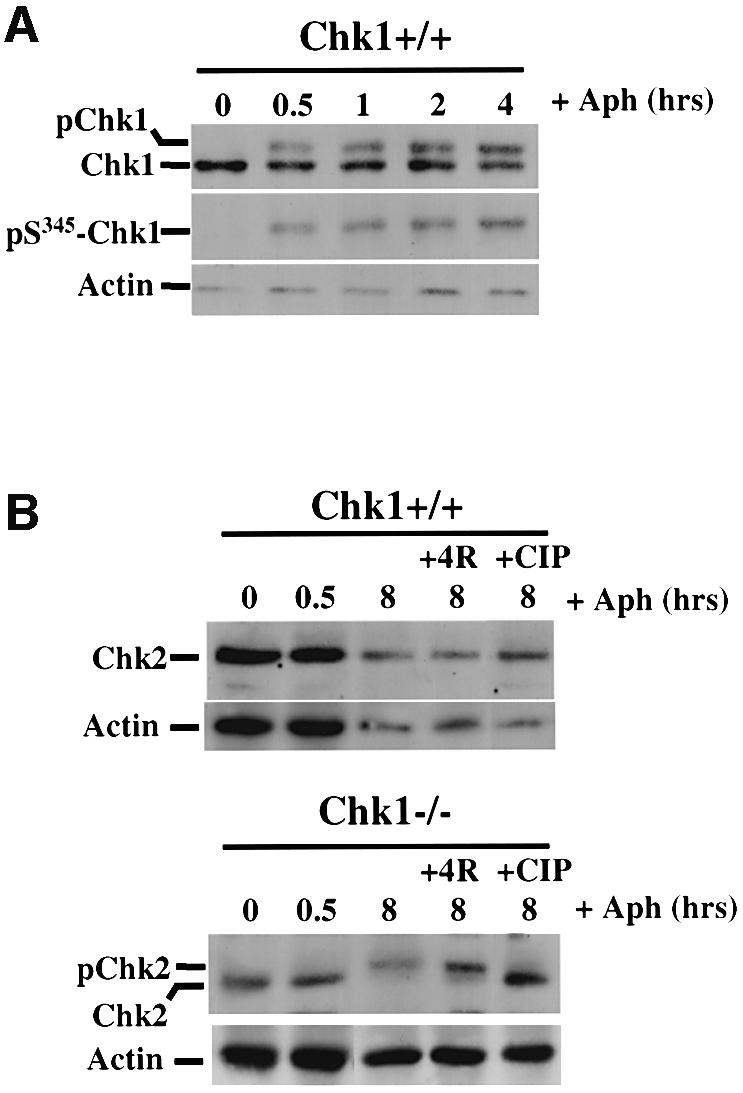

Fig. 5. Chk1-deficient cells fail to enter mitosis during or after release from replication arrest. (A) Western blot analysis of total Chk1 and Chk1 phosphorylated on Ser345, after treatment of Chk1+/+ cells with 20 µM aphidicolin for the indicated times. Actin serves as a loading control. (B) Western blot analysis of Chk2 in Chk1+/+ and Chk1–/– cells after treatment with 20 µM aphidicolin for up to 8 h as indicated, or 4 h after release from 8 h of replication arrest (+4R). The electrophoretic mobility shift seen after 8 h of aphidicolin treatment in Chk1–/– cells was due to Chk2 phosphorylation since it was reversed by treatment of the extract with calf intestinal phosphatase (+CIP). (C) Mitotic indices of Chk1+/+, Chk1–/– and revertant (Rev) cells at the indicated times after release from aphidicolin treatment (8 h, 20 µM); un: untreated cells. A minimum of 200 nuclei was counted for each time point. (D) Representative merged confocal microscopy images of nuclei treated as described in (C) and stained with propidium iodide (red) and anti-phospho-Ser10 histone H3 antibody (green). Mitotic nuclei staining positive for phospho-Ser10 histone H3 are indicated by white arrows.
To determine whether loss of Chk1 leads to premature mitosis under these conditions, Chk1+/+, Chk1–/– and revertant cells were incubated with 20 µM aphidicolin for 8 h and the percentage of phospho-Ser10 histone H3-positive cells determined by fluorescence microscopy. Remarkably, virtually no mitotic cells were observed in Chk1+/+, Chk1–/– or revertant cell cultures after 8 h of aphidicolin treatment (Figure 5C and D, +Aph), indicating that the S–M checkpoint was functional under these conditions in all cell types. Similar results were obtained when cells were treated with 20 µM aphidicolin for up to 16 h and when lower concentrations of drug (2 µM) were employed (data not shown). Thus, in contrast to what has been observed in mouse embryos, loss of Chk1 is not sufficient to abrogate the S–M checkpoint when DNA replication is blocked in somatic DT40 cells.
