

Abstract
Hepatitis B virus assembly begins with the packaging of an RNA pregenome into intracellular nucleocapsids, with subsequent reverse transcription within these nucleocapsids converting the RNA into a characteristic, partially double-stranded DNA, which, alone, is found in enveloped extracellular virions as the viral genome. Using a synchronized replication system for the duck hepatitis B virus, together with a stringent two-step assay for virion secretion, we demonstrate that this selective genome secretion results from an intrinsic secretion competence gained only by the nucleocapsids at the late stage of reverse transcription.
Hepadnaviruses, or hepatitis B viruses (HBVs), are para-retroviruses that harbor a partially double-stranded (ds) DNA genome but replicate this DNA through reverse transcription via an RNA intermediate (the pregenomic RNA or pgRNA) (22). Similar to classical retroviruses, viral assembly begins with the formation of intracellular nucleocapsid particles that specifically package the pgRNA. However, all hepadnaviruses undergo a maturation phase during their morphogenesis that is characterized by the reverse transcription of the pgRNA prior to membrane envelopment and secretion as extracellular virions, rather than subsequent to entry into new target cells, as is the case for classical retroviruses (for a review, see reference 20). Thus, secreted hepadnaviral virions contain only the “mature” ds DNA, whereas intracellular pools of nucleocapsids contain nucleic acid species from all the intermediate stages of reverse transcription ranging from only pgRNA to single (minus)-stranded (ss) DNA as well as ds DNA, a fundamental observation made by Summers and Mason 2 decades ago (22). The phenomenon that only mature DNA-containing nucleocapsids are enveloped and secreted implies that viral DNA synthesis may somehow be coupled to nucleocapsid envelopment. Indeed, the selective secretion of only mature DNA-containing virions could be considered a viral quality control mechanism, wherein only nucleocapsids capable of producing infectious virions would be selected for envelopment and secretion. This quality control mechanism may therefore be at least in part responsible for the extraordinary infectiousness of secreted hepadnaviral virions, whose ratio of infectivity to physical particles approaches unity (5). However, the mechanism underlying this phenomenon of nucleocapsid maturation, manifested as the apparent coupling of viral DNA synthesis to nucleocapsid envelopment, remains enigmatic to this day.
It was proposed early on that a “maturation signal” (22) (e.g., a biochemical modification) may emerge on the mature, ds DNA-containing nucleocapsids, as reverse transcription approaches completion (after extensive second- or plus-strand DNA synthesis). Such a maturation signal would then endow the mature nucleocapsids, alone, with secretion competence, e.g., the ability to interact with the viral surface glycoproteins for envelopment and secretion (Fig. 1A). In this model, “immature” nucleocapsids, containing only pgRNA or less-than-mature DNA species, are intrinsically incapable of being secreted. Although this “intrinsic” maturation signal model remains the most intuitive, direct evidence in its support is still lacking. In principle, at least two other alternative underlying mechanisms may also explain the apparent maturation phenomenon. One such mechanism, which we call the “kinetic” model here, suggests that nucleocapsids at all stages of reverse transcription may in fact have equal intrinsic secretion competence but that the envelopment and secretion process may be slow by comparison to reverse transcription, resulting in the apparent selection of only mature DNA-containing nucleocapsids for secretion (Fig. 1B). In this model, the progression of reverse transcription, with its faster kinetics, would effectively outcompete the secretion of immature nucleocapsids. In other words, the duration of passage of the nucleocapsids through the “secretion pipeline” may enable them to complete reverse transcription before exiting the cell. This model would thus predict that, if reverse transcription within nucleocapsids were to be halted, nucleocapsids containing pgRNA or immature DNA species would be enveloped and secreted along with those containing mature DNA. Another alternative model, which we call the “competition” model, proposes that mature nucleocapsids may be preferentially secreted because they outcompete immature nucleocapsids for a limiting envelopment and secretion machinery, due to their higher secretion efficiency and/or higher steady-state levels. In the context of this competition, even small differences in the intrinsic secretion efficiencies of immature versus mature nucleocapsids could be amplified (Fig. 1C). This model would thus predict that, if mature nucleocapsids were to be eliminated as a potential source of competition, immature nucleocapsids would be able to gain access to the secretion machinery and would be secreted as virions.
FIG. 1.

Models of nucleocapsid maturation. Three alternative models explaining the phenomenon of nucleocapsid maturation in hepadnaviruses, i.e., the apparent selective envelopment and secretion of only mature DNA-containing nucleocapsids. (A) The intrinsic model. An intrinsic property (a “maturation signal”) emerges on mature nucleocapsids (represented by the thick capsid shell) that endows them, alone, with secretion competence, i.e., the ability to interact with the secretion machinery (the thick wavy line with thin studs). Immature (immat) nucleocapsid species, containing pgRNA or immature DNA intermediates of reverse transcription (RT), are not secreted because they lack this intrinsic property. (B) The kinetic model. Each species of nucleocapsid has equal intrinsic secretion competence. However, only mature nucleocapsids are secreted because the kinetics of reverse transcription (shown as thick arrows) is faster than the rate of envelopment and secretion (shown as thin arrows), and, therefore, reverse transcription outcompetes the secretion process for the immature nucleocapsids. In other words, all species of nucleocapsids have had the opportunity to complete reverse transcription by the time that they exit the “secretion pipeline.” (C) The competition model. In the context of competition over a limiting secretion machinery, any increased secretion competence and/or increased abundance (represented by the thickening and darkening of the capsid shell) of mature nucleocapsids over the immature species is amplified. Even if the mature nucleocapsids are only slightly more secretion competent and/or abundant, they may outcompete immature nucleocapsids for this limiting secretion machinery.
