

Abstract
During exocytosis a four-helical coiled coil is formed between the three SNARE proteins syntaxin, synaptobrevin and SNAP-25, bridging vesicle and plasma membrane. We have investigated the assembly pathway of this complex by interfering with the stability of the hydrophobic interaction layers holding the complex together. Mutations in the C-terminal end affected fusion triggering in vivo and led to two-step unfolding of the SNARE complex in vitro, indicating that the C-terminal end can assemble/disassemble independently. Free energy perturbation calculations showed that assembly of the C-terminal end could liberate substantial amounts of energy that may drive fusion. In contrast, similar N-terminal mutations were without effects on exocytosis, and mutations in the middle of the complex selectively interfered with upstream maturation steps (vesicle priming), but not with fusion triggering. We conclude that the SNARE complex forms in the N- to C-terminal direction, and that a partly assembled intermediate corresponds to the primed vesicle state.
Keywords: capacitance measurements, chromaffin cells, exocytosis, SNAP-25, SNARE proteins
Introduction
Formation of a tight, four-helical bundle (the SNARE complex) between the membranes is necessary for membrane fusion. The best-studied SNARE complex, consisting of synaptobrevin 2, syntaxin 1 and SNAP-25, is involved in the exocytosis of neurotransmitter-filled vesicles in the synapse. SNAP-25 contributes two α-helices to the complex and is localized to the plasma membrane by palmitoylation, whereas synaptobrevin and syntaxin are anchored in the vesicular and plasma membrane, respectively, via transmembrane domains (Figure 1). The membrane anchors of synaptobrevin and syntaxin are both placed on the C-terminal side of the assembled complex (Hanson et al, 1997; Sutton et al, 1998). This arrangement gave rise to the idea that SNARE complex formation may occur in a ‘zipper-like' fashion, proceeding in the N- to C-terminal direction (Hanson et al, 1997). In this scenario, the formation energy of the trans-SNARE complex helps overcome the energy barrier for fusion of the membranes. Even though this model is highly attractive and has been investigated in a number of studies, it remains unproven.
Figure 1.
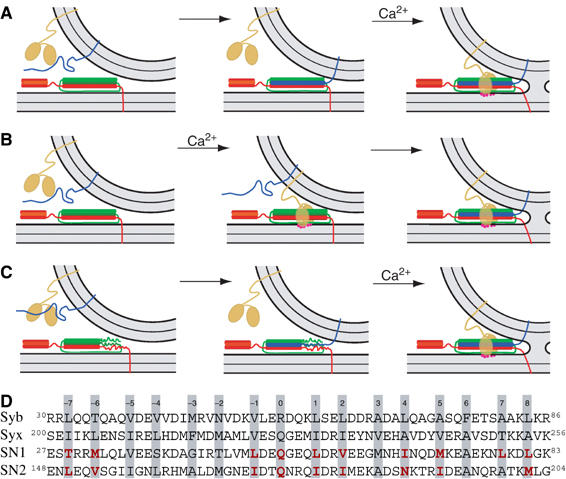
Alternative scenarios for SNARE action. Orange protein: synaptotagmin; blue: synaptobrevin, red: syntaxin; green: SNAP-25; magenta dots: Ca2+. The α-helical and unstructured parts of the cytoplasmic SNARE regions are indicated by cylinders and lines, respectively. In all scenarios we assume that a 1:1 SNAP-25:syntaxin receptor complex comprising both helices of SNAP-25 has already formed on the plasma membrane. (A) SNARE complex assembly is completed before fusion is triggered by Ca2+-binding to synaptotagmin. (B) SNARE assembly is initiated upon Ca2+-binding to synaptotagmin. (C) The SNARE complex is assembled into a half-zippered complex. Calcium-triggering and final SNARE assembly are coupled. (D) Sequence of the SNARE ‘core complex', with mutated layer residues emphasized in red. Syb: synaptobrevin; syx: syntaxin; SN1: N-terminal helix of SNAP-25A; SN2: C-terminal helix of SNAP-25A.
Studies of the soluble part of the SNAREs have shown that SNARE complex formation nucleates rather slowly through an N-terminal interaction between syntaxin and SNAP-25 (Fasshauer and Margittai, 2004). The 1:1 syntaxin:SNAP-25 complex serves as an acceptor site for synaptobrevin binding, but can also bind a second syntaxin in vitro; a step that is unlikely to occur in the cell. Yet, this competing reaction has hindered the determination of the rate and mode of synaptobrevin binding. Structural studies of the homologous yeast SNAREs showed that the binary complex is partly unstructured in the C-terminal end, whereas the ternary complex is fully structured. It was thus suggested that a ternary SNARE complex with a partly unstructured C-terminal end might be an intermediate on the pathway to secretion (Fiebig et al, 1999). Nevertheless, there is no direct evidence showing that the ternary complex can exist in a partly folded state.
SNAREs reconstituted into liposomes can drive membrane fusion, albeit with slow kinetics (Weber et al, 1998). A C-terminal fragment of the synaptobrevin SNARE domain accelerated the fusion rate, and it was suggested that this effect is caused by the induction of structure into a partially formed ternary SNARE complex (Melia et al, 2002). However, the slow fusion rate in vitro may be due to the formation of an unproductive 2:1 syntaxin:SNAP-25 complex that is prevented by the synaptobrevin fragment. Using peptide infusion into permeabilized PC12 cells, it was found that SNARE complex assembly could not be dissociated from Ca2+-triggering (Chen et al, 1999). Infusion of syntaxin fragments supported N- to C-terminal formation of the SNARE complex, but it was argued that a partly formed complex is unlikely to exist (Matos et al, 2003). However, secretion in PC12 cells is very slow (τ∼30 s) compared to neurosecretory cells (τ∼20 ms), possibly because PC12 cells lack a population of vesicles that have matured to be ‘release-ready' (Martin, 2003).
Fusion of release-ready vesicles underlies the first, fast secretory phase in neurosecretory cells and neurons. Kinetic consequences of SNARE cleavage (by neurotoxins), sequestering (by antibodies), elimination (by genetic techniques) or overexpression studies have all indicated that the SNAREs are intimately involved in the last steps of exocytosis (Capogna et al, 1997; Xu et al, 1998, 1999; Hua and Charlton, 1999; Han et al, 2004; Borisovska et al, 2005; Sakaba et al, 2005), and some investigators have suggested the existence of partly folded intermediates (Hua and Charlton, 1999; Xu et al, 1999). However, with these approaches it was not possible to unambiguously identify intermediate steps in the fusion pathway.
Alternative models have been proposed for the SNARE assembly pathway. For example, it was suggested that the C-terminal end of the synaptobrevin SNARE domain is buried in the vesicular membrane, preventing SNARE complex formation (Hu et al, 2002; Kweon et al, 2003). This finding raised the possibility that SNARE complex formation may start from the C-terminal end (Kweon et al, 2003).
In summary, two crucial aspects of SNARE action are still unclear: does the complex indeed assemble in the N- to C-terminal direction, and does assembly occur in a single reaction, or through a partially assembled intermediate? To address these questions, we have now combined analysis of SNARE assembly/stability in vitro with functional analysis of SNAREs in fast neurotransmitter release from chromaffin cells. In these cells vesicle maturation is arrested at the ‘release-ready' state, where the vesicle awaits the calcium trigger for fusion. Using electrophysiological methods it is possible to distinguish between ‘priming reactions', which lead up to this state, and ‘triggering reactions', which lead to fusion from this state. This makes it possible to study the assembly of the SNARE complex, as it applies to these two steps. Three different scenarios can be envisioned: the SNARE complex may form completely either before (Figure 1A) or after (Figure 1B) Ca2+-triggering. These scenarios do not allow the distinction of more than one assembly event, and would thus not provide information about the assembly direction or the existence of intermediates. However, in the third scenario the SNARE complex forms partly during the priming step and then completes assembly simultaneously with the Ca2+-triggering step (Figure 1C). In this case an assembly intermediate coincides with the readily releasable state, and localized interference with complex assembly should allow the determination of the assembly direction, that is, if N- to C-terminal assembly drives secretion (as in Figure 1C), C-terminal interference would affect exocytosis triggering, whereas more N-terminal interventions would affect vesicle priming.