To evaluate the possibility that Chk2 might contribute to the S–M checkpoint in the absence of Chk1, we examined the activation of Chk2 in aphidicolin-treated wild-type and Chk1–/– cells. Activation of Chk2 is associated with a pronounced electrophoretic mobility shift due to regulatory phosphorylation (Matsuoka et al., 1998; Feijoo et al., 2001), which we could monitor by western blotting using a peptide antiserum specific for avian Chk2 (see Materials and methods for details). As shown in Figure 5B, Chk2 was activated very weakly, if at all, after 8 h of aphidicolin treatment in wild-type DT40 cells; however, in Chk1–/– cells, prolonged replication arrest resulted in robust and persistent activation of Chk2 as judged by a phosphatase-reversible gel mobility shift. Although the mechanistic basis of this differential activation remains to be established, it is consistent with the idea that in the absence of Chk1, activation of Chk2 may compensate to delay mitosis whilst DNA replication is incomplete.
We also compared the ability of wild-type and Chk1-deficient cells to recover from replication arrest. When cultures which had been arrested with aphidicolin for 8 h were released from the replication block by washing the drug away, the majority of the Chk1+/+ and revertant cells rapidly progressed through S phase and entered mitosis 4–5 h after release (Figure 5C and D). By 6 h after release, most of the Chk1+/+ and revertant cells had exited mitosis and resumed asynchronous growth, demonstrating that the aphidicolin arrest was fully reversible (Figure 5D). In striking contrast, when Chk1–/– cultures were released, very few mitotic cells were observed at any time up to 16 h post-release (Figure 5C and D; data not shown), indicating that most of the arrested cells were unable to resume normal cell cycle progression after prolonged aphidicolin treatment. Thus, although Chk1 was not required to prevent premature mitosis, these results demonstrated that Chk1–/– cells were severely compromised in their ability to recover from replication arrest.
Chk1-deficient cells fail to maintain the viability of stalled replication forks or suppress futile origin firing during DNA synthesis inhibition
We reasoned that checkpoint defects affecting the replication program per se might account for the inability of Chk1-deficient cells to recover from replication arrest. In yeast, specific checkpoint responses maintain the viability of stalled replication forks and suppress futile origin firing when DNA replication is inhibited (Santocanale and Diffley, 1998; Kim and Huberman, 2001). To determine whether Chk1–/– cells suffered from defects in replication fork function or replication origin control under these conditions, we utilized a dual labelling method using monoclonal antibodies, which discriminate between iodo-deoxyuridine (idU) and chloro-deoxyuridine (CldU) incorporated into DNA (Dimitrova and Gilbert, 1999).
The experimental protocol is illustrated schematically in Figure 6A. Asynchronous cultures of Chk1+/+, Chk1–/– and revertant cells were pre-labelled with IdU for 15 min to label active replication forks, IdU was then removed and the cells were incubated with 20 µM aphidicolin for 0–8 h. Cells were subsequently released from aphidicolin arrest, pulse-labelled with CldU for 15 min, fixed and stained with antibodies specific for IdU (red) and CldU (green). The spatial distribution of incorporated IdU and CldU was then visualized by confocal microscopy.

Fig. 6. Loss of replication fork viability and aberrant origin firing in Chk1–/– cells during replication arrest. (A) Experimental protocol (see text for details). Cultures of Chk1+/+, Chk1–/– and revertant (Rev) cells in exponential growth were pre-labelled with IdU for 15 min and then pulse-labelled with CldU for 15 min either immediately (0) or after 2–8 h of aphidicolin treatment. (B) Confocal microscopy of Chk1+/+, Chk1–/– and revertant (Rev) cells labelled with IdU and CldU either before or after various times of aphidicolin (Aph) arrest. Representative examples of nuclei labelled with IdU (red) and CldU (green) are shown. Early, middle and late refer to the replication patterns of individual nuclei defined according to the initial IdU (red) labelling.
In this procedure, coincident labelling (yellow) identifies replication forks which were active prior to addition of aphidicolin and which remained capable of resuming replication after the period of arrest. Discontinuous labelling identifies replication forks, which were previously active but became incapable of resuming replication during the period of aphidicolin arrest (red alone), and also new replication structures which arose as a result of origin firing (green alone). The temporal position of individual cell nuclei within S phase can also be deduced from the pattern of IdU/CldU incorporation; early-replicating sequences are distributed throughout the nucleus whereas later-replicating sequences are located preferentially at the nuclear periphery and perinucleolar regions (Dimitrova and Gilbert, 1999).