It has been difficult to discriminate between these alternative possibilities, since a mixture of nucleocapsids representing all stages of reverse transcription is normally present intracellularly and since viral DNA synthesis is ongoing while virion secretion occurs. However, recent mutational studies have helped to establish constraints on the possible mechanisms underlying the nucleocapsid maturation phenomenon. In particular, point mutations abolishing the polymerase activity of the viral reverse transcriptase (RT) lead to the generation of nucleocapsids that contain only pgRNA with no viral DNA synthesis. These RNA-containing nucleocapsids, in the complete absence of any reverse transcription or competition from more mature nucleocapsids, remain incompetent for secretion (2, 26). On the other hand, mutations that are thought to diminish the RNase H activity of the RT, which is necessary for the elimination of pgRNA and the generation of ds DNA, seem to cause the accumulation of ss DNA-containing nucleocapsids. These immature, ss DNA-containing nucleocapsids, in the absence of mature, ds DNA-containing nucleocapsids, are enveloped and secreted (2, 26). Interestingly, certain mutations in the viral core protein, which forms the nucleocapsid shell, have also been shown to block reverse transcription at the ss DNA stage. In the case of the avian hepadnavirus, the duck HBV (DHBV), these core mutations prevent virion formation (27). However, analogous core mutations in the human HBV lead to the secretion of immature ss DNA-containing nucleocapsids as virions (13). In addition, when HBV DNA synthesis is blocked with chemical inhibitors of reverse transcription, immature nucleocapsids containing ss DNA may occasionally be secreted as virions (24).
The finding that immature nucleocapsids containing ss DNA can be secreted as virions in the case of the core protein or RNase H mutations may lend support to the kinetic or competition model. However, the interpretation of the core protein mutations is complicated by the fact that these mutations may directly affect the secretion competence of the nucleocapsid particles they form (e.g., by directly affecting the emergence and/or delivery of the putative maturation signal), irrespective of any effect they may have on the nucleic acid content within these particles. In fact, some of these core protein mutations have been shown to destabilize the nucleocapsids as reverse transcription progresses (7). Also, the nucleocapsids containing the putative RNase H mutants may be secreted, not because they represent the immature, ss DNA-containing nucleocapsids, the natural intermediates of reverse transcription, but rather because they presumably contain their newly synthesized ss DNA in an unnatural hybrid with their extant pgRNA and would therefore harbor a ds nucleic acid similar in structure to the natural ds DNA genome. Thus, while previous studies have clearly established that RNA-containing nucleocapsids are indeed intrinsically incompetent for envelopment and secretion, they have not resolved whether nucleocapsids containing immature DNA are secretion competent.
A synchronized DHBV replication system capable of producing staged intracellular nucleocapsids.
In order to test the intrinsic envelopment and secretion competence of nucleocapsids at different stages of reverse transcription (maturation), it is important to establish conditions where no ongoing viral DNA synthesis occurs (which may effectively prevent the secretion of immature nucleocapsids by a kinetic mechanism even if they are intrinsically secretion competent) and where potential competition from the mature nucleocapsids, known to be secretion competent, is absent (Fig. 1). We were able to satisfy these conditions with the application of a synchronized DHBV replication system that we have developed recently (4, 21). In this system, a mutant DHBV genome bearing a mutation causing temperature sensitivity, RT (CA51) (21), has been stably integrated into the genome of the chicken hepatoma cells (LMH) (6). When these stably transfected cells (called 51-3 cells) (4, 21) are cultured at the nonpermissive temperature of 39.5°C, no viral DNA synthesis occurs (Fig. 2). When the cells are shifted to the permissive temperature of 33°C, viral replication can ensue. However, when a reversible RT inhibitor, phosphonoformic acid (PFA; Foscarnet, Sigma), is added to the cells at the time of temperature shift down, the cells are able to assemble large quantities of immature, pgRNA-containing nucleocapsids with no viral DNA synthesis, since PFA blocks reverse transcription without affecting pgRNA packaging (4, 21). After the cells are allowed to accumulate these immature nucleocapsids, PFA is then removed and the preassembled pgRNA-containing nucelocapsids then initiate viral DNA synthesis in a synchronized fashion, first synthesizing immature ss DNA and then mature ds DNA.
FIG. 2.

