

Abstract
Clearance of hepatitis C virus (HCV) infection in humans and chimpanzees is thought to be associated with the induction of strong T-cell responses. We studied four chimpanzees infected with HCV derived from an infectious full-length HCV genotype 1b cDNA. Two of the chimpanzees cleared the infection to undetectable levels for more than 12 months of follow-up; the other two became persistently infected. Detailed analyses of HCV-specific immune responses were performed during the courses of infection in these chimpanzees. Only weak and transient T helper responses were detected during the acute phase in all four chimpanzees. A comparison of the frequency of gamma interferon (IFN-γ)-producing CD4+ and CD8+ T cells in peripheral blood by ELISpot assay did not reveal any correlation between viral clearance and T-cell responses. In addition, analyses of IFN-γ, IFN-α, and interleukin-4 mRNA levels in liver biopsies, presumably indicative of intrahepatic T-cell responses, revealed no distinct pattern in these chimpanzees with respect to infection outcome. The present study suggests that the outcome of HCV infection in chimpanzees is not necessarily attributable to HCV sequence variation and that chimpanzees may recover from HCV infection by mechanisms other than the induction of readily detectable HCV-specific T-cell responses.
Hepatitis C virus (HCV) belongs to the genus Hepacivirus of the family Flaviviridae. Worldwide, an estimated 170 million people are infected with HCV, and it is the most common reason for liver transplantation (1). The most likely outcome of infection is chronicity, which is thought to be attributable to the ability of the virus to rapidly mutate and outpace the host immune response. HCV exists as six different genotypes and more than 30 subtypes and is highly heterogeneous between and within isolates, thus hampering studies on sequence-function relationships.
The outcome of infection with HCV is thought to be determined by the initial character and vigor of the host immune response to the infection, which in turn may be determined by factors such as viral species, viral load, and route of entry. HCV clearance in humans is associated with an early, strong cellular immune response against multiple viral epitopes, and in particular against NS3 (6, 7, 27). After clearance, both CD4+ and CD8+ responses are maintained (26). Loss of a CD4+ response can result in recurrence of HCV infection (10). A nonsustained and/or dysfunctional HCV-specific CD8+ response has been implicated in HCV persistence (11, 19).
Because the majority of patients do not present with acute HCV infection, the early cytotoxic-T-lymphocyte (CTL) response in humans has been difficult to characterize fully. At present, the only animal model for HCV is the chimpanzee, although it has been possible to infect mice harboring chimeric mouse-human livers (23). In chimpanzees, viral clearance has been observed in >60% of infections (2). Like humans, clearance was not associated with a humoral response against the envelope proteins. Other studies have shown clearance in chimpanzees to be associated with a relatively high number of intrahepatic CTL specificities occurring synchronously and early in infection (5). The failure to clear HCV infection may be determined in part through the acquisition of mutations in epitopes recognized by CTLs (8). Such mutations may appear early in infection and persist for years in the viral population.
In order to fully characterize the host response to HCV infection and relate it to the sequence of the viral population, several studies have developed full-length HCV clones, mostly of genotypes 1a and 1b, whose RNA transcripts have demonstrated infectivity after intrahepatic inoculation into chimpanzees (3, 15, 16, 18, 22, 28-31). Such studies have been useful for examining the long-term immune response of the host from the initial point of infection and for examining the molecular evolution of HCV preparations with a known genomic sequence. However, despite these studies, the precise mechanisms of HCV persistence and clearance in the chimpanzee model are still unknown. We previously reported the construction of an infectious HCV genotype 1b clone and showed that it caused persistent infection in two chimpanzees (X0142 and X0234) (28). In this report, we show that the same cloned source of HCV was cleared to undetectable levels in two other chimpanzees (X0132 and X0190). Regardless of the outcome of infection, only a weak and transient T helper response was detected. In addition, there was no apparent correlation between the frequency of circulating gamma interferon (IFN-γ)-producing CD4+ or CD8+ T cells or intrahepatic IFN-α and IFN-γ production and viral clearance. Thus, it is possible that mechanisms other than the induction of T cells may contribute to HCV clearance, at least in the chimpanzee model.
MATERIALS AND METHODS
Full-length HCV plasmid constructs.
The construction of HCV-CG1b was described previously (28). This construct contains the HCV J strain structural region in a BK strain backbone (Fig. 1). HCV-J4CG1 was generated by inserting the J strain structural region between the NruI (nucleotide position 271) and the RsrII (position 3030) sites into the J4 strain backbone (31). HCV-J4CG2 was generated by inserting the J strain structural region between ClaI (position 709) and BssHI (position 2826) into the J4 backbone. HCV-p90/H8 is a derivative of an H77 infectious clone (16) containing the H79 hypervariable region (9).
FIG. 1.

Diagrams of the full-length HCV clones used in these experiments. A gene map of HCV is shown at the top. Numbers refer to nucleotide position at the start of each gene. Untranslated regions are indicated. Regions within the four cloned genomes are shaded according to the HCV strain from which they were derived. Strains are indicated above each region. J, BK, and J4 are of genotype 1b, and H, H77, and H79 are of 1a (Hutchinson) genotype. Restriction sites used to construct the 1b genotype chimeras are shown.
Virus preparation.
HCV was generated from HCV-CG1b by intrahepatic inoculation of chimpanzees with ∼0.5-mg RNA transcripts as described previously (28). Sera were collected at weekly intervals and HCV RNA titers determined by the TaqMan assay (see below). Virus from chimpanzee X0142 at week 2 postinoculation was inoculated into chimpanzees X0234 and X0132. The HCV RNA in this inoculum did not contain any nucleotide differences with respect to the original HCV-CG1b clone (28). To produce a large preparation of virus, chimpanzee X0234 was plasmapheresed at weeks 5 and 7. The titer of HCV RNA in the pooled plasma was 5 × 104 genomes/ml as determined by the TaqMan assay. Sequence analysis of this HCV RNA revealed a single mutation compared with HCV-CG1b in the NS5B gene (A7840G), which caused a lysine-to-arginine substitution at position 2500 of the polyprotein. This plasma pool was the source of the inoculum for chimpanzee X0190.