When Chk1+/+ cells were analysed using this procedure (Figure 6B), only coincident labelling was observed, even when cells were arrested with aphidicolin for up to 8 h, indicating that active replication forks were preserved and that no new origins initiated replication during this time. In striking contrast, coincident labelling was lost in Chk1–/– cells after 4 h of aphidicolin arrest, although previously incorporated IdU (red) persisted and new incorporation of CldU (green) was evident at distinct sites (Figure 6B). Thus, in Chk1-deficient cells a significant proportion of active replication forks became incapable of resuming replication after aphidicolin arrest and this was accompanied by the appearance of new sites of replication. These defects were solely attributable to the absence of Chk1, since the labelling pattern of the Chk1-expressing revertant cells was indistinguishable from wild type (Figure 6B).
Taken together, these observations provide genetic evidence that Chk1 is required for the replication checkpoint functions that maintain viability of stalled replication forks and suppress replication origin firing during replication arrest in somatic DT40 cells.
Loss of Chk1 is associated with incomplete DNA replication and reduced clonogenic cell survival during recovery from replication arrest
To understand how these replication checkpoint defects might affect subsequent cell cycle progression and survival, Chk1+/+, Chk1–/– and revertant cell cultures were arrested with 20 µM aphidicolin for 8 h and then released into fresh medium. Cells were pulse-labelled with BrdU for 45 min prior to harvesting at the indicated time points and BrdU incorporation/DNA content analysed by flow cytometry. As shown in Figure 7A, all cell types were arrested predominantly in early S phase after 8 h of aphidicolin treatment (+Aph). By 3 h after release from aphidicolin arrest, a majority of both Chk1+/+ and revertant cells had entered S phase as judged by BrdU incorporation, and by 24 h post-release the cultures had returned to asynchronous growth and the percentage of cells that were incorporating BrdU was similar to untreated cultures (Figure 7A). In contrast, significantly fewer Chk1–/– cells incorporated BrdU 3 h after release, and although after 24 h the majority of the cells had DNA content between 2N (left arrow) and 4N (right arrow), very few were actively synthesizing DNA as judged by BrdU incorporation. These findings indicate that, despite a partial initial resumption of DNA synthesis, most of the Chk1–/– cells were unable to complete DNA replication after release.

Fig. 7. Chk1-deficient cells fail to complete DNA synthesis after aphidicolin treatment and are hypersensitive to replication arrest. (A) Cell cycle distribution of Chk1+/+, Chk1–/– and revertant (Rev) cells at indicated times after release from aphidicolin (Aph) block (8 h, 20 µM), as measured by BrdU incorporation and DNA content flow cytometry analysis. The percentage (%) of cells incorporating BrdU is shown. Arrows indicate 2N (left) and 4N (right) DNA content. (B) Clonogenic survival of Chk1+/+, Chk1–/– and revertant (Rev) cells after replication arrest. Cells were incubated with 20 µM aphidicolin for the time shown, after which the drug was removed and cell survival determined. Error bars (in general smaller than graph symbols) show the standard deviation of the mean for three experiments.
To determine how this loss of replication capacity might affect cell viability, Chk1+/+, Chk1–/– and revertant cells were exposed to 20 µM aphidicolin for 0–14 h, then the drug was removed and clonogenic survival determined. As shown in Figure 7B, the viability of the Chk1–/– cells (filled circles) declined much more rapidly with time during replication arrest than Chk1+/+ (filled diamonds) or revertant cells (open circles). Interestingly, and in marked contrast to IR (Figure 4B), Annexin V-staining revealed a good correlation between decreased clonogenic survival and increased levels of apoptosis in both Chk1+/+ and Chk1–/– cells after aphidicolin treatment (data not shown). This suggests that radiation and replication arrest trigger cell death in Chk1-deficient cells through distinct molecular mechanisms.
Discussion
By eliminating Chk1 function using gene targeting in DT40 cells, we have established that, as in yeast (Walworth et al., 1993; Walworth and Bernards, 1996), Chk1 is not universally essential for the viability of vertebrate somatic cells under normal growth conditions in culture. Chk1-deficient cells do however proliferate significantly more slowly in the absence of any externally induced DNA damage or replication stress. We believe that this growth defect is primarily attributable to an increase in the incidence of spontaneous apoptotic cell death, which was markedly higher in Chk1-deficient cells, since we were not able to demonstrate any consistent difference in overall cell cycle time between Chk1+/+ and Chk1–/– cells.
The viability of Chk1–/– DT40 cells contrasts strikingly with the absolute requirement for Chk1 during early development in both mouse and Drosophila (Fogarty et al., 1997; Liu et al., 2000; Takai et al., 2000). Although the reasons for this distinction are not yet known, a number of possible explanations can be considered. Chk1-dependent functions may be particularly important during early embryogenesis to resolve spontaneous DNA structure abnormalities arising as a result of the rapid DNA synthesis and short cell cycles of embryonic cells (Edgar, 1995). If so, it is possible that the elevated level of spontaneous cell death in Chk1–/– DT40 cells may represent a less severe manifestation of this phenomenon. Alternatively, Chk1 may have additional specific functions during embryonic development which are no longer required in somatic cells. Finally, DT40 is a tumour cell line which lacks p53 expression (Takao et al., 1999). It seems unlikely that deficiency for p53 alone allows DT40 cells to survive without Chk1, since genetic ablation of p53 function does not rescue the lethality of Chk1-deficient mouse embryonic cells (Liu et al., 2000). Nevertheless, it is conceivable that some other, unknown oncogenic lesion could modify the phenotype of Chk1 deficiency.
