

Abstract
In enzyme catalysis, where exquisitely positioned functionality is the sine qua non, atomic coordinates for a Michaelis complex can provide powerful insights into activation of the substrate. We focus here on the initial proton transfer of the isomerization reaction catalyzed by triosephosphate isomerase and present the crystal structure of its Michaelis complex with the substrate dihydroxyacetone phosphate at near-atomic resolution. The active site is highly compact, with unusually short and bifurcated hydrogen bonds for both catalytic Glu-165 and His-95 residues. The carboxylate oxygen of the catalytic base Glu-165 is positioned in an unprecedented close interaction with the ketone and the α-hydroxy carbons of the substrate (C… O ≈ 3.0 Å), which is optimal for the proton transfer involving these centers. The electrophile that polarizes the substrate, His-95, has close contacts to the substrate's O1 and O2 (N… O ≤ 3.0 and 2.6 Å, respectively). The substrate is conformationally relaxed in the Michaelis complex: the phosphate group is out of the plane of the ketone group, and the hydroxy and ketone oxygen atoms are not in the cisoid configuration. The ɛ ammonium group of the electrophilic Lys-12 is within hydrogen-bonding distance of the substrate's ketone oxygen, the bridging oxygen, and a terminal phosphate's oxygen, suggesting a role for this residue in both catalysis and in controlling the flexibility of active-site loop.
Triosephosphate isomerase (TIM) catalyzes the isomerization between dihydroxyacetone phosphate (DHAP) and d-glyceraldehyde 3-phosphate (GAP). The uphill direction from DHAP to GAP is essential for optimal throughput in the glycolytic pathway. To accomplish this reaction, TIM extracts the pro-R hydrogen from the C1 carbon of DHAP and then stereo-specifically introduces a proton to the C2 carbon (Fig. 1 a and b; ref. 1). Kinetic isotope effects and isotope “washout” experiments suggest that the reaction proceeds through a planar cis-enediol or enediolate intermediate of moderate stability (2, 3), and that the energetic landscape is characterized by several steps of competitive timescales (4). The crucial proton transfers between C1 and C2 atoms of the substrate are most likely carried out by a single base, the side chain carboxylate of Glu-165, whereas His-95 and Lys-12 probably facilitate the transfer of the hydroxyl proton from O1 to O2 (Fig. 1a; ref. 5). Structural studies revealed an α/β fold, now known as the TIM-barrel (6–8), and elucidated the geometry of the catalytic residues at the active site and their interactions with the ligands (9).
Figure 1.

The isomerization reaction catalyzed by triosephosphate isomerase. (a) The reaction pathway from DHAP to GAP, including the putative reaction intermediate, an enediol(ate). The phosphate elimination side reaction produces a toxic compound, methylglyoxal. The catalytic base, Glu-165, extracts the pro-R proton on C1. Stabilization of the enediol(ate) is offered by the neutral His-95, as well as by Lys-12 and Asn-10 (5). (b) The atom-numbering scheme of DHAP and dihedral angles discussed throughout this paper are shown. (c) A molecular model of the previously proposed in-plane arrangement of the phosphate group. (d) The out-of-plane conformation of DHAP observed here in our structure of the Michaelis complex.
TIM is a textbook case in enzymatic enolization chemistry and has become the subject of landmark spectroscopic and computational studies elucidating the details of the mechanism, the protein motions relevant to chemistry, and the design principles that allow efficient and uphill proton transfer in enzyme active sites. Spectroscopic and mutation studies have focused on the polarization of the substrate by catalytic residues as well as on the stabilization of the enendiol(ate) intermediate (10–18). Computational effort has focused on the clarification of steps between the deprotonation at C1 of DHAP by glutamic acid 165 and protonation of C2 to form GAP (19–22), wherein the substrate's oxygens also participate in proton transfer reactions, as well as the contribution of proton tunneling in the reaction mechanism (23).
We initiated this study in the hopes of observing, in full chemical detail, the geometry of the active site of this enzyme, just before chemical attack. With its modest driving force, and with a clear biological significance for the uphill energetic direction, TIM offers a rare opportunity to study an enzyme–substrate Michaelis complex by crystallographic methods (7, 24). The first attempts to observe the productive Michaelis complex were carried out as early as 1976; prolonged soaking of chicken TIM crystals with DHAP resulted in 6-Å diffraction patterns (7). Subsequent experiments used a flow cell to continuously soak unligated yeast TIM crystals with fresh DHAP solution during x-ray diffraction data collection, which extended the resolution to 3 Å (24). Unfortunately, even at that resolution, it was essentially impossible to identify the chemical details of the active site. Here, we report the crystal structure of the TIM:DHAP Michaelis complex at 1.2-Å resolution. The structure clearly defines the binding mode of the substrate and its interactions with the enzyme, allowing for a definitive description of hydrogen bonding, active site and substrate geometry, and mobility of key reacting atoms. The unique features of the Michaelis complex, elucidated in this study, advance our understanding of the chemical activation of DHAP and the initial proton transfer step.