Chimpanzees.
Chimpanzees were maintained at the Southwest Foundation for Biomedical Research, an Association for Assessment and Accreditation of Laboratory Animal and Care-accredited facility, and the study protocol was approved by the Institutional Animal Care and Use Committee at the Foundation and by the Interagency Animal Model Committee at the National Institutes of Health.
After intrahepatic or intravenous inoculation with HCV RNA or HCV, respectively, weekly serum samples from chimpanzees were monitored for alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels by standard assays. HCV RNA was detected by reverse transcription-PCR (RT-PCR; Cobas Amplicor; Roche Molecular Systems, Pleasanton, Calif.) and with anti-HCV antibodies by enzyme immunoassay (EIA 2.0; Abbott Laboratories, Abbott Park, Ill.).
Quantitation of HCV RNA.
HCV RNA was extracted from 100-μl samples of serum by using Trizol (Invitrogen, Carlsbad, Calif.). Quantitative RT-PCR was performed by TaqMan assay (Applied Biosystems, Foster City, Calif.) as described previously (28).
T-cell proliferation assay.
T-cell proliferation was assayed as described previously (26). Peripheral blood mononuclear cells (PBMC) were stimulated with HCV genotype 1b proteins (1 μg/ml): core, helicase, NS3, NS4, NS5A, and NS5B (Mikrogen, Munich, Germany). As controls, separate cultures stimulated with phytohemagglutinin (1 μg/ml; Murex Biotech Ltd., Dartford, England) or medium alone were labeled with [3H]thymidine. The stimulation index (SI) was calculated as the ratio of the mean counts per minute incorporated into the cellular DNA of four replicate cultures in the presence of antigen over that found in the presence of control buffer. An SI of >4.0 was considered positive.
IFN-γ ELISpot assays.
Ninety-six-well plates (Millititer; Millipore/ELISpot, Bedford, Mass.) were coated overnight at 4°C with the primary antibody against human IFN-γ (Endogen, Woburn, Mass.), washed four times with sterile phosphate-buffered saline, and blocked for 1 h at 25°C with RPMI medium and 1% bovine serum albumin (Sigma). Cryopreserved PBMC were thawed and cultured at 140,000 cells/well (the optimal cell concentration as determined by limiting dilution for this assay) in RPMI 1640, 5% fetal bovine serum, and 2 mM l- glutamine with individual HCV proteins (1 μg/ml) or peptides at 10 μg/ml (Table 1) (17, 25). After 30 h, the plates were washed seven times and incubated overnight with 100 μl of biotin-conjugated secondary antibody against IFN-γ (Endogen). After being washed four times and incubated 2 h with streptavidin-alkaline phosphatase (1:2,000 dilution; Dako, Carpinteria, Calif.), the plates were washed again four times with phosphate-buffered saline and developed with freshly prepared NBT/BCIP solution (nitroblue tetrazolium-5-bromo-4-chloro-3-indolylphosphate; Bio-Rad, Richmond, Calif.). The reaction was stopped by rinsing the contents with distilled water, and the spots were counted by using an KS ELISpot-Axioplan 2I (Zeiss, Thornwood, N.Y.).
TABLE 1.
Peptides used to stimulate T cells from chimpanzees
| Peptide group and proteina | Amino acid positionb | Sequence | MHC restrictionc |
|---|---|---|---|
| Group 1 | |||
| Core | 41-49 | GPRLGVRAT | Patr-B13 |
| E2 | 542-550 | TRPPLGNWF | Patr-B13 |
| NS3 | 1357-1365 | VPHPNIEEV | Patr-B13 |
| NS3 | 1379-1386 | IPFYGKAI | Patr-B13 |
| NS3 | 1635-1643 | VTLTHPITK | Patr-A5 |
| NS4B | 1858-1867 | GVAGALVAFK | Patr-A5 |
| NS5B | 2509-2517 | SLTPPHSAK | Patr-A5 |
| NS5B | 2587-2595 | RVCEKMALY | Patr-A5 |
| Group 2 | |||
| Core | 35-44 | YLLPRRGPRL | Patr-B*0101 |
| Core | 131-140 | ADLMGYIPLV | Patr-B*0101 |
| Core | 167-176 | GNLPGCSFSI | Patr-B*0101 |
| E2 | 632-641 | RMYVGGVEHR | Patr-A5 |
| NS3 | 1073-1081 | CVNGVCWTV | Patr-B*0101 |
| NS3 | 1406-1415 | KLVALGINAV | Patr-B*0101 |
| NS3 | 1444-1452 | FTGDFDSVI | Patr-B*0101 |
| NS3 | 1635-1643 | VTLTHPITK | Patr-A5 |
| NS4B | 1807-1816 | LLFNILGGWV | Patr-B*0101 |
| NS4B | 1858-1867 | GVAGALVAFK | Patr-A5 |
| NS4B | 1859-1867 | VAGALVAFK | Patr-A5 |
| NS5B | 2726-2734 | GLQDCTMLV | Patr-B*0101 |
Group 1, peptides used to stimulate T cells from animal X0132; group 2, peptides used to stimulate T cells from animals X0142 and X0234.
Refers to the location in the HCV-CG1b polyprotein (28).
Based on sequence comparison between published Patr and HLA alleles and/or direct Patr restriction testing (25).
Quantification of cytokine mRNA.