Further investigation will be required to distinguish between these possibilities; however, the viability of Chk1-deficient DT40 cells has enabled us for the first time to investigate the role of Chk1 in specific checkpoint responses in vertebrate somatic cells by direct comparison of an isogenic mutant cell line devoid of this function with its wild-type counterpart. It has also, uniquely, allowed us to evaluate the role of Chk1-dependent processes in determining long-term cell survival in response to DNA damage and replication inhibition.
As we have shown, Chk1–/– cells fail to arrest in G2, continue to enter mitosis, and fail to induce inhibitory Tyr15 phosphorylation of Cdc2 or downregulate Cdc2 kinase activity after exposure to IR. These findings complement and extend previous work in Chk1–/– mouse blastocysts and ES cells which also demonstrated a role for Chk1 in the G2/M DNA damage checkpoint (Liu et al., 2000; Takai et al., 2000). In contrast to these earlier studies, however, where checkpoint failure occurred concurrently with apoptotic cell death, we were able to accurately assess the degree to which the G2/M checkpoint was impaired in the absence of Chk1. Remarkably, we were unable to detect any significant arrest response to IR in Chk1-deficient cells using either flow cytometry to document accumulation of cells in G2 or phospho-Ser10 histone H3 staining to specifically quantitate cells entering mitosis (Xu et al., 2002). Thus, our results suggest that Chk1 is solely responsible for the initiation of G2/M arrest in somatic DT40 cells in response to DNA damage induced by IR, and its loss cannot be compensated by any parallel or redundant pathway.
Strikingly, Chk1-deficient cells were more readily killed by IR than wild-type cells demonstrating that, as in yeast, Chk1 controls processes which protect cells against killing by DNA damage (Walworth et al., 1993; Walworth and Bernards, 1996). IR induces apoptosis in wild-type DT40 cells; however, we did not observe any increase in apoptosis after irradiation in Chk1–/– cells that could explain the decrease in clonogenic survival. This somewhat paradoxical finding demonstrates that Chk1 is in fact required for acute radiation-induced apoptosis in these cells. Since DT40 cells lack p53 (Takao et al., 1999), these results implicate Chk1 in signalling apoptosis via a p53-independent mechanism. They also suggest that the radiosensitivity of Chk1–/– cells must be primarily attributable to an increase in the incidence of reproductive or clonogenic, as opposed to apoptotic, cell death.
One important question arising from these observations is whether the increased radiosensitivity of Chk1–/– cells is a direct consequence of G2/M checkpoint failure or of some other mechanism. IR induces predominantly double-strand breaks (DSBs), which can be repaired either via non-homologous end-joining or homologous recombination (Khanna and Jackson, 2001). In many cell types, including DT40, the latter mechanism is thought to predominate and operates preferentially in G2 when recombination between sister chromatids allows for error-free repair (Takata et al., 1998). Prolonged G2 arrest in Chk1+/+ cells after irradiation therefore presumably maximizes the likelihood of successful DSB repair and, consequently, cell survival. In contrast, because Chk1–/– cells fail to arrest in G2, the opportunity for efficient repair would be lost and cells are likely to undergo mitosis with potentially lethal unrepaired DSBs. Division with damage as a consequence of G2/M checkpoint failure may therefore be sufficient to explain the radiosensitivity of Chk1-deficient cells, although we cannot rule out an additional role for Chk1 in regulating DNA repair per se or some other process which favours cell survival.
We also investigated whether loss of Chk1 led to defects in checkpoint responses activated when DNA synthesis is inhibited. Surprisingly, Chk1-deficient DT40 cells did not undergo premature mitosis during prolonged treatment with aphidicolin, indicating that loss of Chk1 is not sufficient to erode the S–M checkpoint in somatic cells. This was unexpected, since loss of S–M checkpoint control, as evidenced by premature entry to mitosis, was previously reported in aphidicolin-treated Chk1–/– blastocytes (Takai et al., 2000). The reason for this difference is currently unclear; one possibility is that distinct or additional checkpoint mechanisms operate in somatic cells to delay mitosis until DNA replication is complete. In this regard it is notable that in fission yeast premature mitosis during replication arrest is observed only in the absence of both Chk1 and Cds1 (Boddy et al., 1998; Lindsay et al., 1998; Zeng et al., 1998). Furthermore, Chk1 is activated during replication arrest only in Cds1 mutants and not in wild-type cells, perhaps because Cds1 is required to prevent the accumulation of aberrant DNA structures derived from stalled replication forks (Lindsay et al., 1998). The selective activation of Chk2 which we observed in Chk1-deficient DT40 cells is strikingly reminiscent of this phenomenon, and suggests that Chk2 may compensate to enforce the S–M checkpoint in the absence of Chk1 during replication arrest. Further work will be required to test this hypothesis.