Materials and Methods
All reagents were purchased from Sigma or Fluka. The purification and crystallization of yeast TIM (W90Y, W157F mutant, containing 5′-fluorotryptophan at W168) has been described (25). The kinetic parameters of the mutant are essentially the same as those of the wild-type enzyme (26). Enzymatic activity was determined by the conversion of GAP to DHAP in the presence of TIM by using an assay linked to glycerol 3-phosphate dehydrogenase, as described by Plaut and Knowles (27). For soaking, unligated crystals were transferred to a solution containing 50 mM Tris (pH 6.8), 50 mM NaCl, 18–20% (wt/vol) PEG 4000, and 30 mM DHAP. The soaking experiments were performed at 4°C to minimize the phosphate-elimination side reaction (28). After 2 days, the recrystallized TIM was transferred to a cryo-protectant solution [soaking solution supplemented with 15% PEG 400 (wt/vol)] and flash-frozen in liquid nitrogen. The concentration of PEG and substrate solution were optimized for successful recrystallization of each individual preparation of unligated enzyme crystals.
An x-ray diffraction data set to 1.2-Å resolution was collected from a monoclinic crystal at 100 K at the Cornell High Energy Synchrotron Source (CHESS) beamline A-1. The diffraction images were processed with the HKL package (29). The crystal belongs to space group P21, isomorphous to that of yeast TIM in the presence of a transition-state analogue (9), with a = 73.62 Å, b = 82.8 Å, c = 38.21 Å, and β = 101.7° (Table 1). In addition, an x-ray diffraction data set to 1.6-Å resolution was collected from an orthorhombic crystal at 100K at CHESS. The crystal belongs to space group P212121, with a = 47.26 Å, b = 62.17 Å, and c = 160.68 Å (Table 1).
Table 1.
Summary of crystallographic information
| Crystal form | Monoclinic | Orthorhombic |
|---|---|---|
| Maximum resolution, Å | 1.2 | 1.6 |
| Number of observations | 285,716 | 231,083 |
| Rmerge, %* | 5.7 | 6.5 |
| Resolution range for refinement | 30–1.2 | 30–1.6 |
| Number of reflections | 134,020 | 62,255 |
| Completeness, % | 95.7 | 97.7 |
| R factor, %† | 12.5 | 20.9 |
| Free R factor, %† | 15.0 | 26.8 |
| rms deviation in bond lengths, Å | 0.013 | 0.016 |
| rms deviation in angle distances, Å | 0.029 | 0.028 |
Rmerge= ∑h ∑i|Ihi − 〈Ih〉|/∑h ∑i Ihi.
R=∑h |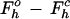 |/ ∑h
|/ ∑h 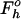 .
.
The structure refinement of the monoclinic form was performed with the program SHELXL (30). The structure of the orthorhombic crystal form was determined by the combined molecular replacement protocol with the program COMO (31, 32) and refined with SHELXL (30). The atomic models were rebuilt against (omit) 2Fo–Fc electron density maps with the program O (33). The refinement statistics are summarized in Table 1.
The active-site loop in subunit A of the orthorhombic form exhibits multiple conformations. This disorder apparently occurs because crystal contacts stabilize the open loop. For this subunit, the position of the substrate and the conformation of the active-site loop are not as well defined as those of subunit B of the orthorhombic form, or of either subunit in the monoclinic form. Refinement of this subunit was carried out allowing for the two conformations of the loop; the occupancy of the two conformations was found to be roughly equal.
Results and Discussion
To elucidate the nature of the reaction complex, we have pursued the structure of the TIM:DHAP complex at higher resolution by using cryo-crystallography to preserve the crystals after soaking. The limitation of earlier soaking experiments was the significant deterioration in the x-ray diffraction quality of the crystals, both during soaking and during data collection. A major concern with soaking experiments in the presence of the substrates is the phosphate elimination side reaction (Fig. 1a). We have performed the experiments at 4°C, where the side reaction is minimized (28). Crystals of unligated yeast TIM [W90Y, W157F, 5′-fluorotryptophan at W168, a fully active mutant used in NMR studies (25, 26)] were soaked in a solution containing DHAP. Prompt deterioration of these crystals was observed upon soaking, and the mosaicity of the crystal increased from 0.3 to 1.4° after soaking for 2 h. This degradation presumably occurs because of the previously reported conformational change for the flexible loop in the active site, as this loop is involved in crystal-packing contacts in the unligated enzyme (9). Nonetheless, a moderate quality diffraction data set to 2.0 Å was collected with cryo-preservation. The crystallographic analysis clearly showed electron density consistent with the presence of substrate in the active site of the enzyme.