RNA was extracted from liver biopsies with Trizol reagent (Invitrogen) and Dounce homogenization. Relative mRNA levels for IFN-γ and interleukin-4 (IL-4) in chimpanzee liver biopsies were determined by using predeveloped TaqMan assay reagent kits for human targets (Applied Biosystems). A specific primer-probe set was designed for measurement of relative mRNA levels of IFN-α. A primer-probe set for the human GAPDH (glyceraldehyde-3-phosphate dehydrogenase) endogenous control was also obtained from Applied Biosystems and used according to the manufacturer's instructions. Reverse transcription of RNA (1 μg) isolated from chimpanzee liver biopsies was carried out by using the first-strand cDNA synthesis kit (Pharmacia) according to the manufacturer's instructions, with 10 pg of random hexamers in a 15-μl reaction. One or three microliters was used to test for IFN-γ, IL-4, and GAPDH in separate tubes. Relative mRNA quantification was calculated by the comparative cycle threshold (CT) method by using the arithmetic formula 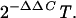 This value calculates the amount of target normalized to an endogenous reference (GAPDH) and relative to a calibrator (a liver biopsy taken prior to challenge) as described in the PE Applied Biosystems User Bulletin #2. Briefly, ΔCT = the difference between threshold cycles for target (cytokine) mRNA and endogenous reference (GAPDH) mRNA. ΔΔCT is the difference between the ΔCT of the target mRNA postchallenge and the ΔCT of the target mRNA prechallenge.
This value calculates the amount of target normalized to an endogenous reference (GAPDH) and relative to a calibrator (a liver biopsy taken prior to challenge) as described in the PE Applied Biosystems User Bulletin #2. Briefly, ΔCT = the difference between threshold cycles for target (cytokine) mRNA and endogenous reference (GAPDH) mRNA. ΔΔCT is the difference between the ΔCT of the target mRNA postchallenge and the ΔCT of the target mRNA prechallenge.
RESULTS
Construction of chimeric HCV 1b cDNA clones.
We previously reported the construction of an infectious HCV 1b clone, pHCV-CG1b (28). This clone consists of a nonstructural backbone derived from BK strain cDNA and structural genes derived from J strain cDNA. We were interested in determining whether an alternate nonstructural backbone would make a difference to the infectivity or virulence of RNA transcripts generated from pHCV-CG1b. To this end, we generated two additional HCV-1b clones—pHCV-J4CG1 and pHCV-J4CG2—containing the J strain structural genes and the infectious J4 strain clone as backbone (Fig. 1) (31). These two clones differ only by the sequences flanking the J strain structural region. Thus, in pHCV-J4CG1 the J strain region includes some sequences from the 5′-untranslated region and the NS2 gene, whereas in pHCV-J4CG2 there are no J strain sequences outside the structural genes. The reason for generating the latter clone was to investigate the importance of the few nucleotide differences between the J strain and J4 strain in the sequences flanking the structural region. Of particular interest were nucleotides 340 and 350, which differ between the J strain and other published infectious clones (28). These nucleotides reside either side of the initiation codon and within stem-loop IV of the HCV internal ribosome entry site (14).
Inoculation and infection course of chimpanzee X0132.
Chimpanzee X0132 was inoculated with RNA transcripts from four different full-length constructs and monitored for signs of HCV infection (Fig. 2A). Initially, RNA was transcribed from HCV-CG1b and inoculated intrahepatically into this chimpanzee. HCV RNA was detected by RT-PCR in serum drawn at week 4, but sera from all other time points were negative by this assay. There was no evidence of hepatitis or anti-HCV antibody response. X0132 was then inoculated with RNA transcripts from the HCV-J4CG1 construct. One positive PCR result was observed 5 weeks postinoculation, but all other samples were negative. HCV RNA was subsequently transcribed from a third construct, pHCV-J4CG2. No HCV RNA was detected after inoculation of this third genotype 1b chimeric clone. To address the possibility that a different HCV genotype would behave differently in chimpanzee X0132, we performed a further inoculation with an infectious clone of genotype 1a, HCV-p90/H8, derived from the original H77 infectious clone (16). After this inoculation, serum samples taken at 5, 6, and 7 weeks postinoculation were positive for HCV RNA by PCR. This viremia was transient, disappearing by week 8. The quantity of HCV RNA in all of the samples that were positive by PCR during the period of the four RNA inoculations was less than 5 × 102 genomes/ml (the limit of detection by TaqMan PCR). Partial sequencing analyses demonstrated that this transient HCV RNA in the serum after the first, second, and fourth inoculations was indeed derived from each of the respective inoculating RNAs. There was no evidence of serum ALT elevation or HCV-specific antibodies in chimpanzee X0132 at any time during the course of these inoculations. In addition, no histological changes were observed in liver biopsies obtained 6, 11, and 15 weeks after the fourth inoculation.
FIG. 2.

Inoculation history and follow-up of chimpanzee X132. (A) Large arrows denote the time of intrahepatic transfections with various HCV RNAs or intravenous inoculation with HCV-CG1b virus. A 102 dilution of serum from X0142 (week 2; HCV RNA titer = 104 genomes/ml) was used as challenge virus, followed by a 101 dilution 5 weeks later (week 87 on chart). Solid bars indicate HCV RNA titers as determined by TaqMan assay; the shaded area indicates the ALT level (normal range for chimpanzees is 21 to 55 U/liter). Above the chart are shown the histology results of the biopsies taken at the times indicated by the small arrows. Intrahepatic IFN-α, IFN-γ, and IL-4 levels determined by TaqMan assay from these biopsies are shown as ratios relative to a prechallenge sample as described in the Materials and Methods. Also indicated are the HCV RT-PCR data (sensitivity = 100 genomes/ml) and the anti-HCV response, as determined by EIA 2.0. (B) T-cell proliferation assay results for various HCV antigens. An SI of >4.0 (dotted line) was considered positive. As positive controls, T cells were stimulated nonspecifically with phytohemagglutinin (SI = 12 to 80). (C and D) IFN-γ ELISpot assay of peripheral blood T-cell responses. SFU, spot-forming units. The numbers of antigen-specific spots minus the background are indicated for various antigens (C) or Patr-restricted peptides (D) at several time points postinoculation. The eight peptides used in the present study are indicated in Table 1; bars are ordered from left to right according to genome location. SFU, spot-forming units. The number of spot-forming units for each antigen or peptide is shown after subtraction of the number of spots in the absence of antigen or peptide. A finding of more than 10 SFU (dotted line) was considered positive.