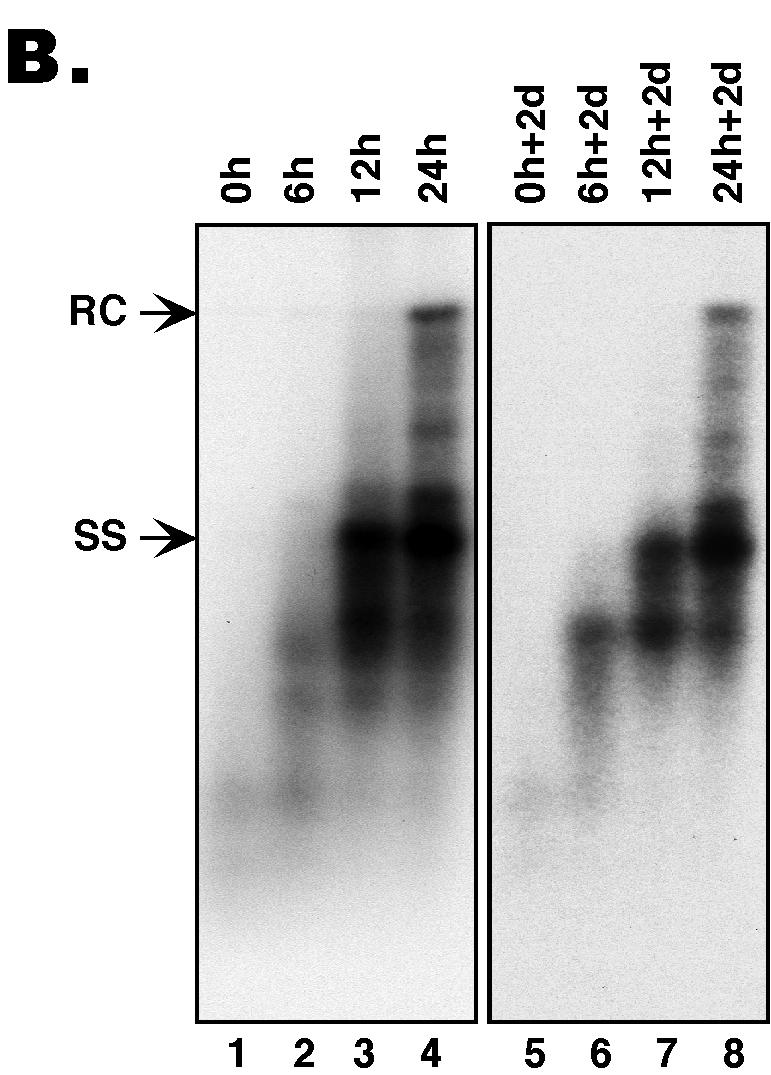
(A) The synchronized replication system. The synchronized DHBV replication system utilizes an LMH-derived cell line, 51-3, that contains a stably integrated DHBV genome harboring a temperature-sensitive (ts) RT mutant, in conjunction with the reversible RT inhibitor, PFA, to synchronize viral replication (4, 21). This system was used to assess the intrinsic secretion competence of immature nucleocapsids, in the absence of ongoing reverse transcription and potential competition from mature nucleocapsids. Cells were expanded and grown to confluence at the nonpermissive temperature (39.5°C) for viral replication and were then shifted to the permissive temperature (33°C) while PFA (final concentration of 1 mM) was added and were maintained at this temperature in the presence of PFA for a period of 3 days to allow for the accumulation of large quantities of pgRNA-containing nucleocapsids. Cells were then washed free of PFA, and DNA synthesis within the nucleocapsids was allowed to proceed in a synchronized fashion for 0, 6, 12, and 24 h. DNA synthesis was then frozen in its progression by the readdition of PFA, and the cells were maintained for an additional 2 days (d) in culture at 33°C in the presence of PFA to allow for the accumulation of secreted virions in the culture supernatants. Culture supernatants were collected both immediately before and after the second stage of PFA treatment to assay for the presence of enveloped virions. (B) Southern blot analysis of intracellular nucleocapsid DNA from the synchronized replication system. Viral DNA was extracted from nucleocapsids that had synthesized viral DNA for 0, 6, 12, and 24 h, respectively, either before (lanes 1 to 4; 0, 6, 12, and 24 h, respectively) or after (lanes 5 to 8; 0 h + 2 days [d], 6 h + 2 days, 12 h + 2 days, and 24 h + 2 days, respectively) the second PFA treatment period (the virion accumulation period) and was detected by Southern blot analysis. RC DNA (RC) is the major form of the mature, ds DNA; ss DNA (SS) is the immature reverse transcription intermediate. These experiments were repeated six times with similar results obtained.
Therefore, as schematized in Fig. 2A, 51-3 cells were expanded and grown to confluence at 39.5°C and then shifted to 33°C, with PFA being added to the culture medium at the time of temperature shift down. The cells were then maintained at 33°C for 3 days in the presence of PFA to allow for the accumulation of pgRNA-containing nucleocapsids. Thereafter, PFA was removed from a subset of the cells in order to release the reverse transcription block so that viral DNA synthesis was allowed to proceed for periods of 6, 12, and 24 h, when nascent ss, full-length ss, and then ds DNA was synthesized, respectively. At each of these time points, medium was removed from the cells and the secretion competence of the nucleocapsids containing either the nascent ss, full-length ss, or ds DNA was assessed by detecting the presence of enveloped viral particles containing these respective DNA species in the culture medium, as described below. In preliminary experiments, we found that the accumulation of secreted virions in the medium lagged behind that of the intracellular nucleocapsids (by 1 or 2 days), regardless of PFA treatment (see below, and data not shown), suggesting that the intracellular nucleocapsids required some time to be enveloped and secreted into the medium after they had gained secretion competence. Therefore, we included a second stage of PFA treatment to again arrest viral reverse transcription and allow the preexisting nucleocapsids (“frozen” at their respective stages of maturation) ample time to be secreted and to accumulate in the medium. Specifically, after viral DNA synthesis was allowed to occur for 0, 6, 12, and 24 h in the absence of PFA, PFA was added back to the culture medium at the respective time points and was maintained for 2 additional days. At the end of this second stage of PFA treatment, the medium was again collected to assay for the presence of enveloped viral particles.