To our surprise, when we soaked unligated crystals for a longer time in the DHAP solution, TIM recrystallized (Fig. 2). Many of the original crystals of the enzyme disappeared after about 24 h of soaking. The new crystals have two different morphologies, plate-shaped and cube-shaped (Fig. 2d). Crystallographic analysis showed that the plate-shaped crystals are isomorphous to the closed form of yeast TIM, in the monoclinic crystal form (8), whereas the cube-shaped crystals are in the orthorhombic system, which has not been reported for yeast TIM. By using a synchrotron radiation source, we were able to collect a 1.2-Å resolution data set on a monoclinic crystal, and a 1.6-Å resolution data set on an orthorhombic crystal (Table 1). We observed similar recrystallization behavior when the free-enzyme crystals were soaked with GAP (data not shown). In addition, we have also obtained micro crystals from cocrystallization experiments with DHAP.
Figure 2.

Recrystallization of TIM upon soaking with DHAP. Time-lapse photographs showing the disappearance of the original free-enzyme crystals and the appearance of cube-shaped (cyan arrow) and plate-shaped (yellow arrow) crystals after soaking for 5 min (a), 1 day (b), 2 days (c), and 4 days (d). The highest resolution data were compatible with a majority species of dihydroxyacetone phosphate.
In these different crystal forms, the structure of the protein is quite similar to the previously reported structures of TIM–ligand complexes. The active-site residues are engaged in similar interactions to those observed for the analog compounds, and the active-site loops of the enzyme are in comparable conformations (Fig. 3a). On the other hand, fine details available from the Michaelis complex reported here are unique to the actual substrate and are extremely informative with respect to the mechanism. Our structural analysis is based on observations from six independent TIM:DHAP Michaelis complexes, in different crystal packing environments, and with different times of exposure to DHAP. The conclusions described here are observed consistently in all these complexes, including the structure obtained after soaking for 2 h. In the discussions below, we will focus on the monoclinic crystal form, obtained at near-atomic resolution (Table 1).
Figure 3.

Structure of yeast TIM:DHAP complex. (a) Triosephosphate isomerase in complex with a tight-binding transition-state analog phosphoglycolohydroxamate [PDB entry 7TIM (9) in cyan and purple for carbon atoms in the enzyme and the analog, respectively] is overlaid with our structure of the actual substrate, DHAP (in yellow and green). The position of the substrate differs in fine details from that of the transition-state analog. (b) Omit electron density for the substrate and catalytic residues Glu-165, His-95, and Lys-12. (c) Omit electron density for the active site in the second molecule. (d) ORTEP representation displaying the anisotropic motion for the active-site atoms. The glutamic acid side chain and the substrate carbons (C3, C2, and less so, C1) as well as the oxygens undergoing isomerization (O1 and O2) are more dynamically flexible than the active-site residues and phosphate group. (e) A schematic drawing of the network of hydrogen bonds in the active site.
Of primary interest is the identification of the major chemical species in the active site. Clearly defined electron density for the bound substrate was observed in all crystal forms. For the monoclinic form, the x-ray diffraction data at near-atomic resolution allowed the identification of this molecule (Fig. 3 b and c). The DHAP molecule has the best fit to the observed electron density and is the most likely molecular species bound in the active site. As an additional verification for the identity of this compound, we carried out a crystallographic refinement without geometric restraints for the substrate in the active site. The resulting conformation had little indication of deviation from planarity for the C2 carbonyl group. The electron density also clearly excludes the possibility that the observed ligand is methylglyoxal and inorganic phosphate, i.e., the side product from phosphate elimination (Fig. 1a). Our structural observations are consistent with both Fourier transform infrared (10) and low temperature SSNMR studies (34), which demonstrated a predominance of DHAP in the active site. Recent solution- and solid-state NMR data (unpublished data) indicate a small population of GAP in comparable samples, possibly bound to the enzyme. Thus, we cannot exclude a small population of GAP or the putative enediol(ate) in the active site (<10%).
