

Abstract
After few days of intense immunoglobulin (Ig) secretion, most plasma cells undergo apoptosis, thus ending the humoral immune response. We asked whether intrinsic factors link plasma cell lifespan to Ig secretion. Here we show that in the late phases of plasmacytic differentiation, when antibody production becomes maximal, proteasomal activity decreases. The excessive load for the reduced proteolytic capacity correlates with accumulation of polyubiquitinated proteins, stabilization of endogenous proteasomal substrates (including Xbp1s, IκBα, and Bax), onset of apoptosis, and sensitization to proteasome inhibitors (PI). These events can be reproduced by expressing Ig-μ chain in nonlymphoid cells. Our results suggest that a developmental program links plasma cell death to protein production, and help explaining the peculiar sensitivity of normal and malignant plasma cells to PI.
Keywords: apoptosis, myeloma, plasma cell, proteasome, unfolded protein response
Introduction
Upon encounter with antigen, B-lymphocytes activate a complex program involving proliferation, generation of memory cells, isotype switch, and affinity maturation. Rapid defense is guaranteed by the differentiation of antibody-secreting plasma cells (Calame et al, 2003; Brewer and Hendershot, 2005). Although a minority of them home in the bone marrow and survive for longer periods, most plasma cells are short-lived and succumb after a few days of intense immunoglobulin (Ig) secretion, particularly those producing IgM (Ho et al, 1986; Manz et al, 2002). Disregulated plasma cell lifespan can cause autoimmune diseases and tumors (Kuehl and Bergsagel, 2002; Hoyer et al, 2004). Although it is clear that extrinsic factors are key to determine the proper environment for long-lived plasma cells, rather little is known on the molecular events that cause apoptosis in short-lived plasma cells. In particular, whether intrinsic factors play a role in limiting plasma cell lifespan is undetermined. An intriguing possibility is that cell death is linked to antibody production.
To explore these issues, we exploited murine primary B cells and a B lymphoma (I.29μ+) that can be induced to differentiate into massive IgM secretion (van Anken et al, 2003). After lipopolysaccharide (LPS) stimulation, I.29μ+ activate a developmental program that induces the stepwise synthesis of different classes of proteins so as to increase the secretory capacity. Endoplasmic reticulum (ER)-resident proteins increase over the entire time of the experiment, whereas IgM subunits increase significantly after 2 or 3 days, to become the dominant molecular species synthesized in the last days of differentiation (van Anken et al, 2003). LPS-stimulated I.29μ+ cells begin to die after day 3 or 4, concomitantly with massive IgM secretion and accumulation of spliced Xbp1 (Xbp1s). This transcription factor is activated during the unfolded protein response (UPR) and is essential for plasma cell development (Reimold et al, 2001; Iwakoshi et al, 2003b). Xbp1s drives the transcription of genes encoding factors involved in secretion (Shaffer et al, 2004; Sriburi et al, 2004; Tirosh et al, 2005) and ER-associated degradation (ERAD) (Yoshida et al, 2003). Promoting the efficiency of protein secretion is of obvious importance for plasma cells, which are specialized in Ig production. Why could ERAD regulation be so important? A considerable fraction of secretory μ-chains are degraded also in hybridoma cells (Fra et al, 1993; Fagioli and Sitia, 2001), probably owing to the inefficient assembly of IgM polymers, the only species that negotiate export from the ER (Sitia et al, 1990; Cals et al, 1996). We reasoned that the exuberant production of IgM that characterizes activated B cells could represent a load on the protein degradation machinery, which is largely dependent on cytosolic proteasomes (Mancini et al, 2000; Fagioli et al, 2001; Goldberg, 2003). This idea stemmed also from the fact that proteasome inhibitors (PI) proved particularly effective in the therapy of multiple myeloma (MM), by inducing apoptosis of malignant cells (Hideshima et al, 2003; Adams, 2004). Therefore, we decided to test a model that, described in broad terms, predicts that apoptosis of short-lived plasma cells and the exquisite sensitivity of plasma cells to PI are promoted by an unbalance between load (protein synthesis) and capacity of the proteolytic machinery (proteasomes). Our results suggest that this may be indeed the case. When Ig synthesis becomes maximal, the abundance of proteasomes decreases in both I.29μ+ and primary B cells. This correlates with reduced proteasome activity, accumulation of polyubiquitinated proteins, enhanced susceptibility to PI, and stabilization of several proteasome substrates important in B-cell physiology (Xbp1s, IκBα, and Bax). Similar events can be reproduced in HeLa cells forced to express μ-chains in large quantities. Hence, we propose that the unbalance between proteasomal load and capacity intrinsically exaggerates ER stress and predisposes plasma cells to apoptosis. These findings help explaining the exquisite sensitivity of normal and malignant plasma cells to PI.