In order to confirm that viral DNA synthesis was in fact arrested at the respective stages during the second period of PFA treatment so that no new DNA synthesis (thus further nucleocapsid maturation) occurred during the time of virion accumulation, viral DNA from intracellular nucleocapsids was extracted by proteinase K digestion in the presence of sodium dodecyl sulfate followed by phenol-chloroform extraction and ethanol precipitation, as previously described (4, 21). Southern blot analysis (3, 17) of intracellular viral DNA harvested from the cells immediately after the first 3-day PFA treatment (to accumulate pgRNA-containing nucleocapsids) (0-h cells [Fig. 2B, lane 1]) or after the second 2-day PFA treatment (0-h + 2-day cells [Fig. 2B, lane 5]) showed little to no viral DNA present at these times. In contrast, after 6, 12, and 24 h of release from the PFA block (Fig. 2B, lanes 2 to 4), as well as 2 days after that block had been reinstated (Fig. 2B, lanes 6 to 8), large amounts of progressively maturer viral DNA species were detected. Specifically, after 6 h, the cells contained primarily incomplete ss viral DNA, after 12 h, both complete and incomplete ss DNA, and by 24 h, a substantial amount of mature ds DNA (the major species of ds DNA being the relaxed circular [RC] form) (19, 23). These results thus confirmed that, during the second stage of PFA treatment (the virion accumulation period), the preexisting nucleocapsids were indeed frozen at their respective stages of maturation that were set before this second PFA treatment, as evidenced by the identical intracellular viral DNA patterns before and after the 2-day PFA treatment (Fig. 2B).
As the viral surface (envelope) proteins are essential for virion secretion (1), another important control experiment was performed to verify that the viral surface proteins were constitutively expressed across the time course of the experiment as anticipated. Western blot analyses demonstrated that both the small and large viral surface proteins were expressed at constant levels over the entire time course and that their expression levels also did not change with PFA treatment or culture temperature (Fig. 3; data not shown). Therefore, our experimental conditions indeed should have allowed us to detect the envelopment and secretion of any immature DNA-containing nucleocapsids with even a very low intrinsic secretion competence, due to the lack of potential competition from mature nucleocapsids and the fact that these immature nucleocapsids were allowed ample time for secretion in the absence of ongoing reverse transcription.
FIG. 3.
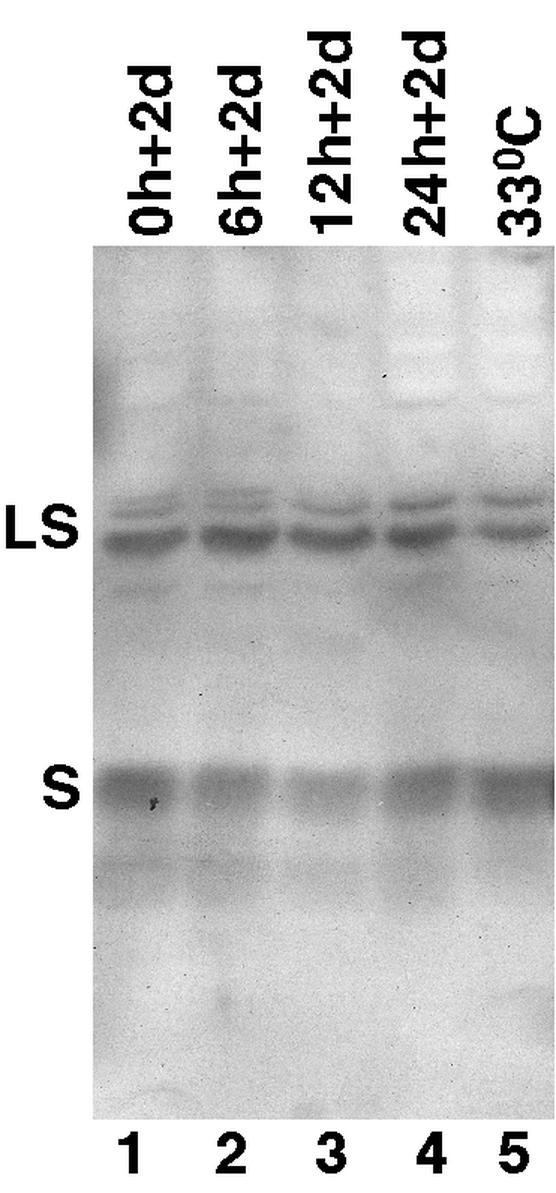
Western blot analysis of viral surface proteins. Cell culture medium was collected from the 51-3 cells at the various time points (Fig. 2) of the synchronized system (lanes 1 to 4) as well as from a steady-state culture maintained at 33°C for 7 days in the absence of PFA (lane 5). The DHBV surface proteins present in the medium samples were then detected by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blot analysis by using a monoclonal antibody specific for the S domain of the DHBV surface proteins (16). The large (LS) and small (S) surface proteins were indicated.
A stringent, two-step assay for virion secretion in cell culture.
Two different assays have been used previously to detect the presence of enveloped virion particles in cell culture medium. Due to the presence of large quantities of “naked” (nonenveloped) nucleocapsids that are released into the medium (by a still ill-defined mechanism) when hepadnaviruses replicate in established hepatoma cell lines, it is imperative that these naked nucleocapsids be removed or separated from the authentic enveloped virions before the detection of viral DNA in the culture medium can be taken as evidence of virion secretion. The classical way of separating naked nucleocapsids from virions is by isopycnic density gradient ultracentrifugation of the culture medium, which takes advantage of the lower density of the enveloped virions compared to the nucleocapsids. Another virion assay, developed more recently (28), takes advantage of the fact that enveloped virion particles, but not naked nucleocapsids, are resistant to protease digestion. Thus, protease digestion followed by nuclease treatment of the culture medium is able to selectively remove viral DNA signals in the medium contributed by the naked nucleocapsids (and potentially, naked DNA as well).