Additional insight into the specific functions of Chk1 during replication arrest was obtained when cells were released from the aphidicolin block. In contrast to their wild-type counterparts, the majority of Chk1-deficient cells subsequently failed to enter mitosis, indicating that Chk1 is required for recovery from replication arrest. Replication arrest is known to be capable of generating DSBs (Carr, 2002); however, since Chk1–/– cells lack DNA damage-induced mitotic delay, it seemed unlikely that accumulation of DNA damage alone could be responsible for failure to enter mitosis.
In fission and budding yeast, checkpoint processes are needed to maintain the viability of stalled replication forks and to suppress futile origin firing when DNA synthesis is inhibited. These replication checkpoint functions depend on the Chk2 homologues, Cds1 and Rad53, respectively (Santocanale and Diffley, 1998; Kim and Huberman, 2001); however, Chk1 has recently been implicated in analogous checkpoint processes in mammalian cells using the inhibitor, UCN-01 (Feijoo et al., 2001). These considerations suggested that degeneration of the replication program itself might impede the recovery of Chk1–/– cells from replication arrest.
As we have shown, Chk1-deficient cells exhibit defects in the regulation and extent of DNA replication both during and after release from replication arrest which strongly supports this interpretation. Thus, double-labelling experiments using CldU and IdU demonstrated that replication forks which were active prior to aphidicolin treatment in Chk1–/– cells became progressively unable to resume replication on release, whilst new sites of replication, which are indicative of futile origin firing, progressively accumulated. These effects were not observed in Chk1+/+ cells, thus providing genetic proof that Chk1 is essential to maintain the viability of stalled replication forks and suppress origin firing when DNA synthesis is inhibited. In vertebrate cells, therefore, both genetic and biochemical evidence (Feijoo et al., 2001) demonstrate that Chk1 is primarily responsible for replication checkpoint functions, which in budding and fission yeast are controlled by the Chk2 homologues Rad53 and Cds1 (Santocanale and Diffley, 1998; Kim and Huberman, 2001).
Because eukaryotic chromosomes have specified replication origins which initiate only once in each cell cycle (Gilbert, 2001), futile origin firing is predicted to deplete the number of viable origins. In the most extreme situation, origin depletion and replication fork collapse might severely compromise or even prevent subsequent DNA replication. Thus, we believe that Chk1–/– cells become trapped with partially replicated DNA after release from prolonged replication arrest because insufficient viable replication origins remain to complete genome duplication, despite an initial firing of the remaining replication origins (as judged by BrdU incorporation) early during recovery. We also believe that this explains why Chk1–/– cells are more sensitive to killing by aphidicolin treatment than wild type, since failure to complete DNA replication is likely to constitute a lethal cell injury. Taken together, these results demonstrate genetically that Chk1 is essential for recovery from replication arrest and that, in contrast to its more circumscribed role in fission yeast (Sanchez et al., 1996), Chk1-dependent replication checkpoint functions are important determinants of vertebrate cell survival under conditions of DNA synthesis inhibition.
In comparison to DNA damage-induced G2/M arrest, which is mediated primarily through Tyr15 phosphorylation of Cdc2 as shown both here and elsewhere (reviewed in O‘Connell et al., 2000), relatively little is known about the molecular mechanisms and targets through which Chk1 controls these replication recovery checkpoint functions in vertebrate cells (Carr, 2002). Chk1-deficient DT40 cells provide a tractable system in which to investigate and dissect the molecular basis of these and potentially other checkpoint responses. In addition, Chk1-deficient DT40 cells provide, for the first time, a means of rigorously evaluating the consequences of checkpoint failure or suppression for tumour cell survival in response to specific perturbations of DNA structure or metabolism. Such studies may aid the development of novel checkpoint suppression strategies to enhance the efficacy of radiation and other DNA-directed anticancer agents based on pharmacological inhibition of Chk1.
Materials and methods
Isolation of avian chk1 cDNA and generation of Chk1-deficient and revertant cells
An avian B cell cDNA library (Stratagene) was screened using a human Chk1 cDNA probe and a full-length natural avian chk1 cDNA was isolated and fully sequenced on both DNA strands. To construct the targeting vectors, oligonucleotide primers designed from the sequence of avian chk1 cDNA were used to PCR amplify segments of the genomic chk1 locus using DT40 DNA as a template. The sequence of the PCR primers is available on request. Two arms of homology of 1.9 kb (left arm) and 1.4 kb (right arm) were synthesized and cloned into Bluescript (Stratagene) creating an intervening BamHI site. BamHI fragments bearing neomycin and puromycin drug selection cassettes (Buerstedde and Takeda, 1991) were then inserted between the chk1 homology arms in opposite transcriptional orientation to the chk1 gene. The targeting constructs were designed to disrupt the structure of the chk1 gene and to delete an essential region (helices αH and αI; Chen et al., 2000) of the Chk1 kinase domain, so that any residual truncated polypeptide encoded by the targeted alleles would be non-functional.