Results
In order to locally destabilize the SNARE complex, we mutated layer residues to alanines, because its small apolar side chain would not introduce steric hindrance of SNARE complex formation or change the surface charge of the complex. This is important, because several other proteins (synaptotagmins, complexins) bind to the surface of the SNARE complex through polar interactions. To evaluate the function of SNAP-25 in a gain-of-function framework, we overexpressed mutated or wild-type (WT) protein in SNAP-25 null embryonic chromaffin cells (Sørensen et al, 2003) and performed parallel measurement of membrane capacitance and amperometric detection of liberated catecholamines. Secretion was elicited by flash photolysis of caged-Ca2+, which led to a step-like increase of [Ca2+]i (see, e.g., Figure 2Aa). Flash-induced secretion occurs in several phases, which are usually assumed to represent sequential events in the exocytotic cascade. A rapid burst of secretion within the first 0.5–1 s after the [Ca2+]i increase is caused by fusion of vesicles that were release-ready before stimulation, whereas the much slower sustained component most likely represents the recruitment (referred to as priming) of new vesicles into the releasable pool (Sørensen, 2004). Kinetic analysis allows determination of the sizes of the two releasable pools (readily releasable pool (RRP) and slowly releasable pool (SRP)), and the rate of the sustained component, all of which report on the priming process, and the fusion time constants, which report on the fusion-triggering process.
Figure 2.
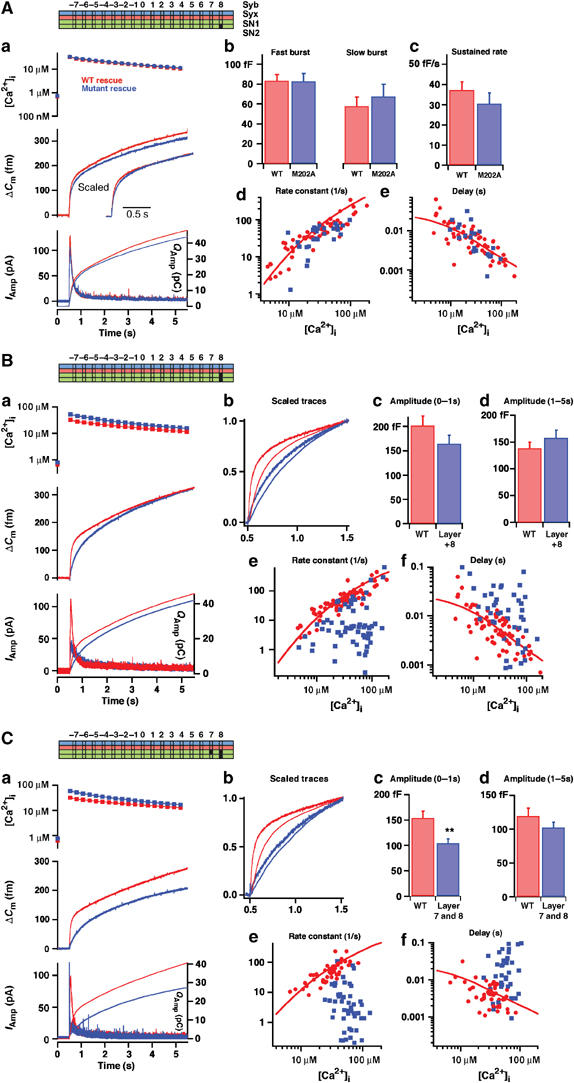
Alanine substitutions in the C-terminal end of the SNARE complex slow down the triggering phase of exocytosis. (Aa) Averaged results for SNAP-25 null cells overexpressing WT SNAP-25 (rescue, black) or SNAP-25 M202A (gray). Top panel: mean±s.e.m. [Ca2+]i before (point on left ordinate axis) and after a flash of UV light liberated calcium from the calcium cage (at 0.5 s). Middle panel: mean capacitance increase; inset: capacitance traces scaled to similar amplitude 1 s after the flash to allow comparison of the fast phase of secretion. Bottom panel: amperometric current (thick traces, left ordinate axis) and cumulative amperometric charge (thin traces, right ordinate axis). Number of cells: N=38 (WT) and N=27 (M202A). (Ab–e) Results of the kinetic analysis of individual capacitance traces (Materials and methods). (Ab) The amplitudes of the fast and slow bursts of secretion correspond to the sizes of the RRP and the SRP, respectively. (Ac) The sustained rate of secretion defines the slope of the near-linear capacitance increase following the burst phase. (Ad) Relationships between the fastest rate constant (1/time constant) resolvable in the exponential fit and the post-flash [Ca2+]i. For WT SNAP-25 rescue this is an increasing relationship. (Ae) Relationship between the secretory delay and the post-flash [Ca2+]i. For WT rescue this is a decreasing relationship. (Ad, e) Lines represent the predictions of a sequential calcium-binding site model of the fast calcium sensor for exocytosis (Voets, 2000) and describe the data in the WT situation. These data show no major change in secretion upon mutation of M202 into alanine. (B) Double mutation in layer +8 (L81A/M202A) slows down the fast phase of secretion. (Ba) Mean results; (Bb) capacitance (thick traces) and cumulative amperometric charge (thin traces) scaled to similar amplitudes at 1 s after the flash, clearly showing the slowdown induced by mutation. (Bc, d) The mean±s.e.m. capacitance increase in 0–1 s and 1–5 s after the flash is given here, because due to the slowdown caused by mutation a kinetic separation of the fast and slow bursts was not always possible. (Be, f) Result of kinetic analysis. The rate constant of the fastest resolvable phase (Be) and the delay (Bf) for WT and mutant experiments are shown. N=51 cells (L81A/M202A) and N=57 cells (WT). (C) Triple mutation in layers +7 and +8 (L78A/L81A/M202A) dramatically slows down the fast phase of secretion. For explanations of (a)–(f), see (B). With the triple mutation, the secretion within the first second of stimulation is depressed, and the secretory rates are even slower and the delays even longer than with the double mutation. N=49 cells (L78A/L81A/M202A) and N=51 cells (WT).
Mutations in the C-terminal end of the complex: electrophysiology
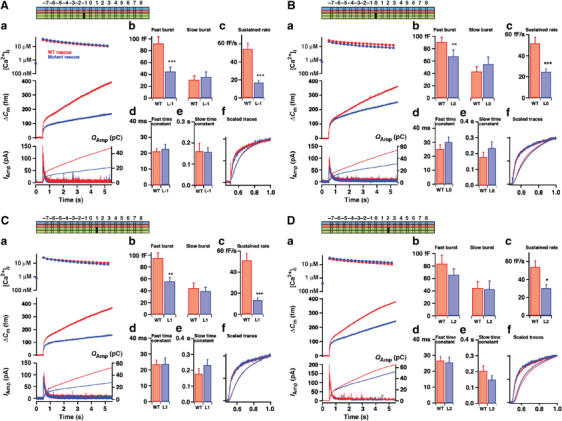
We first targeted the most C-terminal (membrane-proximal) hydrophobic layer of the complex (+8; Figure 1D), which consists of a methionine (Met-202) and a leucine (Leu-81) from SNAP-25, a leucine from synaptobrevin and an alanine from syntaxin. When mutating Met-202 to Ala in SNAP-25, rescued chromaffin cells showed the same level of secretion as in cells rescued with WT SNAP-25 (Figure 2Aa). Kinetic analysis revealed that neither the amplitude of the two exocytotic burst phases nor the sustained rate of release was changed (Figure 2Ab and c), indicating intact priming in these cells. The rate constant of release from the RRP (=1/τ, where τ is the time constant of the fastest exponential) and the time delay (from the flash of UV light until the start of the capacitance increase) were unchanged by this mutation over the range of investigated [Ca2+]i (Figure 2Ad and e), indicating intact exocytosis triggering. When the single mutation Leu-81 to alanine (L81A) was tested, a roughly unchanged exocytosis pattern was found, except for a slight, but significant, decrease in the size of the RRP and a slower rate of exocytosis triggering in some cells (Supplementary Figure 1).