TIM has been a hallmark example of the roles of short hydrogen bonds in promoting enolization reactions and other proton transfers. A strong hydrogen bond between O2 and the NH of His-95 Nɛ2 has been proposed to lower the energy barrier for proton extraction by stabilizing the transition state and intermediate. This electrophile has a crucial kinetic role in facilitating proton transfer, as evidenced by the disastrous effects on kcat when it is replaced by other functionalities (16). Short hydrogen bonds involving His-95 and analog compounds have been characterized with diffraction (9), IR (16), and NMR experiments (13, 17), as well as with computational tools (19). The structures reported herein, at near-atomic resolution, allowed us to define the hydrogen bonding parameters in the presence of the actual substrate for the first time. As for all other TIM structures, the hydrogen bonding context of His-95 suggests that it is neutral, and protonated at the (amino) Nɛ position. Previous NMR studies demonstrated that the imidazole group is neutral at physiological pH values (13). In the Michaelis complex structure, the imino Nδ forms a hydrogen bond with an amidic NH moiety from the protein backbone; Nɛ forms a bifurcated hydrogen bond to O2 (2.6–2.7 Å) and O1 (3.0 Å) of the substrate (Fig. 3 d and e). Prior work had proposed a strengthening of the hydrogen bond as the reaction coordinate advances. Our study suggests, instead, the existence of very compact hydrogen bonds before the reaction begins, presumably relevant for activation of the proton transfer.
Of even more interest is the interaction between the catalytic base and the labile carbon-bound proton. The Oɛ2 oxygen of Glu-165 is in close contact with, and at similar distances to, both the C1 and C2 atoms (Fig. 3 d and e) in all of the active sites studied. The distance of 3.0 Å between the carboxylate's oxygen and both C1 and C2 atoms of DHAP is significantly shorter than typical distances associated with C-H… O hydrogen bonds (35) and compressed below the sum of their van der Waals radii. This close interaction was anticipated and successfully mimicked in transition-state analogs that employ carboxylate or hydroxylamine functionalities in place of the carbon. Nevertheless, although short O… O and O… N distances are common, direct observations of a close C… O contact in a reactive species are quite rare. Glu-165 presumably also interacts with the hydroxyl proton of the substrate. (The substrate's hydroxyl hydrogen in the monoclinic form, as for all of the protons, was not experimentally resolved, and its location was deduced from the heavy atom positions and hydrogen-bonding patterns in the active site.) Why does Glu-165 select C1 as one of its hydrogen bond partners, given the prevalence of OH and NH groups in the active site? This interaction might occur because Glu-165 is sterically limited and sequestered by hydrophobic and bulky Ile-170 on the mobile loop 6 (Fig. 3a). This interaction could also be driven by an electrostatic attraction to the adjacent electropositive C2 carbonyl carbon, rather than by C1-H… O hydrogen bond per se. In some small-molecule crystal structures, solvation by acetone can involve the carbonyl carbon interacting closely with an oxyanionic species; here, the interaction with C2 might similarly stabilize the structure. Whatever the underlying principles, such a bifurcated and compressed arrangement is highly unusual and is presumably a key feature in the enzyme's catalytic power.
The geometry of the attacking carboxylate is primed for proton transfer from C1 and in perfect accord with its proposed role in the mechanism as summarized by Knowles (5). The syn orbital, believed to be more basic, is used for proton abstraction. The pro R proton is somewhat out of the nascent enediolate plane defined by atoms O2-C2-C1-O1; the torsion angle O2-C2-C1-H is 160°. The out-of-plane orientation is assumed to be favorable for extracting the proton and for forming the π-orbital of the enediol(ate). At first glance, the structural information suggests a complete proton transfer coordinate from the C1 to the C2 position, with minimal heavy atom motion, and brings to mind the possibility of tunneling of the proton along the complete reaction coordinate (36). Such a streamlined and concerted picture cannot account for a central experimental evidence: any proposed mechanism must also be reconciled with the modest apparent isotope effect for removal of the H1 proton and with the substantial isotope washout when 1-2H- or 1-3H-DHAP is used as a substrate. These isotope experiments were logically interpreted to mean that the enendiol(ate) intermediate has a finite lifetime on the enzyme (37). In view of these isotope effects, it is more likely that the Michaelis complex we observe will participate in the chemistry of the first proton extraction; additional experiments will be needed to provide details concerning subsequent intermediates and chemical steps.
In subsequent steps, Glu-165 in the wild-type enzyme (18, 19) and the mutants (38) was previously proposed also to interact with both O1 and O2 and act as both a general acid and a base to redistribute protons for these oxygens. Our structure of the Michaelis complex shows much stronger associations of the general base with the substrate's O1 than with O2. For Glu-165 to reach O2 of the substrate and participate in the redistribution of protons in the enolate intermediate, significant restructuring of this compact active site would be needed.