To generate Chk1-deficient DT40 clones, two rounds of targeting were performed to disrupt both chk1 alleles. Cells were electroporated with the neomycin targeting vector using a Gene Pulser (BioRad) at 300 V and 960 µF and drug-resistant clones selected in 1.2 mg/ml G418 (Sigma) to obtain hemizygous Chk1+/– cells. One hemizygous clone was then electroporated with the puromycin targeting vector and selected in 0.4 µg/ml puromycin (Sigma). Chk1–/– clones were obtained with a frequency of ∼25%. To generate the revertant cell line, Chk1–/– cells were transfected with an expression vector encoding avian Chk1 (pcDNA3.1zeo, Invitrogen) and selected in 30 µg/ml zeocin (Invitrogen). Drug-resistant clones were screened for Chk1 expression by western blotting and a cell line with near normal levels of Chk1 identified.
Antibodies
Polyclonal antibodies against Chk1 (FL-476 and N-19) and phospho-Tyr15 of Cdc2 and monoclonal antibodies against Chk1 (G-4), actin (C-2) and Cdc2 (17) were from Santa Cruz Biotechnology. Polyclonal anti-phospho-Ser345 Chk1 was from New England Biolabs and anti-phospho-Ser10 of histone H3 from Upstate Biotechnology. A polyclonal rabbit antiserum specific for avian Chk2 was generated using a synthetic peptide corresponding to the C-terminal 15 amino acids of chicken Chk2 conjugated to keyhole limpet haemocyanin. Rat monoclonal antibody clone BU 1/75 used to detect CldU was from Abcam and mouse monoclonal anti-BrdU antibody reactive with IdU was from Beckton Dickinson.
Cell culture and treatments
DT40 B lymphoma cells were grown in DMEM (Gibco) containing 10% fetal bovine serum, 1% chicken serum, 10–5 M β-mercaptoethanol, penicillin, streptomycin, at 39.5°C. Cells were irradiated with 20 Gy of γ-radiation using an Alcyon II CGR MeV cobalt source, or treated with 20 µM aphidicolin, 1 µg/ml nocodazole and 10 µg/ml of CldU, IdU or BrdU (all from Sigma) as appropriate.
Growth curves and clonogenic cell survival
For growth curve experiments, viable and dead cells were distinguished by trypan blue exclusion and optical microscopy and values represent the mean of three experiments. Clonogenic survival assays were performed using methylcellulose (Sigma) semi-solid medium, essentially as described previously (Takata et al., 1998). Colonies were counted 7 days after seeding.
Apoptosis and kinase assays
An Annexin V apoptosis kit (Pharmigen) was used to detect apoptosis by flow cytometry, following the instructions of the manufacturer. Values indicate the relative change (y%) of the percentage of apoptotic cells in irradiated cultures (i%), compared with untreated cultures (u%): y = [(I – u)/u] × 100%. Immunoprecipitation–kinase assays for Cdc2 were performed essentially as described previously (Clark et al., 2000).
Confocal microscopy
Cells were attached to coverslips coated with poly-l-lysine (Sigma). Metabolic incorporation of CldU and IdU into DNA was detected using discriminating monoclonal antibodies as described previously (Aten et al., 1992).
Accession numbers
The DDBJ/EMBL/GenBank accession number for the avian chk1 cDNA is AF525027.
Acknowledgments
Acknowledgements
We thank W.Clark for help with the initial cloning of the avian chk1 cDNA and construction of the targeting vectors, Drs J.Bartek, J.-M.Buerstedde and C.Smythe for generously providing reagents and advice, and Professor J.A.Wyke and Dr A.M.Carr for constructive comments on the manuscript. This work was supported by the Association for International Cancer Research (AICR; G.Z.), Cancer Research UK (CR-UK; D.A.F.G.). M.D.R. is supported by a Beatson Institute Research Studentship.