The failure to achieve robust infection of X0132 by intrahepatic transfection of transcribed RNA may have been caused by some unique feature of this animal that enabled it to rapidly clear infection by HCV. To test this hypothesis we inoculated X0132 with HCV from X0142 serum taken at week 2. Initially, we used 102 genomes, which had been shown to cause infection in another chimpanzee X0234 (28). After this inoculation, there was no evidence of HCV infection by PCR. We then inoculated with 103 genomes and the chimpanzee became positive for HCV RNA after 1 week, with titers rising to the order of 104 to 105 genomes/ml (Fig. 2A). From weeks 90 to 94 (weeks 3 to 7 postinoculation), ALT levels were elevated. Anti-HCV antibodies were detected in serum from week 93. This antibody response was weak, with titers of less than 1:100 as assessed by endpoint dilution assays performed at various time points after seroconversion (HCV-infected humans have anti-HCV titers typically greater than 1:1,000).
We studied the immune response of X0132 to the different HCV inocula further by performing T-cell proliferation and ELISpot assays at various time points postinoculation. After inoculation with RNA transcripts from the four different HCV clones, the T helper proliferation assay was weakly positive at a number of time points (Fig. 2B). After the first inoculation with RNA from HCV-CG1b, the T helper proliferation assay was positive at week 3 postinoculation for NS4 (SI = 5.9) and at week 4 postinoculation for NS3 (SI = 6.7), NS4 (SI = 4.5), and NS5A (SI = 10.6). After inoculation with HCV-J4CG1 and concurrent with the positive PCR result for serum HCV RNA, there was a mild increase in the stimulation indices for core (SI = 5.0), helicase (SI =4.6), and NS4 (SI = 6.5). After the third inoculation with RNA transcripts from HCV-J4CG2, the T helper proliferation assays were negative for all antigens tested. After the fourth inoculation with RNA transcripts from an HCV genotype 1a clone, T helper proliferation assays were again weakly positive for some antigens at weeks 39 and 49 (weeks 8 and 18 postinoculation). (SI at week 39, helicase = 7.0; SI at week 49, NS3 = 9.6, NS4 = 5.4, NS5A = 9.1, and NS5B = 7.9). Analysis of culture medium in these positive proliferation assays showed production of IFN-γ but not IL-4 (data not shown). After challenge of X0132 with HCV-CG1b virus derived from chimpanzee X0142, a weak and transient T helper response was again observed (Fig. 2B). At week 90 (week 3 postchallenge), a T-cell proliferation assay was positive for NS5A (SI = 5.5) and at week 104, this assay was positive for helicase (SI = 6.2) and NS5B (SI = 11.1). Again, IFN-γ, but not IL-4, was detected in these assays.
ELISpot assays were performed at various time points with HCV antigens for stimulation (Fig. 2C) or CTL epitopes reported for the Patr haplotypes of this chimpanzee (Table 1 and Fig. 2D) (17, 25). The frequency of IFN-γ-producing cells was not increased in any of the samples tested after the four inoculations with HCV RNA. However, after challenge of X0132 with HCV-CG1b virus there was an increase in IFN-γ spots at weeks 93, 97, 99, and 101 (weeks 6, 10, 12, and 14 postchallenge) with various HCV antigens used for stimulation (Fig. 2C) and at weeks 93, 97, and 99 with a panel of eight peptides that map to known CTL epitopes (Table 1 and Fig. 2D). These time points correspond to the final weeks of detectable viremia and the first weeks after viral clearance.
Inoculation of and infection course of chimpanzee X190.
Chimpanzee X0190 was inoculated intravenously with week 5 serum HCV derived from chimpanzee X0234 (titer = 5 × 104 genomes/ml as determined by the TaqMan assay). Sequence analysis of this inoculum showed it to be identical to HCV-CG1b except for a single nucleotide change in NS5B that caused a conserved single amino acid change (K2500R). Initially, X0190 was inoculated with a 1 in 10,000 dilution of the serum. This did not cause a viremia detectable by RT-PCR (sensitivity, 102 genomes) within 5 weeks of inoculation, so we subsequently inoculated the animals at a 1-in-1,000 dilution, equivalent to ca. 50 HCV genomes. The outcome of this inoculation is shown in Fig. 3A.
FIG. 3.

Inoculation history and follow-up of chimpanzee X0190. (A) X0190 was inoculated intravenously with a 104 dilution of serum from X0234 (HCV RNA titer = 5 × 104 genomes/ml), followed 5 weeks later by a 103 dilution of this serum, shown on the chart as week 0. Solid bars indicate the HCV RNA titers in serum; the shaded area indicates the ALT level in serum. Above the chart are shown the histology results from the four biopsies, denoted by arrows at weeks 4, 7, 10 and 13; intrahepatic IFN-α, IFN-γ, and IL-4 levels relative to GAPDH RNA as determined by TaqMan assay; qualitative HCV RT-PCR results (sensitivity = 100 genomes/ml); and anti-HCV EIA 2.0 data. (B) T helper proliferation data for several HCV antigens. An SI of >4.0 (dotted line) was considered positive. (C) IFN-γ ELISpot assay of peripheral blood T-cell responses. SFU, spot-forming units. The number of spot-forming units for each antigen or peptide is shown after subtraction of the number of spots in the absence of antigen or peptide. A finding of more than 10 spot-forming units (dotted line) was considered positive.
HCV RNA was detected in serum from 1 week after inoculation, rising to a maximum titer of 3 × 104 genomes/ml as determined by TaqMan assay. The viremia lasted until week 13, when it became undetectable by RT-PCR or TaqMan assay. HCV RNA was not detected in the 12-month period after clearance. An anti-HCV antibody response was not detected at any time during follow-up, and ALT levels remained within normal limits. Liver biopsy during the viremia revealed evidence of mild hepatitis at week 7 postinoculation. Other biopsies obtained at weeks 4, 10, and 13 showed normal histology.