We have found that, under our experimental conditions, neither of these two assays, when used alone, was able to completely remove or separate the naked nucleocapsids from the virions. Residual amounts of naked nucleocapsids sometimes remained following the protease-DNase digestion, when medium samples from the 51-3 cells (Fig. 4) or a control cell line (D2) (kindly provided by William Mason), which constitutively secretes DHBV at high levels (12) (data not shown), was used. On the other hand, density gradient centrifugation alone, without a prior protease and nuclease digestion, was also not completely reliable as a virion assay in our experiments, as the naked nucleocapsids tended to spread out across the gradient and contaminate the virion fractions at the middle of the gradient, particularly when present in large excess over the virions (data not shown).
FIG. 4.

Intrinsic secretion competence of nucleocapsids of different maturity. Culture medium was collected from the synchronized replication system after viral DNA synthesis was allowed for 0, 6, 12, and 24 h and virion accumulation proceeded for the following 2 days (Fig. 2A); the medium samples are designated 0 h + 2 day (2D) (A), 6 h + 2 day (B), 12 h + 2 day (C), and 24 h + 2 day medium (D), respectively. Viral particles were pelleted from the medium by ultracentrifugation and were subjected to extensive proteolysis and nuclease digestion with protease and DNase I, followed by isopycnic CsCl gradient ultracentrifugation. The direction of sedimentation is indicated. Viral DNA was extracted from the untreatedmedium (lanes 1, M), the digested medium (lanes 2, P/N M), and the gradient fractions and was analyzed by Southern blotting. Sevenfold more medium was loaded in lane 2 than in lane 1. The density of the fractions containing authentic virions spanned 1.15 to 1.18 g/cm3. Note the absence of immature DNA species in the virion fractions at all time points. Residual naked nucleocapsids remaining after pronase-DNase I digestion migrated toward the bottom of the CsCl gradient, well separated from the virions (D). The extra lane on the right of panel D is a longer exposure of the last lane in the panel, showing the presence of immature viral DNA species at the bottom of the CsCl gradient. RC, RC DNA; ss, ss DNA. These experiments were repeated twice with essentially the same results obtained.
Since we were interested in detecting very small amounts of secreted virions in the presence of a large excess of naked nucleocapsids in the culture medium, it was critical that the naked nucleocapsids be completely removed from the virion fractions. Otherwise, residual amounts of naked nucleocapsids would be taken, erroneously, as evidence of envelopment and secretion (albeit at low efficiency), as might have been the case for nucleocapsids containing intermediate reverse transcription products. Therefore, we combined these two procedures by first performing a protease-DNase digestion step, which should help to eliminate most if not all naked nucleocapsids, followed by density gradient centrifugation of the digested medium material, which should be able to separate any residual amounts of remaining naked nucleocapsids from authentic virions.
Thus, viral particles (both enveloped virions and naked nucleopcasid) were first pelleted from clarified culture medium by overnight ultracentrifugation over a cushion of 30% sucrose in TNE (10 mM Tris, pH 8.0, 1 mM EDTA, and 100 mM NaCl) with 0.05% β-mercaptoethanol (β-ME) in a Sorvall Surespin 630 rotor at 30,000 rpm at 4°C. The pellets were resuspended in 1/100 of the original medium volume of TNE-0.05% β-ME by sonication in a cup horn (Fisher 550 Sonic Dismembrator). The resuspended viral pellets were then subjected to extensive protease-DNase I digestion according to a protocol adapted from Yu and Summers (28). Briefly, 0.75 mg of pronase (Boehringer Mannheim)/ml (final concentration) was added to the viral resuspension and the samples were incubated at 37°C for 60 min. Nuclease digestion was then conducted by adding a 6 mM final concentration of magnesium acetate and a 1-mg/ml final concentration of DNase I (Roche), incubating at 37°C for 30 min, and then adding an additional 1 mg of DNase I/ml and incubating for an additional 30 min at 37°C. Reactions were terminated by adding 15 mM EDTA, pH 8.0. The protease-DNase digested samples were subsequently subjected to the second step of isopycnic density gradient centrifugation in order to separate any naked nucleocapsids remaining after the protease-DNase digestion from the authentic virions. The digested samples were transferred to ultracentrifuge tubes, and an equal volume of a 2× CsCl solution (49.74% [wt/vol] CsCl in TNE-0.05% β-ME) was added so that the density of the solution was 1.18 g/cm3. The solutions were ultracentrifuged in a Beckman SW55 Ti rotor at 54,000 rpm, 4°C, for approximately 96 h. Fractions were then collected into siliconized tubes. Viral DNA was then extracted from the undigested medium, the protease-DNase-digested medium, and the fractions collected from the CsCl density gradient and was detected by Southern blot analysis as described above for intracellular viral DNA.
As will be evident below, we found that this two-step procedure was very effective in completely removing the naked nucleocapsids from the virions (Fig. 4 and data not shown). Therefore, we adopted the two-step procedure as a stringent virion secretion assay, which, when applied to the synchronized replication system described above, allowed us to confidently detect the presence of even low levels of enveloped and secreted immature nucleocapsids, if they were indeed secretion competent.