To elicit a stronger phenotype, we combined the two mutations (Figure 2B). The double mutation L81A/M202A led to a noticeable slowdown of the fastest phase of secretion (Figure 2Ba). The amplitude of secretion within the first 1 s or within the time period 1–5 s after the flash was, however, not significantly changed (Figure 2Bc and d). We analyzed the fastest resolvable rate constant of secretion and the secretory delay. This revealed substantially slower triggering kinetics with the double mutation, compared to WT (Figure 2Be and f). Alternatively, as a model-independent way of analyzing triggering speed, we scaled the capacitance increase and the integrated amperometric charge in mutant and WT to the same amplitude at 1 s after the flash. This confirmed the slowdown of the fast-phase secretion by the double mutation (Figure 2Bb).
We next combined the L81A/M202A mutation with a mutation in layer +7:L78A. The triple mutation L78A/L81A/M202A displayed substantial slowdown of secretion (Figure 2Ca), and also reduced secretion at 1 s after the flash (Figure 2Cc). This effect was more severe than for the double mutation, which was reflected in slower rates and longer delays (Figure 2Ce and f). Slowdown of secretion was confirmed in the amperometric trace (Figure 2Cb).
The flash photolysis data pointed to a more severe phenotype of the triple mutation compared to the double mutation. To investigate this further, we stimulated the cells using slowly increasing calcium concentrations (calcium ramps). The simultaneously measured capacitance change indicated that secretion was shifted to later times (or higher calcium concentrations) with the double mutation, but even more so with the triple mutation (Supplementary Figure 2), indicating a graded effect of accumulating mutations in the C-terminal end of the SNARE complex.
With the triple mutation our possibilities for making alanine substitutions in the last layers were exhausted, since the other amino acids from SNAP-25 in layers +7 and +6 are already alanines (Figure 1D). To further destabilize the last layers, we deleted the last nine amino acids (Δ9 mutant) in the C-terminal helix of SNAP-25, corresponding to the Botulinum Neurotoxin A (BoNT/A) cleavage product. For comparison we also deleted the last 26 amino acids (Δ26 mutant), corresponding to cleavage by BoNT/E. When raising [Ca2+]i to 20–30 μM, neither of these two mutants supported secretion, when compared to SNAP-25 null cells (Figure 3A). The Δ26 deletion actually acted as a dominant negative, depressing secretion to below SNAP-25 null levels. In the same cells we performed a second stimulation, increasing the post-flash [Ca2+]i to >100 μM (Figure 3B). Under these conditions a large increase in cell membrane capacitance is normally observed, which does not correlate with the amperometric current and has been attributed to the fusion of another population of vesicles that does not contain catecholamines (Xu et al, 1998). Therefore, under these conditions we evaluated the integrated amperometric current as a measure of catecholamine secretion (Materials and methods). The released amperometric charge was significantly higher in the Δ9 mutation than in SNAP-25 null cells, whereas the Δ26 was still dominant negative when compared to the null (Figure 3B, lower panel). Therefore, the Δ9 mutation can drive secretion of catecholamine-containing vesicles; however, it does so at much higher [Ca2+]i—even higher than the L78A/L81A/M202A triple mutation. The Δ26 mutation, on the other hand, seems unable to support secretion at all.
Figure 3.
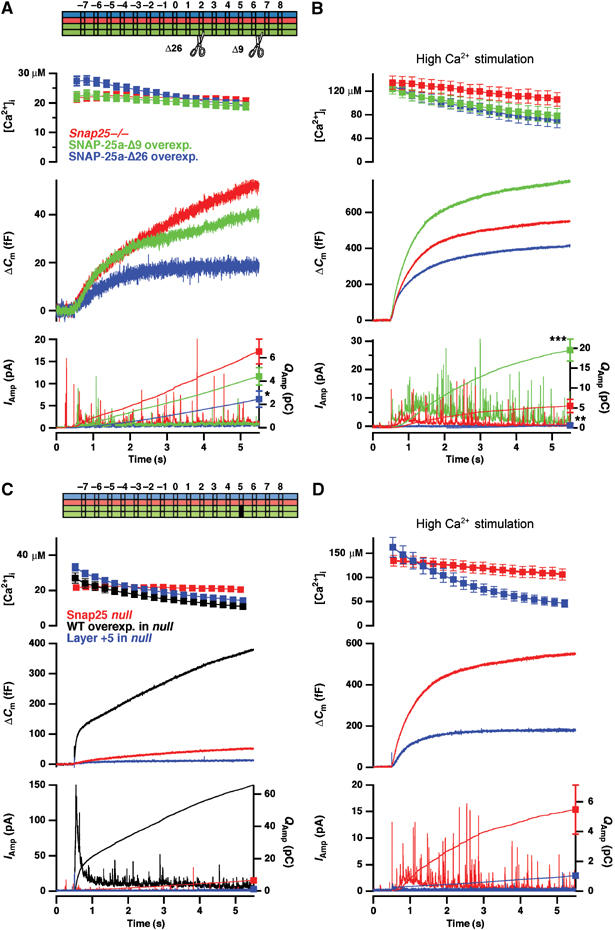
C-terminal deletions of SNAP-25, or mutation in layer +5, display dramatic phenotypes. (A) Averaged results of deleting the C-terminal 9 (Δ9) or 26 (Δ26) last amino acids compared to SNAP-25 null cells. Capacitance measurements revealed no rescue of secretion by the deletions. Bottom panel: The amperometric currents were corrected for artifacts (Materials and methods) in order to allow quantitative analysis. Note that the amplitude of individual spikes is reduced, due to averaging. The Δ26 mutation significantly reduced the amperometric charge released during the 5-s period. N=33 cells (null); N=27 cells (Δ9) and N=16 cells (Δ26). (B) The same cells as in (A) were stimulated with a stronger uncaging flash, resulting in post-flash [Ca2+]i>100 μM (top panel). Fusion of catecholamine-containing vesicles was evaluated using the cumulative amperometric charge (bottom panel), revealing that at these higher [Ca2+]i the Δ9 mutation supported significantly more secretion than the SNAP-25 null, whereas the Δ26 was still dominant negative. (C) Double mutation in layer +5 abolishes secretion (blue traces). Null cells from the same preparations rescued with SNAP-25 WT displayed normal secretion (black traces). (D) A second flash to increase [Ca2+]i to >100 μM also did not recover secretion in the mutant. Compared to SNAP-25 null cells (red traces), the layer +5 mutation was dominant negative. N=18 cells (layer +5), N=26 cells (WT rescue). The results from SNAP-25 null cells were obtained in separate experiments (presented in Figure 5B). *: P<0.05; **: P<0.01; ***: P<0.001.
We then returned to alanine substitutions, and introduced mutations into layer +5 (M71A/I192A). This mutation completely eliminated secretion (Figure 3C and D). In 18 cells from three preparations expressing this mutant, only in one cell were a few amperometric spikes detected upon stimulation—secretion was also not restored when increasing [Ca2+]i to >100 μM (Figure 3D). We noticed that these cells appeared to be smaller than normal, and this was confirmed by capacitance measurements (layer +5: 4.0±0.2 pF, N=18; WT: 7.9±0.4 pF, N=26, P<0.001). A similar mutation in layer +4 (I67A/N188A) also did not rescue secretion to anywhere near WT levels, even though amperometric spikes were seen frequently upon stimulation (total capacitance increase at 5 s after a flash: 35±7.7 fF, N=18 cells in the mutant compared to 370±44 fF, N=24 cells in WT rescued cells, P<0.001). We also overexpressed the Δ26, layer +5 and layer +4 mutations in WT cells expressing endogenous SNAP-25, where all mutations led to a near-elimination of secretion (not shown). Thus, these mutations are dominant negative, indicating that they can still interact with their cognate SNARE partners (syntaxin, synaptobrevin), but that the formed complexes are nonproductive for fusion.