T helper proliferation assays (Fig. 3B) showed a weak and transient response against some HCV antigens at weeks 4, 12, and 16 postinoculation (SI values at week 4, NS3 = 5.4; at week 12, helicase = 6.8 and NS4 = 4.3; and at week 16, NS3 = 4.1). ELISpot analysis of peripheral blood T-cell responses showed an increase in frequency of IFN-γ producing cells at weeks 6, 12, and 14 postinoculation, with various HCV antigens for stimulation (Fig. 3C). However, there was no correlation between these increases and viral clearance, which occurred at week 13. Because the Patr haplotype information was not available for this chimpanzee, the ELISpot analysis for CD8+ response was not performed.
Inoculation and infection course of chimpanzees X0142 and X0234.
Chimpanzee X0142 was inoculated intrahepatically with RNA transcripts from HCV-CG1b as described previously (28). Chimpanzee X0234 was inoculated intravenously with the week 2 postinoculation serum from X0142. The courses of infection for both chimpanzees have been reported (28). Both animals developed persistent infection during 26 months (X0142) and 17 months (X0234) of follow-up. Anti-HCV antibodies were detected in X0142 from week 24 and in X0234 from week 31, but the titers were less than 1:100 throughout the course of follow-up.
Chimpanzees X0142 and X0234 were persistently infected with HCV-CG1b, and both developed only a weak, transient T helper response as described previously (28). To characterize the type of T-cell response (Th1 versus Th2), cell culture medium from the T helper proliferation assay was analyzed for IFN-γ and IL-4 levels. IFN-γ was produced in the range of 500 to 4,000 pg/ml, whereas IL-4 was not detected in these samples.
Figure 4 and 5 show further analyses of the immune response in X0142 and X0234. An IFN-γ ELISpot assay was performed with HCV structural and nonstructural antigens to analyze CD4+ T-cell responses and a panel of 12 peptides mapping to known CTL epitopes (Table 1) (17, 25) to analyze CD8+-T-cell responses. In X0142 (Fig. 4), a significant number of spots was noted with PBMC isolated at week 36 (Fig. 4B), a finding corresponding to the increase in SI for various antigens in the T helper proliferation assay (Fig. 4A). CD8+ responses were observed with the peptide panel at weeks 36, 60, and 70 to 75, with the highest activities against some of the peptides at week 36, which corresponded to the positive CD4+ responses (Fig. 4C). Similarly, in X0234 the IFN-γ ELISpot assay with various HCV antigens for stimulation was positive at weeks 5, 7, and 11 (Fig. 5B), coinciding with the time that the T helper proliferation assay was weakly positive (Fig. 5A). The assay was also positive at weeks 5 and 7 by using the CD8 specific peptide panel (Fig. 5C). Sequence analysis of serum HCV RNA from both X0142 and X0234, reported previously (28), revealed two mutations arising de novo within these peptide sequences in the HCV RNAs derived from X0142 from weeks 60 and 78 postinoculation. One of these was a D1447N substitution, potentially affecting the NS3-1444 peptide binding; the other was V1635I, potentially affecting NS3-1635. The ELISpot assays with the NS-1444 peptide were persistently negative during the course of infection, whereas the assays with the NS-1635 peptide were positive at week 36 and then at weeks 70 and 75.
FIG. 4.

T-cell responses of chimpanzee X0142 after inoculation with HCV-CG1b RNA. Chimpanzee X0142 was inoculated with HCV-CG1b RNA as described previously (28) and became persistently infected with HCV from week 1 postinoculation. (A) T-cell proliferative responses for several HCV antigens. An SI of >4.0 was considered positive. Above the chart are indicated the biopsies (arrows) obtained at weeks 6, 8, 12, 18, 66, and 72 and the intrahepatic IFN-α, IFN-γ, and IL-4 levels for these biopsies relative to GAPDH RNA, as determined by TaqMan assay. Also indicated are the qualitative HCV RNA data. (B and C) IFN-γ ELISpot assay results with various HCV antigens (B) or a panel of 12 peptides that map to known CTL epitopes (C) for stimulation. The peptides used in the present study are indicated in Table 1; bars are ordered from left to right according to genome location. SFU, spot-forming units. The number of spot-forming units for each antigen or peptide is shown after subtraction of the number of spots in the absence of antigen or peptide. A finding of more than 10 spot-forming units (dotted line) was considered positive.
FIG. 5.

T-cell responses of chimpanzee X0234 after inoculation with HCV-CG1b virus. Chimpanzee X0234 was inoculated with serum from week 2 postinoculation of chimpanzee X0142 (28). (A) T-cell proliferative responses for several HCV antigens. An SI of >4.0 was considered positive. Above the chart are indicated biopsy results obtained preinoculation and at weeks 6, 8, 10, and 12 postinoculation, and the intrahepatic IFN-α, IFN-γ, and IL-4 levels for these biopsies are indicated as a ratio of cytokine RNA to GAPDH RNA, as determined by TaqMan assay. Also shown are qualitative HCV RNA data. (B and C) IFN-γ ELISpot assay with various HCV antigens (B) or a panel of 12 peptides that map to known CTL epitopes (C) for stimulation. The peptides used in the present study are indicated in Table 1; bars are ordered from left to right according to genome location. SFU, spot-forming units. The number of spot-forming units for each antigen or peptide is shown after subtraction of the number of spots in the absence of antigen or peptide. A finding of more than 10 SFU (dotted line) was considered positive.
Comparison of intrahepatic IFN-α, IFN-γ, and IL-4 RNA levels in chimpanzees.