Only mature nucleocapsids were secretion competent, whereas immature nucleocapsids were intrinsically secretion incompetent.
Using the two-step virion secretion assay, we sought to determine at what stage of reverse transcription the nucleocapsids became enveloped and secreted into the medium of the synchronized system. Culture medium of the 51-3 cells was collected at each of the four time points (0, 6, 12, and 24 h after releasing the PFA block) (Fig. 2A) when the intracellular nucleocapsids contained, respectively, only pgRNA, the nascent ss DNA, full-length ss DNA (but little or no ds DNA), and finally, the mature ds DNA. Furthermore, a second round of medium was collected, as described earlier (Fig. 2), after a second stage of PFA treatment that was employed to allow the nucleocapsids, arrested at various stages of maturation, ample time to be enveloped and secreted (the 2-day virion accumulation period). Viral particles in the culture supernatant were pelleted by ultracentrifugation and were then subjected to extensive protease and nuclease treatment to eliminate the majority of the naked nucleocapsids and were then further purified by isopycnic CsCl gradient ultracentrifugation to separate authentic virions from any remaining nucleocapsids (as described above). Similar results were obtained with respect to the profile of viral DNA detectable in the culture medium, with or without the additional 2-day period of virion accumulation, although, without it, the amount of viral DNA detected was substantially lower (data not shown). Therefore, only the results obtained from the culture medium after the virion accumulation period are shown in Fig. 4.
Southern blot analysis of viral DNA showed that the concentrated raw culture medium contained a DNA profile (Fig. 4, lanes 1) that was essentially the same as the intracellular core DNA pattern at each time point (Fig. 2B), reflecting the substantial presence of naked nucleocapsids in the culture medium. After extensive protease and nuclease treatment, much of the viral DNA signal had been eliminated (Fig. 4, lanes 2), whereas the mature ds DNA (mainly the RC species) contained in the enveloped virions was significantly enriched, reflecting the preferential degradation of the naked nucleocapsids harboring immature DNA species. Still, significant amounts of immature DNA species remained, which might have suggested that immature nucleocapsids containing these DNA species could be secreted at low efficiency. However, when the digested medium samples were then subjected to isopycnic CsCl gradient ultracentrifugation, a strong, discrete DNA peak emerged at the characteristic virion density only in the 24-hour time point sample (Fig. 4D). Moreover, this authentic virion DNA consisted exclusively of only the most mature viral DNA species. The immature DNA species that had remained in the digested medium samples were recovered from denser fractions near the bottom of the gradient, as expected of naked nucleocapsids (Fig. 4D). This indicated that the detection of the immature DNA species in the digested medium samples before CsCl centrifugation reflected incomplete digestion of naked nucleocapsids by the protease and nuclease. We noticed that the recovery of these immature DNA species was less than complete, likely due to the tendency of naked nucleocapsids to aggregate and to adhere to the centrifuge tubes in the CsCl gradient without any detergent, resulting in their apparent loss (14; J. Hu, unpublished observation).
Importantly, trace amounts of mature RC DNA, present at earlier time points and enriched in the digested medium, could be recovered quantitatively in the virion peaks from the CsCl gradient (Fig. 4A and B). The small number of virions secreted into the medium at these earlier time points resulted from minute leak-through reverse transcription at the nonpermissive temperature (4, 21), leading to the formation of trace quantities of mature, secretion competent, RC DNA-containing nucleocapsids (Fig. 2B). The detection of such a minute number of secreted virions at the earlier time points, in the presence of a huge excess of naked nucleocapsids containing immature DNA species, served as an important internal control for the detection of even very low levels of authentic virions at these early time points. It effectively excluded the possibility that a small number of virions containing immature DNA species might have been present but were lost due to overdigestion (removal of virions in addition to nucleocapsids) with the protease and nuclease. Additionally, a control digestion consisting of DHBV-infected duck serum (containing only virions and no naked capsids) mixed together with intracellular, immature nucleocapsids (harvested from 51-3 cells at the 6-h time point, Fig. 2B, lane 2) showed no evidence of overdigestion under identical conditions, as would have been indicated by a decrease in the serum-derived virion DNA signal (data not shown). Thus, in summary, the complete absence of any detectable amounts of immature DNA species in the virion density regions of the isopycnic gradient at any time points thus demonstrated, conclusively, that nucleocapsids containing these DNA species were indeed incapable of being secreted as enveloped virions, even in the absence of ongoing reverse transcription or potential competition from mature nucleocapsids.