In conclusion, we have identified a number of C-terminal mutations leading to graded effects on exocytosis triggering, establishing the sequence: layer +5 mutation<Δ9<L78A/L81A/M202A<L81A/M202A<L81A≈WT. Subsequently, we turned to biochemical measurements in order to investigate the in vitro properties of the SNARE complex.
The SNARE complex contains two functional domains that can fold/unfold independently
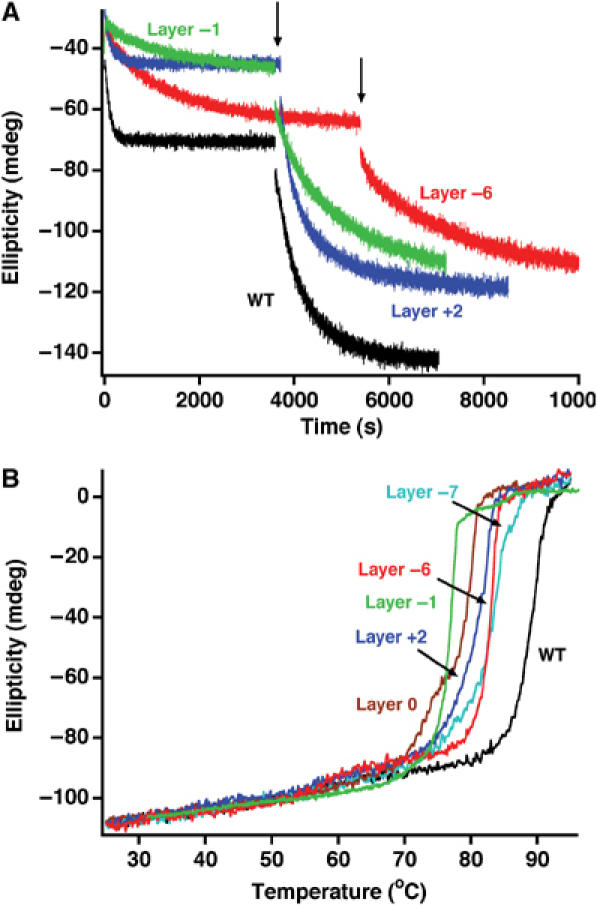
SNARE assembly/disassembly exhibits pronounced hysteresis, indicative of a kinetic barrier between the assembled and unfolded states (Fasshauer et al, 2002). This makes it impossible to reach equilibrium within an experimentally accessible time scale, but at the same time it opens a rare possibility for studying both the assembly and disassembly reactions without interconversions. Hence, thermal unfolding experiments give insights into the stability of the four-helix bundle, whereas the assembly reaction can be measured at room temperature, where disassembly is exceedingly slow. Since the formation of the complex is accompanied by a large increase in α-helical structure, CD spectroscopy can be used to monitor complex assembly and disassembly.
N-terminal truncations of SNAP-25 severely impair assembly of a ternary SNARE complex, whereas C-terminal deletions (Δ9 and Δ26) do not (Fasshauer and Margittai, 2004), indicating that N-terminal nucleation is rate limiting for assembly. In line with these findings, all C-terminal alanine mutants were able to normally assemble into a binary syntaxin/SNAP-25, as well as into a ternary complex (Figure 4A). Next, thermal denaturation experiments on preassembled ternary SNARE ‘core complexes' were carried out. The WT complex unfolded in a single transition at ∼89°C (Figure 4B). The melting curve of the SNARE complex containing the single layer +8 mutation (L81A) was almost undistinguishable from WT. When the double (L81A/M202A) or the triple (L78A/L81A/M202A) SNAP-25 mutations were analyzed, a small deviation from a single-step unfolding behavior was observed. Remarkably, a very clear two-step unfolding process was detected for the Δ9 deletion and the layer +5 mutation (M71A/I192A) (Figure 4B). The two-step unfolding process suggests that the C-terminal portion of the SNARE bundle can unfold independently of the N-terminal portion. This is the first clear structural evidence that two distinct domains reside in the extended SNARE bundle. The first unfolding step occurred at different temperatures, establishing the sequence: layer +5 mutation<Δ9<L78A/L81A/M202A<L81A/M202A<L81A≈WT. Thereby, the temperature of the first unfolding step scales with the severity of the mutation for exocytosis, suggesting that the defect on secretion depends on the destabilization of the C-terminal end of the SNARE complex. However, the layer +4 mutation (I67A/N188A) did not exhibit two-step unfolding, but rendered the entire four-helix bundle less stable (∼81°C; not shown). It is possible that the layer +4 is so far removed from the C-terminal end that its mutation compromises the whole complex, rather than just the C-terminal end.
Figure 4.
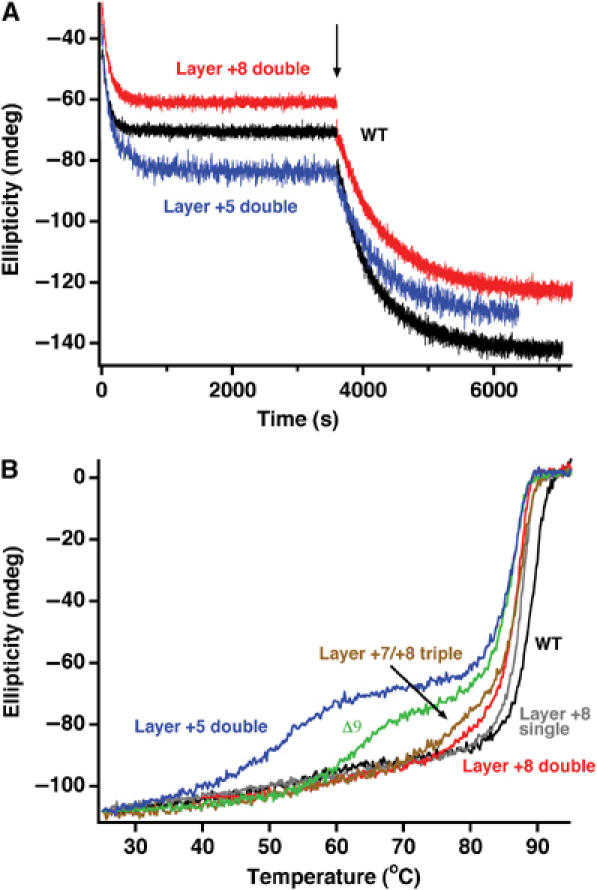
C-terminal SNAP-25 mutations selectively destabilize the C-terminal region of the SNARE complex. (A) The assembly rates of C-terminal SNAP-25 mutants into a binary or ternary SNARE complex were similar compared to WT SNAP-25. SNAP-25 and the SNARE motif of syntaxin were mixed and the increase in α-helical signal at 222 nm upon complex formation was followed by CD spectroscopy. Synaptobrevin was added after binary assembly (indicated by an arrow) and the formation of the ternary complex followed over time. (B) Thermal melts of SNARE complexes containing SNAP-25 variants. Complexes containing a deletion of the last nine C-terminal amino acids (Δ9) or a double alanine mutation in layer +5 unfolded in two separate steps; also, a triple mutation in the last C-terminal layers deviated from a single unfolding transition. The first unfolding step probably represents the autonomous unfolding of the C-terminal region of the SNARE four-helix bundle. The unfolding temperature of the remaining SNARE bundle was only slightly reduced compared to the WT complex.
The mutations analyzed indicate that exocytosis triggering is affected by mutations that specifically destabilize the C-terminal end of the SNARE complex, and that this end of the complex can fold/unfold independently of the rest. This suggests that SNARE assembly—probably of its C-terminal end—is intimately linked to exocytosis triggering and does not occur in an upstream priming step.