As an indirect measure of intrahepatic cellular immune response, IFN-α, IFN-γ, and IL-4 RNA quantities were determined in liver biopsies from the four chimpanzees in the present study. Total RNA was extracted from liver biopsies and IFN-α, IFN-γ, and IL-4 RNA was measured by the TaqMan assay and normalized among samples by using the GAPDH mRNA values. Intrahepatic cytokine levels are reported relative to a prechallenge sample. In chimpanzee X0132, an increase in IFN-α level to 9.0 was noted after the inoculation with HCV-p90/H8 RNA, and an increase to 10.3 occurred after inoculation with HCV-CG1b virus. There was no increase in IFN-γ levels in biopsies taken after the fourth inoculation with HCV-p90/H8 RNA (Fig. 2A), but the relative level increased to 11.9 during the viremia caused by the challenge inoculation with HCV-CG1b virus. In chimpanzee X0190, biopsies taken at weeks 4, 7, and 13 postinoculation showed a mild increase to 4.8 in intrahepatic IFN-γ at week 10 only (Fig. 3A). In both X0142 and X0234 there was a transient increase in relative intrahepatic IFN-α levels (to 23.8 at week 8 for X0142 and to 12.9 at week 6 for X0234) and IFN-γ mRNA levels (Fig. 4A and 5A). Notably, for X0142, relative amounts of IFN-γ of 24.0 and 45.0 were seen at weeks 12 and 18, respectively. The maximum level of IFN-γ detected in X0234 was 6.9 at week 12 postinoculation. Hepatic IL-4 levels were mostly undetectable or unchanged in all chimpanzees except X0142, which showed an increase to 12.7 at week 18, coincidental with elevation of IFN-γ.
DISCUSSION
This report described comparative studies of chimpanzees inoculated with a homogeneous infectious HCV of genotype 1b. We showed that the outcome of infection varied with respect to viral persistence, and this might be a consequence of an as-yet-undetermined host response.
In prior studies, two of the chimpanzees (X0142 and X0234) developed persistent HCV infection after inoculation with an infectious genotype 1b clone (28). In this study, four separate inoculations of chimpanzee X0132 with similar HCV RNA transcripts resulted in only weak and transient HCV infections (Fig. 2). There was no evidence of an increase in IFN-γ-producing PBMC or intrahepatic IFN-γ or IL-4 mRNA levels, suggesting that there was little or no induction of HCV-specific CTLs. These data suggested that X0132 was somehow able to overcome HCV infection despite mounting only a weak immune response. Subsequently, X0132 was challenged with diluted serum from X0142 but did not develop a detectable viremia (Fig. 2). This dilution had been sufficient to cause chronic infection in a different chimpanzee, X0234 (28). Further inoculation of X0132 with 10 times more virus caused viremia with titers reaching 2 × 105 genomes/ml and an antibody response that was detected 4 weeks after inoculation. In the experiments with X0142 and X0234 (28) and in other reports of HCV infection of chimpanzees, an antibody response was generally not detected until at least 3 to 4 months after inoculation. The relatively early appearance of anti-HCV antibodies suggests that the prior four RNA transfections, while not causing typical viremia or detectable seroconversion, were sufficient to prime the immune system. X0132 subsequently cleared the viremia to levels undetectable by RT-PCR. There was evidence of CD8 immune response prior to viral clearance, including an increase in IFN-γ-producing cells infiltrating the liver. These data suggest that HCV-specific CD8 cells might have contributed to viral clearance during this rechallenge. However, it is not completely surprising that the chimpanzee, after repeated challenge with various HCV RNAs and then infectious HCV serum, developed a detectable CD8 response.
Like X0132, Chimpanzee X0190 also cleared HCV infection in the absence of a strong T helper response (Fig. 3). Indeed, there was no evidence of increased peripheral IFN-γ-producing cells and only a mild increase in intrahepatic IFN-γ levels during the viremia. In light of such a weak CD4 response, it is unlikely that a vigorous CD8 response exists, although this possibility cannot be excluded.
Analysis of the HCV-specific immune responses of chimpanzees X0142 and X0234, both of which developed persistent viremia, did not reveal any major difference with the two chimpanzees that cleared infection. Both animals X0142 and X0234 had a weak and transient T helper cell response, and both showed mild increases in peripheral, IFN-γ-producing CD4+ and CD8+ cells (Fig. 4 and 5). Interestingly, X0142 showed higher intrahepatic IFN-α and IFN-γ mRNA levels than any of the other chimpanzees, even though there was no reduction in viral load or ALT elevation during follow-up. Furthermore, previous analysis of HCV RNA derived from X0142 and X0234 at various time points postinoculation revealed several mutations (28), but only two of them identified in HCV RNA from X0142 occurred within known CTL epitopes. Furthermore, these two mutations occurred late during the course of infection (after 60 weeks). This is in contrast to a recent study suggesting that one of the mechanisms by which HCV persists is through the generation of CTL escape variants (8). In that study, whether the escape phenomenon was indeed responsible for the failure of the host to clear acute HCV infection or a result of continuous immunological pressure after the persistent infection was established is not completely clear. In addition, it is not unexpected that chimpanzees with resolved hepatitis C had no discernible CTL mutations because the virus had little chance to mutate.
Although our study did not present a comprehensive analysis of all possible CTL responses, the testing of multiple epitopes that have been shown to be restricted by the Patr haplotypes of each chimpanzee failed to reveal any difference in the peripheral CD8+ responses in these chimpanzees. Furthermore, we did not examine the intrahepatic T-cell response in these chimpanzees, but we reason that hepatic IFN-γ and IL-4 levels should be indicative of any intrahepatic T-cell response, as shown in other viral hepatitis models (13). Therefore, CTL escape is probably not a principal mechanism for viral persistence in our study. However, not until all HCV CTL epitopes are fully characterized in this system will we be able to completely define the role of CD8+ T cells in this model system.
Studies of acute hepatitis C in humans have suggested the importance of robust CD4+ and/or CD8+ responses in viral clearance (7, 20, 24, 27), which contrasts with our observations. However, these study subjects were infected with heterogeneous viral strains which may differ with respect to disease activities, pathogenesis, and immunological responses. Furthermore, all of the studies were done on symptomatic infection, and many were based on patients who had already seroconverted to anti-HCV, which is a relatively late stage of acute infection (24). In only one report were individuals with acute HCV infection studied at the early stage (27). This study showed equally robust CD4+ responses in two of four patients who progressed to chronic infection as in the one patient who cleared HCV infection. Previous reports in which a correlation between viral clearance and CD8+ response was shown used polyclonal HCV genotype 1a inocula and studied chimpanzees with acute hepatitis (5, 8). Moreover, only intrahepatic CD8+-T-cell lines that required long-term antigen-specific expansion were analyzed. In contrast, our study used a monotypic HCV 1b source, and the chimpanzees had asymptomatic infection without ALT elevation. Finally, it is entirely possible that the chimpanzees and humans may behave differently in response to HCV infection. It is well known that chimpanzees do not develop active chronic liver disease and respond differently to other viruses, such as human immunodeficiency virus. The chimpanzees also appear to have a higher rate of viral clearance from HCV infection than humans (2). If we can unravel the molecular and immunological basis of this difference, we may be able to use this information to target a protective response against HCV in humans for both prevention and therapy.