These results have therefore effectively excluded the kinetic or competition model as possible explanations for the nucleocapsid maturation phenomenon. Instead, they strongly support the intrinsic, or maturation signal, model as the underlying mechanism of the maturation phenomenon. These results have also further defined the precise stage during the process of nucleocapsid maturation when the maturation signal emerges: only the nucleocapsids that contained nearly complete ds DNA were secreted as virions, indicating that the appearance of the maturation signal is coupled to the late stage of reverse transcription, as observed during natural hepadnavirus infections (10, 23). The rather homogeneous nature of the DNA genome in the secreted virions suggests that the maturation signal may emerge abruptly (rather than gradually), as viral DNA synthesis approaches completion, at least for DHBV. Alternatively, the maturation signal may actually emerge earlier during plus-strand DNA synthesis but a delay between the emergence of the maturation signal and the complete envelopment of the nucleocapsid (which would prevent further DNA elongation owing to the unavailability of nucleotide substrates to the enveloped particles) may permit the near-completion of plus-strand DNA synthesis. The second possibility may accommodate the different genome structures between DHBV and HBV, with the former containing a nearly complete ds DNA genome (10) while the latter contains a partially ds DNA genome with an incomplete plus strand heterogeneous in size ranging from approximately 50% to nearly complete (18, 23). Thus, it is possible that the maturation signal emerges shortly after the plus-stand DNA synthesis is half completed for both HBV and DHBV; however, plus-strand DNA synthesis may be slower, or the envelopment process faster, in HBV than in DHBV, thus leading to the presence of the incomplete plus strands in HBV virions,
Our results may also shed new light on the previous observation that inactivation of the RNase H activity of the viral RT leads to the secretion of ss DNA-containing virions (2, 26). We suggest that the apparent secretion of these immature particles may result from the presence in those nucleocapsids of a ds, DNA-RNA hybrid consisting of the ss DNA hybridized to the pgRNA, which would normally be degraded by the RNase H activity during ss DNA synthesis. Together, these results imply that the trigger for the emergence of the maturation signal on the nucleocapsids is a ds nucleic acid, which is normally the ds DNA genome but can be mimicked by a DNA-RNA hybrid, as in the case of the RNase H mutant (or possibly even by a ds RNA molecule were that to be packaged into the nucleocapsids). This suggests that it may be important to determine the state of the pgRNA in the nucleocapsids when immature nucleocapsids containing ss DNA are apparently secreted as virions, as in the case of some HBV core protein mutants (13). It appears that, even during a natural infection, some virions may contain a DNA-RNA hybrid genome (11), possibly due to a defect in pgRNA degradation.
Little is presently known about the nature of the maturation signal. However, the viral core protein, which forms the nucleocapsid shell, appears to be a logical candidate for the bearer of such a signal. It is perfectly situated to transmit the information regarding the nature of the nucleic acid from within the nucleocapsid, across the capsid shell, to its exterior, so that this information may be recognized by the secretion machinery, including the viral envelope proteins. This notion has gained support from mutational analyses of the core protein. Thus, both naturally occurring (9) and engineered HBV core protein mutations (8) have been found that allow complete viral DNA synthesis (including the mature ds DNA) but, nevertheless, block virion secretion. In addition, other core protein mutants can induce the envelopment and secretion of immature nucleocapsids containing ss DNA without affecting intracellular viral DNA synthesis (29, 30). How the maturation signal may emerge on the core protein is presently unknown, but a posttranslational modification of the core protein, which is coupled to the synthesis of the mature ds DNA can, in principle, generate such a signal. Indeed, it was reported early on that the virion-derived core protein is hypophosphorylated compared to the core protein derived from intracellular nucleocapsids (15), suggesting that dephosphorylation of the core protein occurs sometime during the maturation and secretion process. Other candidates include a conformational/structural change of the nucleocapsids, perhaps induced by the dephosphorylation of the core protein. Future studies are required to carefully examine the nucleocapsids harvested across the whole maturation process in order to correlate any biophysical and biochemical properties of these nucleocapsids with their acquisition of secretion competence.
The conveyance of information regarding the interior of a capsid particle across the capsid shell to its exterior can be considered a special case of signal transduction that is not limited to the hepadnaviruses. For example, it has been reported recently that the human immunodeficiency virus proviral DNA structure (specifically the presence of a central DNA flap), which is contained in a capsid-like nucleoprotein complex, is somehow sensed by the host cell nuclear import machinery, leading to enhanced nuclear import of the viral DNA (31). The hepadnavirus DNA genome also needs to be imported into the nucleus to be converted to the covalently closed circular DNA (25), the viral transcriptional template, both after its initial entry into the cell and during the subsequent covalently closed circular DNA amplification, which is thought to be necessary for viral persistence. It is thus conceivable that hepadnaviruses may have evolved the nucleocapsid maturation mechanism not only to quality control virus exit from the producing cell but also to ensure that the viral genome can gain access to the cell nucleus, which is critical for establishing and maintaining viral replication. It will be important, in that light, to determine the nuclear entry competence of immature as well as of mature hepadnavirus DNA.
Acknowledgments
We thank Christoph Seeger for providing the 51-3 cells and for helpful discussions, William Mason for the D2 cells, and William Mason and Jesse Summers for a critical reading of the manuscript.
This work was supported by a Public Health Service grant (R01 AI43453) from the National Institutes of Health.