Expression and targeting of mutated SNAP-25 to the plasma membrane
An alternative explanation for the effects of our mutations could be defective expression or targeting to the plasma membrane. We therefore investigated membrane targeting and the overexpression level of SNAP-25 mutations using isolated plasma membrane sheets. These data confirmed that similar amounts of SNAP-25 mutants were targeted to the plasma membrane (Figure 5). The expression level was ∼10-fold over the level in WT cells (Figure 5C). We have previously shown that overexpression of SNAP-25 for 6–10 h using Semliki Forest Virus (SFV) does not impair secretion in chromaffin cells (Sørensen et al, 2003).
Figure 5.
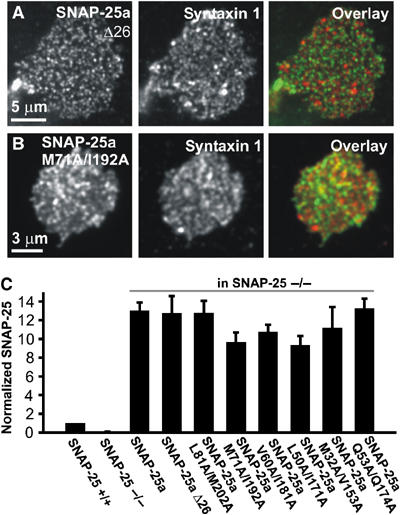
Mutations do not affect membrane targeting of overexpressed SNAP-25. SNAP-25 and syntaxin 1 specific immunostaining in plasma membrane sheets generated from SNAP-25 null chromaffin cells expressing (A) SNAP-25a Δ26 and (B) SNAP-25a M71A/I192A. SNAP-25 and syntaxin 1 are only partially colocalized (Lang et al, 2001). Note that the higher magnification of the lower picture can result in the impression that the colocalization was higher. (C) Mean±s.e.m. SNAP-25 immunofluorescence from 4–10 WT and SNAP-25 null animals expressing SNAP-25a constructs for 8 h. The SNAP-25 expressing SFV increased the level of membrane-bound SNAP-25 to roughly 10-fold the level in WT cells. Mutation of the interaction layers, or C-terminal deletion, did not affect plasma membrane targeting or expression level.
Alanine substitutions in the center of the SNARE complex affect the priming reaction
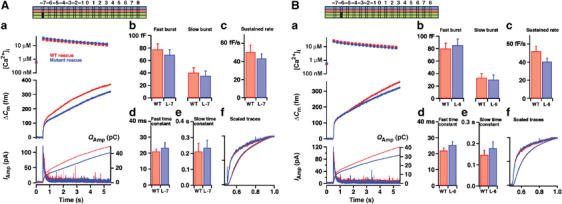
In the next experimental series, we introduced alanines pairwise into the central SNARE layers. The zero layer is special, because here a charged arginine from synaptobrevin interacts with three glutamines (Figure 1D). Nevertheless, alanine substitutions into layers −1 (L50A/I171A), 0 (Q53A/Q174A), +1 (L57A/I178A) and +2 (V60A/I181A) had very similar effects on exocytosis (Figure 6). In all cases the only parameters significantly changed were the sustained component of release (all mutations; Figure 6Ac, Bc, Cc and Dc) and the size of the fast exocytotic burst (significantly affected in layers −1, 0, and +1, but not in layer +2, even though the same tendency was seen; Figure 6Ab, Bb, Cb and Db). In contrast, the time constants of the fast and slow burst of release were unchanged (Figure 6Ad, e, Bd, e, Cd, e and Dd, e). Inspection of capacitance and amperometric traces scaled to WT amplitude confirmed that the kinetics of fast secretion were unchanged (Figure 6Af, Bf, Cf and Df). Thus, with mutations in the central part of the SNARE complex, the sustained component of release is slowed down, which is an indication that priming of new vesicles for release is impaired. The reduction in the size of the fast burst leads to the same conclusion: fewer vesicles had been made ready for fast release before stimulation. However, the kinetics of fusion of those vesicles that were already primed at the time of stimulation remained unaffected. These data extend previous overexpression experiments in bovine chromaffin cells targeting the zero layer (Wei et al, 2000) by showing that this effect is shared by neighboring layers, and is seen with mutations that do not impose steric hindrance of SNARE complex formation.
Figure 6.
Mutation in the middle layers slows down the vesicular priming rate. Double mutation in layer −1 (L50A/I171A) (Aa), layer 0 (Q53A/Q174A) (Ba), layer +1 (L57A/I178A) (Ca) and layer +2 (V60A/I181A) (Da) decreased the mean secretion. (Ab, c, Bb, c, Cb, c, Db, c) the sustained rate of release was significantly decreased in all cases, whereas the amplitude of the fast burst was significantly depressed in layer −1, 0 and +1 mutations. (Ad, e, Bd, e, Cd, e, Dd, e) Time constants of fast and slow burst fusion were unchanged. (Af, Bf, Cf, Df) The mean capacitance and cumulative amperometric charge scaled to WT amplitude revealed no differences in the triggering phases of secretion. N=21 cells (layer −1), N=25 cells (WT); N=31 cells (layer 0), N=31 cells (WT); N=25 cells (layer +1), N=26 cells (WT); N=24 cells (layer +2), N=26 cells (WT). *: P<0.05; **: P<0.01; ***: P<0.001.
Alanine substitutions in the N-terminal end of the SNARE complex fail to modify secretion
Interestingly, alanine substitutions in the outermost N-terminal layers, −7 (T29A/L150A) and −6 (M32A/V153A), only caused a small depression of mean secretion (Figure 7). Upon kinetic analysis, however, neither the burst components nor the sustained rate of release were significantly reduced (Figure 7Ab–c and Bb–c). Most importantly, the triggering phase of secretion was unchanged by both mutations (Figure 7Ad–e and Bd–e). This conclusion is also reached when inspecting scaled capacitance and amperometric traces (Figure 7Af and Bf). Thus, mutations in the very N-terminal layers did not significantly affect exocytosis.
Figure 7.
N-terminal alanine substitutions do not affect secretion. Double mutation in layer −7 (T29A/L150A) (Aa) or layer −6 (M32A/V153A) (Ba) left mean secretion only slightly depressed. (Ab, c, Bb, c) The sizes of both burst components and the sustained rates of secretion were not significantly changed. (Ad, e, Bd, e) The time constants of fast and slow burst components were also unchanged. (Af, Bf) The mean capacitance and cumulative amperometric charge scaled to WT amplitude at 1 s after the flash revealed no differences in the fast triggering phase of secretion. N=28 cells (layer −7), N=24 cells (WT); N=31 cells (layer −6), N=35 cells (WT).
Formation rate and stability of SNARE complexes with central and N-terminal mutations
We next investigated the assembly of SNARE complexes with mutations in the N-terminal end and in the middle. The double mutations in the N-terminal end of the complex (layers −6 and −7) strongly inhibited the formation of both the binary syntaxin/SNAP-25 complex and the ternary complex with synaptobrevin (layer −6; see Figure 8A). At least part of this effect extended into the central portion of the bundle; for example, the layer −1 mutant slowed the binary and ternary SNARE interaction (Figure 8A). It should be stressed that the measurements in Figure 8A do not allow statements about the kinetics of synaptobrevin binding to the syntaxin:SNAP-25 1:1 complex, because of the spontaneous formation of a 2:1 syntaxin:SNAP-25 complex in vitro. Thermal denaturation experiments revealed that the mutations decreased the stability of the ternary SNARE complex compared to WT; however, two-step disassembly was not observed, except for a tendency in the layer 0 mutation (Figure 8B).
Figure 8.