HCV persistence is probably a multifactorial process, arising because of an inadequate immune response of the host early in infection and being maintained by the ability of the virus to evade host responses later in infection. In humans, the quasispecies nature of the virus, and in particular the hypervariable N-terminal region of the E2 envelope protein, are thought to assist in immune evasion. However, in chimpanzees, persistence can occur without hypervariability in this region of E2 (2). Evidently, the generation of CTL escape variants is an important mechanism of persistence but, considering the data presented here, it is unlikely to be the only mechanism. As has been postulated in humans, there are likely to be several pathways to persistence in acute HCV infection (27). In our study, weak cellular responses were observed regardless of the infection outcome and, overall, we could not find a correlation between viral clearance and T-cell response. It is interesting to reason that components of innate immunity, such as NKT or NK cells, might play a principal role in viral clearance in these chimpanzees. There is compelling evidence that these immune mechanisms are crucial in controlling viral infection, especially during the acute phase of infection (4, 12, 21). Further experiments are needed to identify the precise mechanisms that determine the outcome of infection in this system.
Acknowledgments
We thank Ronda Sapp and Anthony Davis for excellent technical assistance and the staff in the NIH Clinical Center Department of Transfusion Medicine for virologic and serologic testing. We thank Renee Tucker of the Southwest Foundation for Biomedical Research for providing clerical support for this study.
We also thank Luiz Barbosa of NHLBI for providing partial support for the study under contract NO1-HB-27091.
REFERENCES
- 1.Alter, H. J., and L. B. Seeff. 2000. Recovery, persistence, and sequelae in hepatitis C virus infection: a perspective on long-term outcome. Semin. Liver Dis. 20:17-35. [DOI] [PubMed] [Google Scholar]
- 2.Bassett, S. E., D. L. Thomas, K. M. Brasky, and R. E. Lanford. 1999. Viral persistence, antibody to E1 and E2, and hypervariable region 1 sequence stability in hepatitis C virus-inoculated chimpanzees. J. Virol. 73:1118-1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beard, M. R., G. Abell, M. Honda, A. Carroll, M. Gartland, B. Clarke, K. Suzuki, R. Lanford, D. V. Sangar, and S. M. Lemon. 1999. An infectious molecular clone of a Japanese genotype 1b hepatitis C virus. Hepatology 30:316-324. [DOI] [PubMed] [Google Scholar]
- 4.Brown, M. G., A. O. Dokun, J. W. Heusel, H. R. C. Smith, D. L. Beckman, E. A. Blattenberger, C. E. Dubbelde, L. R. Stone, A. A. Scalzo, and W. M. Yokoyama. 2001. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science 292:934-937. [DOI] [PubMed] [Google Scholar]
- 5.Cooper, S., A. L. Erickson, E. J. Adams, J. Kansopon, A. J. Weiner, D. Y. Chien, M. Houghton, P. Parham, and C. M. Walker. 1999. Analysis of a successful immune response against hepatitis C virus. Immunity 10:439-449. [DOI] [PubMed] [Google Scholar]
- 6.Cucchiarini, M., A. R. Kammer, B. Grabscheid, H. M. Diepolder, T. J. Gerlach, N. Gruner, T. Santantonio, J. Reichen, G. R. Pape, and A. Cerny. 2000. Vigorous peripheral blood cytotoxic T-cell response during the acute phase of hepatitis C virus infection. Cell. Immunol. 203:111-123. [DOI] [PubMed] [Google Scholar]
- 7.Diepolder, H. M., R. Zachoval, R. M. Hoffmann, E. A. Wierenga, T. Santantonio, M. C. Jung, D. Eichenlaub, and G. R. Pape. 1995. Possible mechanism involving T-lymphocyte response to non-structural protein 3 in viral clearance in acute hepatitis C virus infection. Lancet 346:1006-1007. [DOI] [PubMed] [Google Scholar]
- 8.Erickson, A. L., Y. Kimura, S. Igarashi, J. Eichelberger, M. Houghton, J. Sidney, D. McKinney, A. Sette, A. L. Hughes, and C. M. Walker. 2001. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity 15:883-895. [DOI] [PubMed] [Google Scholar]
- 9.Farci, P., A. Shimoda, D. Wong, T. Cabezon, D. De Gioannis, A. Strazzera, Y. Shimizu, M. Shapiro, H. J. Alter, and R. H. Purcell. 1996. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc. Natl. Acad. Sci. USA 93:15394-15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerlach, J. T., H. M. Diepolder, M. C. Jung, N. H. Gruener, W. W. Schraut, R. Zachoval, R. Hoffmann, C. A. Schirren, T. Santantonio, and G. R. Pape. 1999. Recurrence of hepatitis C virus after loss of virus-specific CD4+ T-cell response in acute hepatitis C. Gastroenterology 117:933-941. [DOI] [PubMed] [Google Scholar]
- 11.Gruener, N. H., F. Lechner, M. C. Jung, H. Diepolder, T. Gerlach, G. Lauer, B. Walker, J. Sullivan, R. Phillips, G. R. Pape, and P. Klenerman. 2001. Sustained dysfunction of antiviral CD8+ T lymphocytes after infection with hepatitis C virus. J. Virol. 75:5550-5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guidotti, L. G., and F. V. Chisari. 2001. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu. Rev. Immunol. 19:65-91. [DOI] [PubMed] [Google Scholar]