REFERENCES
- 1.Bruss, V., and D. Ganem. 1991. The role of envelope proteins in hepatitis B virus assembly. Proc. Natl. Acad. Sci. USA 88:1059-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerelsaikhan, T., J. Tavis, and V. Bruss. 1996. Hepatitis B virus nucleocapsid envelopment does not occur without genomic DNA synthesis. J. Virol. 70:4269-4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu, J., and C. Seeger. 1996. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc. Natl. Acad. Sci. USA 93:1060-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu, J., D. O. Toft, and C. Seeger. 1997. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J. 16:59-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jilbert, A. R., D. S. Miller, C. A. Scougall, H. Turnbull, and C. J. Burrell. 1996. Kinetics of duck hepatitis B virus infection following low dose virus inoculation: one virus DNA genome is infectious in neonatal ducks. Virology 226:338-345. [DOI] [PubMed] [Google Scholar]
- 6.Kawaguchi, T., K. Nomura, Y. Hirayama, and T. Kitagawa. 1987. Establishment and characterization of a chicken hepatocellular carcinoma cell line, LMH. Cancer Res. 47:4460-4464. [PubMed] [Google Scholar]
- 7.Köck, J., S. Wieland, H. E. Blum, and F. von Weizsäcker. 1998. Duck hepatitis B virus nucleocapsids formed by N-terminally extended or C-terminally truncated core proteins disintegrate during viral DNA maturation. J. Virol. 72:9116-9120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koschel, M., D. Oed, T. Gerelsaikhan, R. Thomssen, and V. Bruss. 2000. Hepatitis B virus core gene mutations which block nucleocapsid envelopment. J. Virol. 74:1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le Pogam, S., T. T.-T. Yuan, G. K. Sahu, S. Chatterjee, and C. Shih. 2000. Low-level secretion of human hepatitis B virus virions caused by two independent, naturally occurring mutations (P5T and L60V) in the capsid protein. J. Virol. 74:9099-9105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lien, J.-M., D. J. Petcu, C. E. Aldrich, and W. S. Mason. 1987. Initiation and termination of duck hepatitis B virus DNA synthesis during virus maturation. J. Virol. 61:3832-3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller, R. H., C. T. Tran, and W. S. Robinson. 1984. Hepatitis B virus particles of plasma and liver contain viral DNA-RNA hybrid molecules. Virology 139:53-63. [DOI] [PubMed] [Google Scholar]
- 12.Moraleda, G., T. T. Wu, A. R. Jilbert, C. E. Aldrich, L. D. Condreay, S. H. Larsen, J. C. Tang, J. M. Colacino, and W. S. Mason. 1993. Inhibition of duck hepatitis B virus replication by hypericin. Antivir. Res. 20:235-247. [DOI] [PubMed] [Google Scholar]
- 13.Nassal, M. 1992. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J. Virol. 66:4107-4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oberhaus, S. M., and J. Newbold. 1993. Detection of DNA polymerase activities associated with purified duck hepatitis B virus core particles by using an activity gel assay. J. Virol. 67:6558-6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pugh, J., A. Zweidler, and J. Summers. 1989. Characterization of the major duck hepatitis B virus core particle protein. J. Virol. 63:1371-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pugh, J. C., and H. Simmons. 1994. Duck hepatitis B virus infection of Muscovy duck hepatocytes and nature of virus resistance in vivo. J. Virol. 68:2487-2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pugh, J. C., K. Yaginuma, K. Koike, and J. Summers. 1988. Duck hepatitis B virus (DHBV) particles produced by transient expression of DHBV DNA in a human hepatoma cell line are infectious in vitro. J. Virol. 62:3513-3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robinson, W. S., D. A. Clayton, and R. L. Greenman. 1974. DNA of a human hepatitis B virus candidate. J. Virol. 14:384-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sattler, F., and W. S. Robinson. 1979. Hepatitis B viral DNA molecules have cohesive ends. J. Virol. 32:226-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seeger, C., and J. Hu. 1997. Why are hepadnaviruses DNA and not RNA viruses? Trends Microbiol. 5:447-450. [DOI] [PubMed] [Google Scholar]
- 21.Seeger, C., E. H. Leber, L. K. Wiens, and J. Hu. 1996. Mutagenesis of a hepatitis B virus reverse transcriptase yields temperature-sensitive virus. Virology 222:430-439. [DOI] [PubMed] [Google Scholar]
- 22.Summers, J., and W. S. Mason. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403-415. [DOI] [PubMed] [Google Scholar]
- 23.Summers, J., A. O'Connell, and I. Millman. 1975. Genome of hepatitis B virus: restriction enzyme cleavage and structure of DNA extracted from Dane particles. Proc. Natl. Acad. Sci. USA 72:4597-4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tencza, M. G., and J. E. Newbold. 1997. Heterogeneous response for a mammalian hepadnavirus infection to acyclovir: drug-arrested intermediates of minus-strand viral DNA synthesis are enveloped and secreted from infected cells as virion-like particles. J. Med. Virol. 51:6-16. [PubMed] [Google Scholar]
- 25.Tuttleman, J. S., C. Pourcel, and J. Summers. 1986. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 47:451-460. [DOI] [PubMed] [Google Scholar]
- 26.Wei, Y., J. E. Tavis, and D. Ganem. 1996. Relationship between viral DNA synthesis and virion envelopment in hepatitis B viruses. J. Virol. 70:6455-6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu, M., and J. Summers. 1991. A domain of the hepadnavirus capsid protein is specifically required for DNA maturation and virus assembly. J. Virol. 65:2511-2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu, M., and J. Summers. 1994. Multiple functions of capsid protein phosphorylation in duck hepatitis B virus replication. J. Virol. 68:4341-4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan, T. T.-T., G. K. Sahu, W. E. Whitehead, R. Greenberg, and C. Shih. 1999. The mechanism of an immature secretion phenotype of a highly frequent naturally occurring missense mutation at codon 97 of human hepatitis B virus core antigen. J. Virol. 73:5731-5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuan, T. T.-T., P.-C. Tai, and C. Shih. 1999. Subtype-independent immature secretion and subtype-dependent replication deficiency of a highly frequent, naturally occurring mutation of human hepatitis B virus core antigen. J. Virol. 73:10122-10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zennou, V., C. Petit, D. Guetard, U. Nerhbass, L. Montagnier, and P. Charneau. 2000. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell 101:173-185. [DOI] [PubMed] [Google Scholar]