Assembly and stability of SNARE complexes with N-terminal and central double alanine layer mutations. (A) Mutations in the N-terminal and central layers perturbed the interaction of syntaxin and SNAP-25 (layers −6, −1 and +2 are shown). Assembly into the ternary complex was also very slow with the layer −6 mutation, and moderately slowed down by the layer −1 mutation. (B) Central layer mutants (layers −1, 0 and +2) rendered the entire ternary SNARE complex somewhat less stable than N-terminal layer mutants (layers −7 and −6). Note that the 0-layer mutation appears to cause a slight two-step unfolding process (Tm1≈73°C, Tm2≈79°C), which is not seen when mutating neighboring layers.
The biochemical data thus show that all our alanine mutations destabilized the SNARE complex (Figure 7). At the same time, mutations in layers −6 and −7 led to a dramatic inhibition of in vitro SNARE complex formation, even though secretion was intact. This reveals the existence of a kinetic step in SNARE complex assembly that is rate limiting in vitro but not in vivo (see Discussion).
The energy available for fusing the membranes
The data presented above support the two-step assembly model in Figure 1C, in which fusion coincides with SNARE complex assembly across the C-terminal layers. This raises the question whether C-terminal assembly can exert enough force to fuse the membranes. This cannot be answered directly, since the energy required to fuse membranes is unknown. Furthermore, the formation energy of the SNARE complex cannot be measured biochemically, because of the folding/unfolding hysteresis (Fasshauer et al, 2002). Instead, we used free energy perturbation simulations to assess the change in thermodynamic stability introduced by some of the C-terminal mutations. This gives a lower estimate of the total energy that can be made available by assembly across the last two layers. An assumption behind these calculations is that the mutations do not cause major structural changes. Based on the unfolding experiments in Figure 4B, this assumption appears reasonable when calculations are made at physiological temperatures and restricted to mutations in the last two hydrophobic layers.
Simulations were carried out by mutating the amino acids to alanines ‘in silico', yielding free energy differences for the mutation in both the folded and unfolded states (ΔG). The difference between the two (ΔΔG) provides an estimate for the amount of destabilization of the complex. The results in Table I show that the single, double and triple C-terminal mutations progressively destabilize the complex. The ΔΔG for the triple mutation of 45 kJ/mol indicates that assembly across the last layers may make substantial amounts of energy available for fusing of the membranes.
Table 1.
Free energy perturbation calculations
| ΔG (forward) (kJ/mol) | ΔG (backward) (kJ/mol) | ΔG (average) (kJ/mol) | ΔΔG (kJ/mol) | |
|---|---|---|---|---|
| Tripeptide | ||||
| L78A | 1.7 | 5.7 | 3.7±2.0 | |
| L81A | 3.2 | 4.0 | 3.6±0.4 | |
| M202A | 7.1 | 6.3 | 6.7±0.4 | |
| Protein | ||||
| M202A | 23.3 | 11.5 | 17.4±5.9 | 10.8±6.3 |
| L81A/M202A | 46.7 | 31.3 | 39.0±7.7 | 28.7±8.5 |
| L78A/L81A/M202A | 66.2 | 52.6 | 59.4±6.8 | 45.4±9.6 |
| Free energy changes, ΔG, between WT and mutants for the unfolded and folded state, respectively, and differences, ΔΔG, as an estimate of the effect of the mutation on the thermodynamic stability of the complex. An estimate for the uncertainty is obtained by comparing forward and backward ΔG values. The single, double and triple mutants display a progressive amount of destabilization with respect to the WT protein. | ||||
Discussion
Sequential N- to C-terminal ‘zipping-up' of the SNARE complex
In this study, we have interfered systematically with the hydrophobic layers of the SNARE complex. C-terminal mutations led to a slowdown of the fast phase of exocytosis, indicating that the C-terminal region of the complex is intimately coupled to exocytosis triggering. Thermal denaturation experiments of SNARE complexes formed with C-terminal mutations revealed a two-step unfolding process (Figure 4B), indicating that these mutations specifically loosened the C-terminal end without affecting the remaining structure of the complex. This provides the first biochemical evidence that two functional domains reside in the extended SNARE bundle. Such a feature is a prerequisite for stepwise SNARE assembly (Figure 1C). Free energy perturbation calculations showed that assembly of the C-terminal end of the complex might release substantial amounts of energy, which could be coupled to membrane fusion when the complex forms in trans. Weakening of the C-terminal portion would diminish this energy and eventually, with more disruptive mutations, SNARE assembly would surrender to the repulsive forces between two apposing membranes.
Mutations in the middle of the complex led to a specific slowdown of vesicle priming, but did not affect the fusion rate constant of primed vesicles. The simplest explanation for this finding is that chromaffin cells have a certain number of vesicles with SNARE complexes in a state of assembly extending beyond the central layers. Consequently, during fusion triggering only the C-terminal layers have to assemble (Figure 1C), rendering the fusion of primed vesicles very fast, and not limited by the preceding slow N-terminal assembly process. Collectively, our findings support the ‘SNARE zipper model' (Hanson et al, 1997).
A two-step SNARE assembly process was previously suggested to underlie priming and triggering in neurosecretory cells, based on an assay where recombinant SNARE fragments were introduced into cracked PC12 cells (Chen et al, 2001). These authors found that ATP-dependent priming created a binding site for syntaxin, and thus suggested that the precomplex is formed by synaptobrevin and SNAP-25. Yet, the proposed scenario does not satisfyingly explain why the synaptobrevin helix is as efficient as the syntaxin helix in inhibiting secretion. We now know that the 1:1 syntaxin/SNAP-25 complex can bind to both syntaxin and synaptobrevin (Fasshauer and Margittai, 2004). Thus, if ATP-dependent ‘priming' involves the formation of a 1:1 syntaxin/SNAP-25 complex, these observations are in agreement with our model in Figure 1C. Inherent in this explanation is the assumption that ATP-dependent ‘priming' is upstream of the formation of the readily releasable state of the vesicle (this process is referred to as ‘priming' in this article). This is in agreement with the proposition that PC12 cells lack a stabilized pool of readily releasable vesicles (Martin, 2003).
Another group used intramolecular FRET of an engineered SNAP-25 (termed SCORE) expressed in PC12 cells to monitor the formation of the syntaxin:SNAP-25 dimer (An and Almers, 2004). They concluded that the N-terminal SNARE domain of SNAP-25 joins syntaxin during priming, forming a two-helical bundle, which serves as an acceptor for the C-terminal helix of SNAP-25 and synaptobrevin during triggering. It is possible that this assay has resolved another SNARE intermediate; however, a two-helix bundle would need to be stabilized by other factors, which so far remain unidentified. In the light of our data, it appears unlikely that the C-terminal SNAP-25 helix would join the complex during triggering, but it is conceivable that a two-helix bundle could precede the formation of the three-helical acceptor complex. Other possibilities are that WT SNAP-25 might have complemented the SCORE construct, resulting in a three-helix bundle that would have gone undetected, or that the SCORE construct might have unusual properties because of the presence of a fluorophore in the linker domain.
Mutation in the C-terminal end of the SNARE complex
The identification of a double alanine mutation in layer +5, which led to pronounced two-step unfolding in thermal melts, should make it possible to study the partly assembled ternary complex in detail using structural methods. The lack of two-step unfolding in the neighboring layer +4 mutation may mean that layer +4 is close to the interface between zipped and unzipped layers in the partially assembled complex. From the scenario in Figure 1C, it might then be expected that the layer +4 mutation has more upstream effects on secretion than the layer +5 mutation. However, since both mutations virtually abolished secretion, it is not possible to determine whether they do so by interfering with different steps (priming or triggering). It is striking that the most deleterious mutation of a SNARE protein ever described (the layer +5 mutation) was obtained by the relatively conservative substitution of alanines for two larger hydrophobic amino acids in the interior of the complex. This underlines the extreme importance of the assembly of the C-terminal end of the complex for secretion. This critical region has also been identified in other studies. Thus, genetic screens in Caenorhabditis elegans identified mutations in layers +4, +5 and +6 (Nonet et al, 1998; Saifee et al, 1998). In Drosophila, a double mutation of syntaxin in layers +4 and +5 strongly reduced release (Fergestad et al, 2001), whereas more N-terminal layer mutations led to milder phenotypes (Wu et al, 1999; Rao et al, 2001). We suggest that this area defines the interface between two functional domains in the SNARE complex.