- 13.Guidotti, L. G., R. Rochford, J. Chung, M. Shapiro, R. Purcell, and F. V. Chisari. 1999. Viral clearance without destruction of infected cells during acute HBV infection. Science 284:825-829. [DOI] [PubMed] [Google Scholar]
- 14.Honda, M., E. A. Brown, and S. M. Lemon. 1996. Stability of a stem-loop involving the initiator AUG controls the efficiency of internal initiation of translation on hepatitis C virus RNA. RNA 2:955-968. [PMC free article] [PubMed] [Google Scholar]
- 15.Hong, Z., M. Beaudet-Miller, R. E. Lanford, B. Guerra, J. Wright-Minogue, A. Skelton, B. M. Baroudy, G. R. Reyes, and J. Y. Lau. 1999. Generation of transmissible hepatitis C virions from a molecular clone in chimpanzees. Virology 256:36-44. [DOI] [PubMed] [Google Scholar]
- 16.Kolykhalov, A. A., E. V. Agapov, K. J. Blight, K. Mihalik, S. M. Feinstone, and C. M. Rice. 1997. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 277:570-574. [DOI] [PubMed] [Google Scholar]
- 17.Kowalski, H., A. L. Erickson, S. Cooper, J. D. Domena, P. Parham, and C. M. Walker. 1996. Patr-A and B, the orthologues of HLA-A and B, present hepatitis C virus epitopes to CD8+ cytotoxic T cells from two chronically infected chimpanzees. J. Exp. Med. 183:1761-1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lanford, R. E., H. Lee, D. Chavez, B. Guerra, and K. M. Brasky. 2001. Infectious cDNA clone of the hepatitis C virus genotype 1 prototype sequence. J. Gen. Virol. 82:1291-1297. [DOI] [PubMed] [Google Scholar]
- 19.Lechner, F., N. H. Gruener, S. Urbani, J. Uggeri, T. Santantonio, A. R. Kammer, A. Cerny, R. Phillips, C. Ferrari, G. R. Pape, and P. Klenerman. 2000. CD8+ T lymphocyte responses are induced during acute hepatitis C virus infection but are not sustained. Eur. J. Immunol. 30:2479-2487. [DOI] [PubMed] [Google Scholar]
- 20.Lechner, F., D. K. Wong, P. R. Dunbar, R. Chapman, R. T. Chung, P. Dohrenwend, G. Robbins, R. Phillips, P. Klenerman, and B. D. Walker. 2000. Analysis of successful immune responses in persons infected with hepatitis C virus. J. Exp. Med. 191:1499-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu, Z. X., S. Govindarajan, S. Okamoto, and G. Dennert. 2000. NK cells cause liver injury and facilitate the induction of T-cell-mediated immunity to a viral liver infection. J. Immunol. 164:6480-6486. [DOI] [PubMed] [Google Scholar]
- 22.Major, M. E., K. Mihalik, J. Fernandez, J. Seidman, D. Kleiner, A. A. Kolykhalov, C. M. Rice, and S. M. Feinstone. 1999. Long-term follow-up of chimpanzees inoculated with the first infectious clone for hepatitis C virus. J. Immunol. 73:3317-3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mercer, D. F., D. E. Schiller, J. F. Elliott, D. N. Douglas, C. Hao, A. Rinfret, W. R. Addison, K. P. Fischer, T. A. Churchill, J. R. Lakey, D. L. Tyrrell, and N. M. Kneteman. 2001. Hepatitis C virus replication in mice with chimeric human livers. Nat. Med. 7:927-933. [DOI] [PubMed] [Google Scholar]
- 24.Missale, G., R. Bertoni, V. Lamonaca, A. Valli, M. Massari, C. Mori, M. G. Rumi, M. Houghton, F. Fiaccadori, and C. Ferrari. 1996. Different clinical behaviors of acute hepatitis C virus infection are associated with different vigor of the antiviral cell-mediated immune response. J. Clin. Investig. 98:706-714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizukoshi, E., M. Nascimbeni, J. B. Blaustein, K. Mihalik, C. M. Rice, T. J. Liang, S. M. Feinstone, and B. Rehermann. 2002. Molecular and immunological significance of chimpanzee major histocompatibility complex haplotypes for hepatitis C virus immune response and vaccination studies. J. Virol. 76:6093-6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takaki, A., M. Wiese, G. Maertens, E. Depla, U. Seifert, A. Liebetrau, J. L. Miller, M. P. Manns, and B. Rehermann. 2000. Cellular immune responses persist and humoral responses decrease two decades after recovery from a single-source outbreak of hepatitis C. Nat. Med. 6:578-582. [DOI] [PubMed] [Google Scholar]
- 27.Thimme, R., D. Oldach, K. M. Chang, C. Steiger, S. C. Ray, and F. V. Chisari. 2001. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J. Exp. Med. 194:1395-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomson, M., M. Nascimbeni, S. Gonzales, K. Murthy, B. Rehermann, and T. J. Liang. 2001. Emergence of a distinct pattern of viral mutations in chimpanzees infected with a homogeneous inoculum of hepatitis C virus genotype 1b. Gastroenterology 121:1226-1233. [DOI] [PubMed] [Google Scholar]
- 29.Yanagi, M., R. H. Purcell, S. U. Emerson, and J. Bukh. 1999. Hepatitis C virus: an infectious molecular clone of a second major genotype (2a) and lack of viability of intertypic 1a and 2a chimeras. Virology 262:250-263. [DOI] [PubMed] [Google Scholar]
- 30.Yanagi, M., R. H. Purcell, S. U. Emerson, and J. Bukh. 1997. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc. Natl. Acad. Sci. USA 94:8738-8743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yanagi, M., M. St. Claire, M. Shapiro, S. U. Emerson, R. H. Purcell, and J. Bukh. 1998. Transcripts of a chimeric cDNA clone of hepatitis C virus genotype 1b are infectious in vivo. Virology 244:161-172. [DOI] [PubMed] [Google Scholar]