References
- Ajiro K., Yoda,K., Utsumi,K. and Nishikawa,Y. (1996) Alteration of cell cycle-dependent histone phosphorylations by okadaic acid. Induction of mitosis-specific H3 phosphorylation and chromatin condensation in mammalian interphase cells. J. Biol. Chem., 271, 13197–13201. [DOI] [PubMed] [Google Scholar]
- al-Khodairy F., Fotou,E., Sheldrick,K.S., Griffiths,D.J., Lehmann,A.R. and Carr,A.M. (1994) Identification and characterization of new elements involved in checkpoint and feedback controls in fission yeast. Mol. Biol. Cell, 5, 147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aten J.A., Bakker,P.J., Stap,J., Boschman,G.A. and Veenhof,C.H. (1992) DNA double labelling with IdUrd and CldUrd for spatial and temporal analysis of cell proliferation and DNA replication. Histochem. J., 24, 251–259. [DOI] [PubMed] [Google Scholar]
- Baber-Furnari B.A., Rhind,N., Boddy,M.N., Shanahan,P., Lopez-Girona,A. and Russell,P. (2000) Regulation of mitotic inhibitor Mik1 helps to enforce the DNA damage checkpoint. Mol. Biol. Cell, 11, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy M.N., Furnari,B., Mondesert,O. and Russell,P. (1998) Replication checkpoint enforced by kinases Cds1 and Chk1. Science, 280, 909–912. [DOI] [PubMed] [Google Scholar]
- Buerstedde J.M. and Takeda,S. (1991) Increased ratio of targeted to random integration after transfection of chicken B cell lines. Cell, 67, 179–188. [DOI] [PubMed] [Google Scholar]
- Busby E.C., Leistritz,D.F., Abraham,R.T., Karnitz,L.M. and Sarkaria,J.N. (2000) The radiosensitizing agent 7-hydroxystaurosporine (UCN-01) inhibits the DNA damage checkpoint kinase hChk1. Cancer Res., 60, 2108–2112. [PubMed] [Google Scholar]
- Carr A.M. (2002) Checking that replication breakdown is not terminal. Science, 297, 557–558. [DOI] [PubMed] [Google Scholar]
- Chen P. et al. (2000) The 1.7 Å crystal structure of human cell cycle checkpoint kinase Chk1: implications for Chk1 regulation. Cell, 100, 681–692. [DOI] [PubMed] [Google Scholar]
- Christensen P.U., Bentley,N.J., Martinho,R.G., Nielsen,O. and Carr,A.M. (2000) Mik1 levels accumulate in S phase and may mediate an intrinsic link between S phase and mitosis. Proc. Natl Acad. Sci. USA, 97, 2579–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark W. and Gillespie,D.A. (1997) Transformation by v-Jun prevents cell cycle exit and promotes apoptosis in the absence of serum growth factors. Cell Growth Differ., 8, 371–380. [PubMed] [Google Scholar]
- Clark W., Black,E.J., MacLaren,A., Kruse,U., LaThangue,N., Vogt,P.K. and Gillespie,D.A. (2000) v-Jun overrides the mitogen dependence of S phase entry by deregulating retinoblastoma protein phosphorylation and E2F–pocket protein interactions as a consequence of enhanced cyclin E-cdk2 catalytic activity. Mol. Cell. Biol., 20, 2529–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova D.S. and Gilbert,D.M. (1999) The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Mol. Cell, 4, 983–993. [DOI] [PubMed] [Google Scholar]
- Edgar B. (1995) Diversification of cell cycle controls in developing embryos. Curr. Opin. Cell Biol., 7, 815–824. [DOI] [PubMed] [Google Scholar]
- Enoch T., Carr,A.M. and Nurse,P. (1992) Fission yeast genes involved in coupling mitosis to completion of DNA replication. Genes Dev., 6, 2035–2046. [DOI] [PubMed] [Google Scholar]
- Feijoo C., Hall-Jackson,C., Wu,R., Jenkins,D., Leitch,J., Gilbert,D.M. and Smythe,C. (2001) Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J. Cell Biol., 154, 913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty P., Campbell,S.D., Abu-Shumays,R., Phalle,B.S., Yu,K.R., Uy,G.L., Goldberg,M.L. and Sullivan,W. (1997) The Drosophila grapes gene is related to checkpoint gene chk1/rad27 and is required for late syncytial division fidelity. Curr. Biol., 7, 418–426. [DOI] [PubMed] [Google Scholar]
- Furnari B., Blasina,A., Boddy,M.N., McGowan,C.H. and Russell,P. (1999) Cdc25 inhibited in vivo and in vitro by checkpoint kinases Cds1 and Chk1. Mol. Biol. Cell, 10, 833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert D.M. (2001) Making sense of eukaryotic DNA replication origins. Science, 294, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z., Kumagai,A., Wang,S.X. and Dunphy,W.G. (2000) Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev., 14, 2745–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna K.K. and Jackson,S.P. (2001) DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet., 27, 247–254. [DOI] [PubMed] [Google Scholar]