Previous data on bovine chromaffin cells overexpressing the Δ9 deletion were interpreted as a selective loss of the RRP, whereas the other releasable vesicle pool—the SRP—persisted (Wei et al, 2000). Here we showed that, when overexpressed in SNAP-25 null cells, the Δ9 deletion cannot support secretion at 20–30 μM [Ca2+], but higher calcium concentrations can partly overcome the defect. This finding is in agreement with data obtained in neurons after BoNT/A cleavage (Capogna et al, 1997; Trudeau et al, 1998; Gerona et al, 2000; Sakaba et al, 2005). Molecularly, this was explained by defective Ca2+-dependent synaptotagmin binding to SNARE complexes containing truncated SNAP-25 (Gerona et al, 2000). In the present investigation, we have arranged the Δ9-mutation into a sequence of mutations causing progressive destabilization of the C-terminal end of the complex. This makes it likely that the destabilization of the C-terminal end of the SNARE complex is causative also for the effect of the Δ9 deletion. However, if final zipping of the last couple of SNARE layers happens simultaneously with Ca2+-binding to synaptotagmin (Figure 1C), allosteric coupling between the two processes could make zipping of the SNARE complex dependent on binding of synaptotagmin to the SNARE complex and vice versa. Thus, the two explanations for the effect of the Δ9 deletion do not exclude each other.
Mutation in the N-terminal end of the SNARE complex
In vitro, the nucleation step between syntaxin and SNAP-25 is rate-limiting for SNARE assembly and can be disturbed easily by N-terminal mutations (Figure 7A). Probably, the reason for the slow in vitro nucleation is that this step requires the coordinated parallel alignment of three helices. However, N-terminal alanine mutations were without significant effect on exocytosis (Figure 7). In the cell, therefore, this step is not rate limiting. This is not surprising, since priming of new vesicles for release requires only seconds, whereas assembly of SNAREs in vitro requires many minutes (Figure 8A). It is usually assumed that specific ‘priming factors' drives priming. We suggest that what they do is to catalyze the initial N-terminal SNARE complex assembly.
Whereas mutations in the outermost N-terminal layers were without significant effect, mutations in the central layers slowed down vesicle priming. One possibility is that the N-terminal binding of synaptobrevin may be more robustly catalyzed, and followed by spontaneous zipping across the central layers, which may be more susceptible to disruption. Another possibility is that initial synaptobrevin binding involves a longer stretch of the syntaxin:SNAP-25 precomplex, so that the perturbation of individual layers is insufficient to elicit a clear phenotype.
How to render SNARE-driven membrane fusion calcium-dependent
Two interesting open questions are: what keeps the half-assembled SNARE complex from ‘zipping' all the way up prematurely, and how is this last assembly step coupled to the calcium trigger for fast exocytosis (presumably synaptotagmin 1)? The concept of a ‘clamp on exocytosis' has been put forward by several authors, and it has been suggested that this halt may be exerted by synaptotagmin; however, there is no indication that synaptotagmin can do this (discussed by Koh and Bellen, 2003). Another possibility is that the SNARE complex may be intrinsically unable to assemble all the way when it faces the energy barrier created by the repulsion of the two membranes. In this situation, ‘zipping' would come to a stop. Additional energy input would then be required to allow assembly to go forward and fuse the membranes. This energy could be provided by calcium-loaded synaptotagmin binding to the target membrane, to SNAREs, or to both. This would then lead to calcium-dependent fusion, driven by the collective effort of the SNAREs and synaptotagmin.
Materials and methods
Chromaffin cell preparation, mutagenesis and expression
SNAP-25 null embryos (E17–E19) were recovered by Cesarean section and chromaffin cells prepared as described (Sørensen et al, 2003). Mutations were introduced into a SNAP-25a containing pSFV1 plasmid (pSFV1 SNAP-25a-IRES-EGFP) using PCR mutagenesis and all constructs were sequenced. Following production of SFV, chromaffin cells were infected at days 2–4 after isolation and the cells were used 6–10 h after infection.
Semiquantification of membrane-bound SNAP-25
Plasma membrane sheets from mouse embryonic chromaffin cells were prepared as described (Nagy et al, 2005). SNAP-25 and syntaxin 1 were immunodetected as described previously (Lang et al, 2001). The amount of membrane-bound SNAP-25 was semiquantified by measuring the average intensity of a sheet (Nagy et al, 2005). At least 10 membrane sheets from each animal were analyzed and the mean value for each animal used to calculate the population mean and s.e.m. (number of animals=4–10).
Electrophysiology and electrochemistry
Patch-clamp capacitance measurements, measurements of intracellular calcium concentration, flash photolysis of caged calcium and amperometry were carried out as described (Sørensen et al, 2003). Capacitance and amperometric measurements were carried out in parallel, to ensure that the fusion of catecholamine-containing vesicles was being monitored. For some of the more severe mutations, we used the integrated amperometric charge as a quantitative measurement of secretion. Since secretion was low in these cases, it was necessary to correct the amperometric measurements for current artifacts caused by illumination of the carbon fiber by the flash light and the light used to measure calcium. This was done by removing the cell using the patch pipette after each experiment, followed by stimulation of the carbon fiber with the standard protocol. The resulting amperometric trace was subtracted from the measurements. The remaining artifact amounts to an integrated current within ±2 pC over the course of 5 s.
Kinetic analysis of capacitance data
When possible, capacitance traces were fitted with a sum of exponential functions, in order to separate the sizes of the two burst components (appear as amplitudes of the exponentials and are denoted as ‘fast burst' and ‘slow burst' in the figures) from the kinetics of fusion triggering (appear as time constants of the exponentials), as described previously (Sørensen et al, 2003). Owing to the slowdown caused by some mutations, a kinetic separation of fast and slow burst was not always possible; for this reason, we sometimes resorted to evaluating the amplitude of the capacitance increase at 1 and 5 s. after the flash (denoted as ‘Amplitude(0–1 s)' and ‘Amplitude (1–5 s)' in Figure 2). Data are given as mean±s.e.m. and Mann–Whitney U-test was used to test the statistical difference: *P<0.05; **P<0.01; ***P<0.001.
Protein purification
The basic SNARE expression constructs in a pET28a vector, cysteine-free SNAP-25A (C84S, C85S, C90S, C92S; res. 1–206), the syntaxin 1A SNARE motif (res. 180–262) and synaptobrevin 2 (res. 1–96), have been described before (Fasshauer and Margittai, 2004). For all truncations and point mutations in SNAP-25A, the cysteine-free variant was used as template. The recombinant SNARE proteins were isolated from Escherichia coli and purified by Ni-NTA affinity chromatography followed by ion exchange chromatography on an Äkta system (Amersham), essentially as described (Fasshauer and Margittai, 2004). All ternary SNARE complexes were assembled overnight and purified using a Mono Q-column. Protein concentration was determined by absorption at 280 nm.
CD spectroscopy measurements
CD measurements were performed using a Jasco model J-720 instrument. All experiments were carried out in 20 mM sodium phosphate or 20 mM TRIS, pH 7.4, in the presence of 100 mM NaCl and 1 mM DTT. For thermal denaturation experiments, about 10 mM purified ternary SNARE complexes were heated in Hellma quartz cuvettes with a path length of 0.1 cm. The ellipticity at 222 nm was recorded between 25 and 95°C at a temperature increment of 30°C/h. Kinetic measurements were carried out in 1 cm quartz cuvettes at 25°C. About 2 mM of the individual proteins were mixed and the increase in α-helical signal was followed at 222 nm.