Well defined electron density with low Debye Waller factors were observed for most of the residues in the active site, including the catalytic residues His-95 and Lys-12, suggesting that they are highly ordered (Fig. 3 b and c). In comparison, the side chain of the catalytic base, Glu-165, has higher mobility, as evidenced by its larger temperature factors (Fig. 3d). Glu-165 was previously known to be mobile, and its mobility is believed to be essential for the mechanism. The side chain of Glu-165 swings by about 2 Å upon closure of the adjacent active-site loop, allowing Glu-165 to contact the bound ligand. Moreover, the placement of this side chain seemed to depend on the selection of active-site ligand (39, 40). In the Michaelis complex, one oxygen of the carboxylate interacts almost symmetrically with C1 and C2 (Fig. 3d). Interestingly, the maximal motion of this oxygen appears to parallel the line connecting the substrate's C1 and C2 atoms (Fig. 3d). In other words, the conformational flexibility apparently may allow the catalytic base to slide between the protonation sites. Debye-Waller factors for the phosphate moiety are low, but those of the substrate's reacting oxygens and carbons, O2, O1, C1, C2, and C3, are higher (Fig. 3d). The mobility in Glu-165 and the substrate is consonant with the prior observation that the chemical reaction can proceed in some crystalline forms.
The bound conformation of DHAP is informative about the notion of substrate geometry and strain in the reaction pathway. We use the term strain to describe conformational destabilization in the ground state or Michaelis complex that coerces the substrate to resemble the transition state of the reaction or otherwise activates the substrate toward chemistry (41). Typically, substrate strain is proposed to selectively reduce the activation barrier of the reaction. TIM has often been invoked as an illustration of the possible association of protein mobility with strain (42). The O2-C2-C3-O3 torsion angle (χ3, Fig. 1b) describing the phosphate group orientation with respect to the ketone group of DHAP was proposed to be strained: with the phosphate group out-of-plane, the substrate is expected to be at an energetic minimum; with the phosphate group in-plane, steric and electrostatic clashes are anticipated. The putative strain most likely would be induced in connection with the interactions of the phosphate group with active-site mobile loop 6, once the loop is closed. This proposed strain provided a central hypothesis about TIM's ability to suppress the toxic side reaction. If the substrate is bound with its phosphate group in the plane of the ketone, minimizing orbital overlap between the phosphate's bridging oxygen and the enediolate π-orbital, the β-elimination of phosphate was expected to be reduced. In contrast, a relaxed, out-of-plane, anti-periplanar arrangement was expected to facilitate the elimination reaction (43, 44). Control over the undesired side reaction is an important element of this mechanism; the enediol(ate) intermediate readily decomposes to methylglyoxal in solution (Fig. 1a), as well as on a structurally related enzyme, methylglyoxal synthase (MGS), with remarkably similar active-site residues and interactions (45). The strain hypothesis gained more attention when structural studies of TIM in complex with transition-state analogs showed an in-plane arrangement of the phosphate (9, 44). On the other hand, more recently, the in-plane conformation of these analogs also was observed in complex with MGS (45, 46), thereby casting doubts on the relevance of this hypothesis.
The bound conformation of DHAP observed in our Michaelis complex has the phosphate group out of the plane of the C2 carbonyl group of DHAP (the O2-C2-C3-O3 torsion angle, χ3, is on average 100°, Fig. 3e). As this conformation was seen in several active-site structures in several crystal-packing environments, our structures demonstrate that the phosphate group is not strained in the Michaelis complex. Possibly the substrate conformation acquires strain only midway through the reaction, and the relaxed arrangement observed here might hold only for the Michaelis species. Of course such a mechanism would differ from strain as defined above, because the deformation energy would effectively add to the activation barrier rather than subtract from it. Mechanistically, this proposal is also unclear, because the active-site loop is already closed in our structure; therefore, other structural element(s) capable of coercing the substrate toward an in-plane conformation at the transition state would have to be identified. Therefore, we believe that another mechanism is most likely needed to explain the suppression of phosphate elimination, possibly from geometric or electrostatic considerations.