- Kim S.M. and Huberman,J.A. (2001) Regulation of replication timing in fission yeast. EMBO J., 20, 6115–6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A., Guo,Z., Emami,K.H., Wang,S.X. and Dunphy,W.G. (1998) The Xenopus Chk1 protein kinase mediates a caffeine-sensitive pathway of checkpoint control in cell-free extracts. J. Cell Biol., 142, 1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay H.D., Griffiths,D.J., Edwards,R.J., Christensen,P.U., Murray,J.M., Osman,F., Walworth,N. and Carr,A.M. (1998) S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes Dev., 12, 382–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q. et al. (2000) Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev., 14, 1448–1459. [PMC free article] [PubMed] [Google Scholar]
- Lopez-Girona A., Furnari,B., Mondesert,O. and Russell,P. (1999) Nuclear localization of Cdc25 is regulated by DNA damage and a 14-3-3 protein. Nature, 397, 172–175. [DOI] [PubMed] [Google Scholar]
- Lopez-Girona A., Tanaka,K., Chen,X.B., Baber,B.A., McGowan,C.H. and Russell,P. (2001) Serine-345 is required for Rad3-dependent phosphorylation and function of checkpoint kinase Chk1 in fission yeast. Proc. Natl Acad. Sci. USA, 98, 11289–11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S., Huang,M. and Elledge,S.J. (1998) Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science, 282, 1893–1897. [DOI] [PubMed] [Google Scholar]
- Michael W.M., Ott,R., Fanning,E. and Newport,J. (2000) Activation of the DNA replication checkpoint through RNA synthesis by primase. Science, 289, 2133–2137. [DOI] [PubMed] [Google Scholar]
- Murakami H. and Okayama,H. (1995) A kinase from fission yeast responsible for blocking mitosis in S phase. Nature, 374, 817–819. [DOI] [PubMed] [Google Scholar]
- O’Connell M.J., Walworth,N.C. and Carr,A.M. (2000) The G2-phase DNA-damage checkpoint. Trends Cell Biol., 10, 296–303. [DOI] [PubMed] [Google Scholar]
- Rhind N. and Russell,P. (1998) Tyrosine phosphorylation of cdc2 is required for the replication checkpoint in Schizosaccharomyces pombe. Mol. Cell. Biol., 18, 3782–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N. and Russell,P. (2000) Chk1 and Cds1: linchpins of the DNA damage and replication checkpoint pathways. J. Cell Sci., 113, 3889–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N., Furnari,B. and Russell,P. (1997) Cdc2 tyrosine phosphorylation is required for the DNA damage checkpoint in fission yeast. Genes Dev., 11, 504–511. [DOI] [PubMed] [Google Scholar]
- Sanchez Y., Desany,B.A., Jones,W.J., Liu,Q., Wang,B. and Elledge,S.J. (1996) Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science, 271, 357–360. [DOI] [PubMed] [Google Scholar]
- Santocanale C. and Diffley,J.F. (1998) A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature, 395, 615–618. [DOI] [PubMed] [Google Scholar]
- Senderowicz A.M. (2000) Small molecule modulators of cyclin-dependent kinases for cancer therapy. Oncogene, 19, 6600–6606. [DOI] [PubMed] [Google Scholar]
- Takai H., Tominaga,K., Motoyama,N., Minamishima,Y.A., Nagahama,H., Tsukiyama,T., Ikeda,K., Nakayama,K. and Nakanishi,M. (2000) Aberrant cell cycle checkpoint function and early embryonic death in Chk1(–/–) mice. Genes Dev., 14, 1439–1447. [PMC free article] [PubMed] [Google Scholar]
- Takao N. et al. (1999) Disruption of ATM in p53-null cells causes multiple functional abnormalities in cellular response to ionizing radiation. Oncogene, 18, 7002–7009. [DOI] [PubMed] [Google Scholar]
- Takata M., Sasaki,M.S., Sonoda,E., Morrison,C., Hashimoto,M., Utsumi,H., Yamaguchi-Iwai,Y., Shinohara,A. and Takeda,S. (1998) Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J., 17, 5497–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth N.C. and Bernards,R. (1996) rad-dependent response of the chk1-encoded protein kinase at the DNA damage checkpoint. Science, 271, 353–356. [DOI] [PubMed] [Google Scholar]
- Walworth N., Davey,S. and Beach,D. (1993) Fission yeast chk1 protein kinase links the rad checkpoint pathway to cdc2. Nature, 363, 368–371. [DOI] [PubMed] [Google Scholar]
- Xu B., Kim,S.T., Lim,D.S. and Kastan,M.B. (2002) Two molecularly distinct G(2)/M checkpoints are induced by ionizing irradiation. Mol. Cell. Biol., 22, 1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y., Forbes,K.C., Wu,Z., Moreno,S., Piwnica-Worms,H. and Enoch,T. (1998) Replication checkpoint requires phosphorylation of the phosphatase Cdc25 by Cds1 or Chk1. Nature, 395, 507–510. [DOI] [PubMed] [Google Scholar]
- Zhao H. and Piwnica-Worms,H. (2001) ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol., 21, 4129–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]