Energy perturbation calculations
See Supplementary data.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Materials and Methods
Acknowledgments
We thank Reinhard Jahn, Thorsten Lang and Erwin Neher for critically commenting on the manuscript and for ongoing discussions, and Dirk Reuter and Wolfgang Berning-Koch for expert technical assistance. This work was supported by the Boehringer Ingelheim Foundation (IM), the Deutsche Forschungsgemeinschaft (DFG): SFB523/TP4, SO 708/1-1, and GRK521 (JBS).
References
- An SJ, Almers W (2004) Tracking SNARE complex formation in live endocrine cells. Science 306: 1042–1046 [DOI] [PubMed] [Google Scholar]
- Borisovska M, Zhao Y, Tsytsyura Y, Glyvuk N, Takamori S, Matti U, Rettig J, Südhof T, Bruns D (2005) v-SNAREs control exocytosis of vesicles from priming to fusion. EMBO J 24: 2114–2126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capogna M, McKinney RA, O'Connor V, Gähwiler BH, Thompson SM (1997) Ca2+ or Sr2+ partially rescues synaptic transmission in hippocampal cultures treated with botulinum toxin A and C, but not tetanus toxin. J Neurosci 17: 7190–7202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YA, Scales SJ, Patel SM, Doung YC, Scheller RH (1999) SNARE complex formation is triggered by Ca2+ and drives membrane fusion. Cell 97: 165–174 [DOI] [PubMed] [Google Scholar]
- Chen YA, Scales SJ, Scheller RH (2001) Sequential SNARE assembly underlies priming and triggering of exocytosis. Neuron 30: 161–170 [DOI] [PubMed] [Google Scholar]
- Fasshauer D, Antonin W, Subramaniam V, Jahn R (2002) SNARE assembly and disassembly exhibit a pronounced hysteresis. Nat Struct Biol 9: 144–151 [DOI] [PubMed] [Google Scholar]
- Fasshauer D, Margittai M (2004) A transient N-terminal interaction of SNAP-25 and syntaxin nucleates SNARE assembly. J Biol Chem 279: 7613–7621 [DOI] [PubMed] [Google Scholar]
- Fergestad T, Wu MN, Schulze KL, Lloyd TE, Bellen HJ, Broadie K (2001) Targeted mutations in the syntaxin H3 domain specifically disrupt SNARE complex function in synaptic transmission. J Neurosci 21: 9142–9150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiebig KM, Rice LM, Pollock E, Brunger AT (1999) Folding intermediates of SNARE complex assembly. Nat Struct Biol 6: 117–123 [DOI] [PubMed] [Google Scholar]
- Gerona RRL, Larsen EC, Kowalchyk JA, Martin TFJ (2000) The C terminus of SNAP25 is essential for Ca2+-dependent binding of synaptotagmin to SNARE complexes. J Biol Chem 275: 6328–6336 [DOI] [PubMed] [Google Scholar]
- Han X, Wang CT, Bai J, Chapman ER, Jackson MB (2004) Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis. Science 304: 289–292 [DOI] [PubMed] [Google Scholar]
- Hanson PI, Heuser JE, Jahn R (1997) Neurotransmitter release—four years of SNARE complexes. Curr Opin Neurobiol 7: 310–315 [DOI] [PubMed] [Google Scholar]
- Hu K, Carroll J, Fedorovich S, Rickman C, Sukhodub A, Davletov B (2002) Vesicular restriction of synaptobrevin suggests a role for calcium in membrane fusion. Nature 415: 646–650 [DOI] [PubMed] [Google Scholar]
- Hua SY, Charlton MP (1999) Activity-dependent changes in partial VAMP complexes during neurotransmitter release. Nat Neurosci 2: 1078–1083 [DOI] [PubMed] [Google Scholar]
- Koh TW, Bellen HJ (2003) Synaptotagmin I, a Ca2+ sensor for neurotransmitter release. Trends Neurosci 26: 413–422 [DOI] [PubMed] [Google Scholar]
- Kweon DH, Kim CS, Shin YK (2003) Regulation of neuronal SNARE assembly by the membrane. Nat Struct Biol 10: 440–447 [DOI] [PubMed] [Google Scholar]
- Lang T, Bruns D, Wenzel D, Riedel D, Holroyd P, Thiele C, Jahn R (2001) SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J 20: 2202–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TF (2003) Tuning exocytosis for speed: fast and slow modes. Biochim Biophys Acta 1641: 157–165 [DOI] [PubMed] [Google Scholar]
- Matos MF, Mukherjee K, Chen X, Rizo J, Südhof TC (2003) Evidence for SNARE zippering during Ca2+-triggered exocytosis in PC12 cells. Neuropharmacology 45: 777–786 [DOI] [PubMed] [Google Scholar]
- Melia TJ, Weber T, McNew JA, Fisher LE, Johnston RJ, Parlati F, Mahal LK, Söllner TH, Rothman JE (2002) Regulation of membrane fusion by the membrane-proximal coil of the t-SNARE during zippering of SNAREpins. J Cell Biol 158: 929–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Milosevic I, Fasshauer D, Muller EM, de Groot BL, Lang T, Wilson MC, Sorensen JB (2005) Alternative splicing of SNAP-25 regulates secretion through nonconservative substitutions in the SNARE domain. Mol Biol Cell 16: 5675–5685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet ML, Saifee O, Zhao H, Rand JB, Wei L (1998) Synaptic transmission deficits in Caenorhabditis elegans synaptobrevin mutants. J Neurosci 18: 70–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SJ, Stewart BA, Rivlin PK, Vilinsky I, Watson BO, Lang C, Boulianne G, Salpeter MM, Deitcher DL (2001) Two distinct effects on neurotransmission in a temperature-sensitive SNAP-25 mutant. EMBO J 20: 6761–6771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saifee O, Wei L, Nonet ML (1998) The Caenorhabditis elegans unc-64 locus encodes a syntaxin that interacts genetically with synaptobrevin. Mol Biol Cell 9: 1235–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Stein A, Jahn R, Neher E (2005) Distinct kinetic changes in neurotransmitter release after SNARE protein cleavage. Science 309: 491–494 [DOI] [PubMed] [Google Scholar]
- Sørensen JB (2004) Formation, stabilisation and fusion of the readily-releasable pool of secretory vesicles. Pflügers Arch 448: 347–362 [DOI] [PubMed] [Google Scholar]
- Sørensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E (2003) Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell 114: 75–86 [DOI] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT (1998) Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature 395: 347–353 [DOI] [PubMed] [Google Scholar]
- Trudeau LE, Fang Y, Haydon PG (1998) Modulation of an early step in the secretory machinery in hippocampal nerve terminals. Proc Natl Acad Sci USA 95: 7163–7168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets T (2000) Dissection of three Ca2+-dependent steps leading to secretion in chromaffin cells from mouse adrenal slices. Neuron 28: 537–545 [DOI] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachi M, Parlati F, Söllner TH, Rothman JE (1998) SNAREpins: minimal machinery for membrane fusion. Cell 92: 759–772 [DOI] [PubMed] [Google Scholar]
- Wei S, Xu T, Ashery U, Kollewe A, Matti U, Antonin W, Rettig J, Neher E (2000) Exocytotic mechanism studied by truncated and zero layer mutants of the C-terminus of SNAP-25. EMBO J 19: 1279–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MN, Fergestad T, Lloyd TE, He Y, Broadie K, Bellen HJ (1999) Syntaxin 1A interacts with multiple exocytic proteins to regulate neurotransmitter release in vivo. Neuron 23: 593–605 [DOI] [PubMed] [Google Scholar]
- Xu T, Binz T, Niemann H, Neher E (1998) Multiple kinetic components of exocytosis distinguished by neurotoxin sensitivity. Nat Neurosci 1: 192–200 [DOI] [PubMed] [Google Scholar]
- Xu T, Rammner B, Margittai M, Artalejo AR, Neher E, Jahn R (1999) Inhibition of SNARE complex assembly differentially affects kinetic components of exocytosis. Cell 99: 712–722 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Materials and Methods