The other strain in the Michaelis complex that could be expected is a cisoid configuration, bringing O1 into the plane of the ketone, which may promote the formation of the putative enediol(ate) intermediate. However, our structure showed that the torsion angle O2-C2-C1-O1 (χ1, Fig. 1b) ranges from 19 to 43° in the different active sites that we studied here (Fig. 3e); the enzyme does not appear to bias DHAP toward a fully planar conformation, nor is the labile proton fully in-line with the approaching base. It is possible that a small rotation of the C1-C2 bond will take place as the reaction progresses, both reducing the torsion angle O2-C2-C1-O1 and placing the pro-R proton in line with Glu-165 Oɛ2. (Stated differently, the value of the torsion angle O2-C2-C1-HR might change from about 160 to 120° just before the reaction.) Indeed, the thermal anisotropy in O1 for all of the active sites is rather high, whereas that of C1 is relatively low (Fig. 3d).
One of the fascinating aspects of this enzyme is the coordination of larger scale protein motion to chemistry. Prior evidence from over 40 crystal structures suggests that the enzyme can exist in two different limiting conformations, with the majority of the barrel quite stationary, but with a flexible loop (loop 6, residues 167–176) alternating between an open and closed state (9, 47–49). The closed form is observed essentially whenever the enzyme is ligated, as in the present study. Evidently, the open form allows substrate binding and release, whereas the closed form aligns the active-site residues to initiate the reaction and stabilize the reaction intermediates. Biochemical and genetic evidence suggests that the loop, and its precise sequence, is critical for function (42). Kinetic and spectroscopic measurements indicate that the time scale of loop motion matches that of product release and turnover, suggesting that the reaction intermediate is effectively protected from water throughout its lifetime (25, 50). How is the loop opening coordinated with the completion of the chemical reaction?
Lys-12 is believed to have a crucial participation in the reaction, playing a role in binding, chemistry, and conformational gating. This residue potentially has the capability of coordinating chemistry to protein motion. Mutational changes in Lys-12 have important effects on both Km and kcat (11). Presumably, the effect on Km occurs because the ammonium group's charge is essential for phosphate binding. The effect on kcat might occur if the ammonium group participates in catalysis by polarizing the substrate's oxygen, or even acting as a general acid to protonate O2. The close proximity of Lys-12 to the phosphate's bridging oxygen and O2 was noted in a previous theoretical discussion of the TIM mechanism (19) and was there proposed to play an important electrostatic role for activation. Based on TIM-inhibitor structures, this residue is 3.5–4.5 Å from the bridging oxygen and about 2.8–3.1 Å from O2. In our structures, Lys-12 is only 3.1–3.2 Å from the bridging oxygen and 3.0 Å from O2. The shorter distance to the bridging oxygen of the bound substrate as compared with the mimic compounds is apparently a consequence of the phosphate group's being out-of-plane, rather than nearly in-plane as it is for most inhibitors. The ammonium group is also in water-mediated contact with the phosphate's O4 in all ligated TIM complexes, irrespective of the nature of the bound ligand. The phosphate group's interactions with Lys-12 and with amides from loops 6 and 7 are believed to be a dominant contribution to the substrate's binding energy. If Lys-12 were to participate in proton transfer to O2, or to change its hydrogen bonding geometry during the final proton transfers, then these stabilizing interactions would be expected to be disrupted, presumably causing the loop to open and the substrate to be released. Thus, the intriguing possibility exists that the proximity of Lys-12 to O2, O3, and O4 engenders this residue with a unique possibility to orchestrate loop opening and substrate release consequent to completion of the chemical reaction.
In summary, structural information from the productive TIM:DHAP complex offers significant constraints for the catalytic mechanism of the enzyme. The bound substrate's relaxed conformation, the compressed interaction with the active-site acids and bases, and the limited in situ dynamical degrees of freedom are clearly revealed. In conjunction with future computational studies, these coordinates are expected to be relevant for addressing the issue of activation of DHAP, stabilization of the endiol(ate), and proton transfers involving the oxygen atoms. With the detailed interactions between the authentic substrate and the active-site proton transfer players in hand, we may ultimately unravel the course of events on this enzyme.
Acknowledgments
We thank Youngchul Park, Yihua Huang, and Hao Wu for help with data collection at Cornell High Energy Synchrotron Source. We thank Dr. Jeremy R. Knowles for his pioneering work and insight into triosephosphate isomerase reaction mechanism. This work was supported by Columbia University (to L.T.) and National Institutes of Health Grant GM 66388 (to A.E.M.).
Abbreviations
- TIM
triosephosphate isomerase
- DHAP
dihydroxyacetone phosphate
- GAP
d-glyceraldehyde 3-phosphate
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1NEY and 1NF0).
References
- 1.Bloom B, Topper Y J. Science. 1956;124:982–983. doi: 10.1126/science.124.3229.982. [DOI] [PubMed] [Google Scholar]
- 2.Rose I A. Brookhaven Symp Biol. 1962;15:293–309. [PubMed] [Google Scholar]
- 3.Albery W J, Knowles J R. Biochemistry. 1976;15:5627–5631. doi: 10.1021/bi00670a031. [DOI] [PubMed] [Google Scholar]
- 4.Blacklow S C, Raines R T, Lim W A, Zamore P D, Knowles J R. Biochemistry. 1988;27:1158–1167. doi: 10.1021/bi00404a013. [DOI] [PubMed] [Google Scholar]
- 5.Knowles J R. Nature. 1991;350:121–124. doi: 10.1038/350121a0. [DOI] [PubMed] [Google Scholar]
- 6.Banner D W, Bloomer A C, Petsko G A, Phillips D C, Pogson C I, Wilson I A. Nature. 1975;255:609–614. doi: 10.1038/255609a0. [DOI] [PubMed] [Google Scholar]
- 7.Phillips D C, Rivers P S, Sternberg M J, Thornton J M, Wilson I A. Biochem Soc Trans. 1977;5:642–647. doi: 10.1042/bst0050642. [DOI] [PubMed] [Google Scholar]
- 8.Lolis E, Alber T, Davenport R C, Rose D, Hartman F C, Petsko G A. Biochemistry. 1990;29:6609–6618. doi: 10.1021/bi00480a009. [DOI] [PubMed] [Google Scholar]
- 9.Davenport R C, Bash P A, Seaton B A, Karplus M, Petsko G A, Ringe D. Biochemistry. 1991;30:5821–5826. doi: 10.1021/bi00238a002. [DOI] [PubMed] [Google Scholar]
- 10.Belasco J G, Knowles J R. Biochemistry. 1980;19:472–477. doi: 10.1021/bi00544a012. [DOI] [PubMed] [Google Scholar]
- 11.Lodi P J, Chang L C, Knowles J R, Komives E A. Biochemistry. 1994;33:2809–2814. doi: 10.1021/bi00176a009. [DOI] [PubMed] [Google Scholar]
- 12.Lodi P J, Knowles J R. Biochemistry. 1993;32:4338–4343. doi: 10.1021/bi00067a024. [DOI] [PubMed] [Google Scholar]
- 13.Lodi P J, Knowles J R. Biochemistry. 1991;30:6948–6956. doi: 10.1021/bi00242a020. [DOI] [PubMed] [Google Scholar]
- 14.Komives E A, Lougheed J C, Liu K, Sugio S, Zhang Z, Petsko G A, Ringe D. Biochemistry. 1995;34:13612–13621. doi: 10.1021/bi00041a041. [DOI] [PubMed] [Google Scholar]
- 15.Komives E A, Lougheed J C, Zhang Z, Sugio S, Narayana N, Xuong N H, Petsko G A, Ringe D. Biochemistry. 1996;35:15474–15484. doi: 10.1021/bi961556v. [DOI] [PubMed] [Google Scholar]
- 16.Komives E A, Chang L C, Lolis E, Tilton R F, Petsko G A, Knowles J R. Biochemistry. 1991;30:3011–3019. doi: 10.1021/bi00226a005. [DOI] [PubMed] [Google Scholar]
- 17.Harris T K, Abeygunawardana C, Mildvan A S. Biochemistry. 1997;36:14661–14675. doi: 10.1021/bi972039v. [DOI] [PubMed] [Google Scholar]
- 18.Harris T K, Cole R N, Comer F I, Mildvan A S. Biochemistry. 1998;37:16828–16838. doi: 10.1021/bi982089f. [DOI] [PubMed] [Google Scholar]
- 19.Cui Q, Karplus M. J Am Chem Soc. 2002;124:3093–3124. doi: 10.1021/ja0118439. [DOI] [PubMed] [Google Scholar]
- 20.Aqvist J, Fothergill M. J Biol Chem. 1996;271:10010–10016. doi: 10.1074/jbc.271.17.10010. [DOI] [PubMed] [Google Scholar]
- 21.Alagona G, Ghio C, Kollman P A. J Am Chem Soc. 1995;117:9855–9862. [Google Scholar]
- 22.Feierberg I, Aqvist J. Theor Chem Acc. 2002;108:71–84. [Google Scholar]
- 23.Cui Q, Karplus M. J Phys Chem B. 2002;106:1768–1798. [Google Scholar]
- 24.Alber T, Banner D W, Bloomer A C, Petsko G A, Phillips D, Rivers P S, Wilson I A. Philos Trans R Soc London B. 1981;293:159–171. doi: 10.1098/rstb.1981.0069. [DOI] [PubMed] [Google Scholar]
- 25.Rozovsky S, Jogl G, Tong L, McDermott A E. J Mol Biol. 2001;310:271–280. doi: 10.1006/jmbi.2001.4673. [DOI] [PubMed] [Google Scholar]
- 26.Sampson N S, Knowles J R. Biochemistry. 1992;31:8482–8487. doi: 10.1021/bi00151a014. [DOI] [PubMed] [Google Scholar]
- 27.Plaut B, Knowles J R. Biochem J. 1972;129:311–320. doi: 10.1042/bj1290311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Campbell I D, Jones R B, Kiener P A, Waley S G. Biochem J. 1979;179:607–621. doi: 10.1042/bj1790607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 30.Sheldrick G M, Schneider T R. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 31.Tong L. Acta Crystallogr A. 1996;52:782–784. [Google Scholar]
- 32.Jogl G, Tao X, Xu Y W, Tong L. Acta Crystallogr D. 2001;57:1127–1134. doi: 10.1107/s0907444901006783. [DOI] [PubMed] [Google Scholar]
- 33.Jones T A, Zou J Y, Cowan S W, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 34.Tomita Y A. Ph.D. thesis. New York: Columbia Univ.; 1996. [Google Scholar]
- 35.Derewenda Z S, Lee L, Derewenda U. J Mol Biol. 1995;252:248–262. doi: 10.1006/jmbi.1995.0492. [DOI] [PubMed] [Google Scholar]
- 36.Alston W C, Kanska M, Murray C J. Biochemistry. 1996;35:12873–12881. doi: 10.1021/bi960831a. [DOI] [PubMed] [Google Scholar]
- 37.Bloom B, Topper Y J. Nature. 1958;4616:1128–1129. doi: 10.1038/1811128b0. [DOI] [PubMed] [Google Scholar]
- 38.Nickbarg E B, Davenport R C, Petsko G A, Knowles J R. Biochemistry. 1988;27:5948–5960. doi: 10.1021/bi00416a019. [DOI] [PubMed] [Google Scholar]
- 39.Noble M E, Wierenga R K, Lambeir A M, Opperdoes F R, Thunnissen A M, Kalk K H, Groendijk H, Hol W G. Proteins. 1991;10:50–69. doi: 10.1002/prot.340100106. [DOI] [PubMed] [Google Scholar]
- 40.Kursula I, Partanen S, Lambeir A M, Antonov D M, Augustyns K, Wierenga R K. Eur J Biochem. 2001;268:5189–5196. doi: 10.1046/j.0014-2956.2001.02452.x. [DOI] [PubMed] [Google Scholar]
- 41.Jencks W P. Catalysis in Chemistry and Enzymology. New York: Dover; 1987. [Google Scholar]
- 42.Pompliano D L, Peyman A, Knowles J R. Biochemistry. 1990;29:3186–3194. doi: 10.1021/bi00465a005. [DOI] [PubMed] [Google Scholar]
- 43.Deslongchamps P, Atlani P, Frehel D, Malaval A. Can J Chem. 1972;50:3405–3408. [Google Scholar]
- 44.Lolis E, Petsko G A. Biochemistry. 1990;29:6619–6625. doi: 10.1021/bi00480a010. [DOI] [PubMed] [Google Scholar]
- 45.Marks G T, Harris T K, Massiah M A, Mildvan A S, Harrison D H T. Biochemistry. 2001;40:6805–6818. doi: 10.1021/bi0028237. [DOI] [PubMed] [Google Scholar]
- 46.Saadat D, Harrison D H. Biochemistry. 2000;39:2950–2960. doi: 10.1021/bi992666f. [DOI] [PubMed] [Google Scholar]
- 47.Alber T, Gilbert W A, Ringe P D, Petsko G A. Mobility and Function in Proteins and Nucleic Acids. London: Pitman; 1983. pp. 4–24. [Google Scholar]
- 48.Wierenga R K, Noble M E, Postma J P, Groendijk H, Kalk K H, Hol W G, Opperdoes F R. Proteins. 1991;10:33–49. doi: 10.1002/prot.340100105. [DOI] [PubMed] [Google Scholar]
- 49.Joseph D, Petsko G A, Karplus M. Science. 1990;249:1425–1428. doi: 10.1126/science.2402636. [DOI] [PubMed] [Google Scholar]
- 50.Rozovsky S, McDermott A E. J Mol Biol. 2001;310:259–270. doi: 10.1006/jmbi.2001.4672. [DOI] [PubMed] [Google Scholar]