Results
In LPS-activated I.29μ+ cells, apoptosis correlates with exuberant IgM production
I.29μ+ are B lymphoma cells that can differentiate and acquire a plasmacytoid phenotype if stimulated with LPS (van Anken et al, 2003). After 3–4 days of LPS treatment, concomitantly with massive IgM synthesis and secretion, I.29μ+ cells undergo apoptosis, thus mimicking the fate of short-lived plasma cells. Indeed, after 3–4 days of stimulation, the frequency of apoptotic cells increases sharply (Figure 1A). Apoptotic cells contain high amounts of intracellular μ-chains, correlating cell death with IgM production (Figure 1B). Following an initial acceleration in cell division, apoptotic cells accumulated. The onset of apoptosis correlated with a decrease in G2/M cells (Supplementary Figure 1A). LPS-treated I.29μ+ cells divided more rapidly at day 1 and less at days 3–4 (Supplementary Figure 1B). Caspases 3, 8, and 9 were strongly activated after day 3 (Supplementary Figure 1C).
Figure 1.
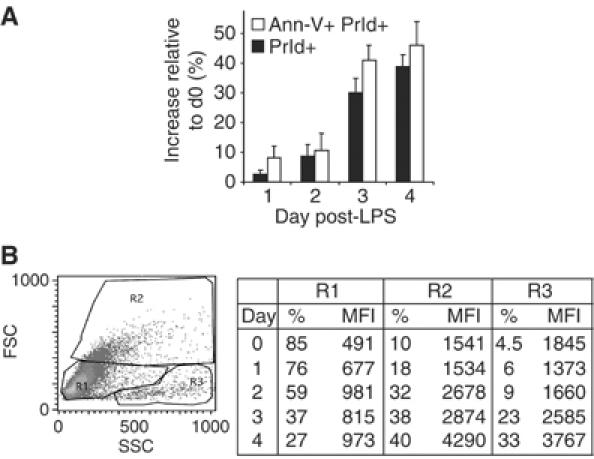
LPS-stimulated I.29μ+ cells undergo apoptosis. (A) Cell death was assessed in LPS-stimulated I.29μ+ cells by Annexin V (Ann-V) and propidium iodide (PrId) staining, and expressed as variation with respect to unstimulated cells. Mean±s.d. of three independent experiments. (B) Flow cytometry statistics of μ-chain content in LPS-stimulated I.29μ+ cells. R1, R2 and R3 were enriched in resting, activated and dead cells, respectively (van Anken et al, 2003 and our unpublished data). Mean fluorescent intensity (MFI) is indicated.
Decreased proteasomal levels and activity in differentiating I.29μ+ cells
In both B and plasma cells, a fraction of secretory and membrane IgM are rapidly degraded (Sitia et al, 1987; Fra et al, 1993), mostly by cytosolic proteasomes (Mancini et al, 2000; Fagioli and Sitia, 2001; Fagioli et al, 2001; Rabinovich et al, 2002). The existence of mechanisms that adapt proteasome biogenesis to the degradative demand has been postulated (Meiners et al, 2003). Therefore, as the massive increase in Ig synthesis induced in I.29μ+ cells by LPS (van Anken et al, 2003) represents an increased proteolytic demand, we expected proteasomes to increase in abundance. Surprisingly instead, two constitutive proteasomal catalytic subunits (Y and X) significantly decreased after LPS stimulation (Figure 2A and B). Also the interferon-γ (IFNγ)-induced subunits LMP2 and MECL-1 (Cascio et al, 2001) decreased, albeit to a lesser extent, whereas LMP7 and Z remained essentially constant.
Figure 2.
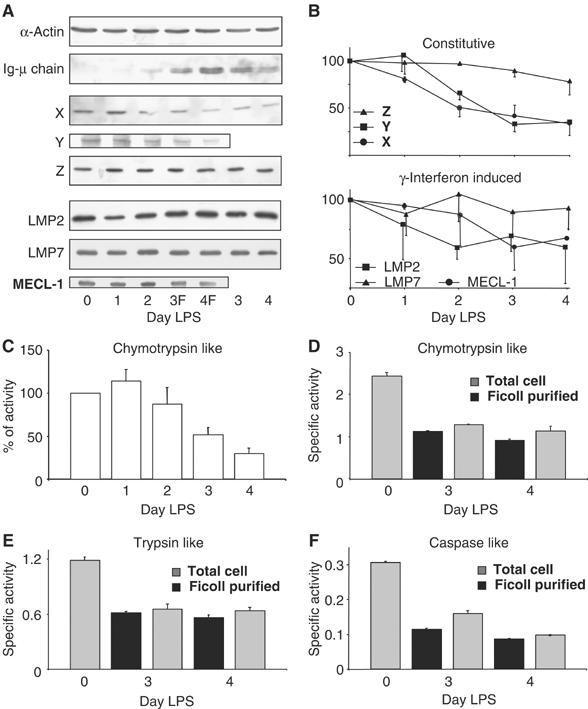
Decrease in proteasome function during terminal I.29μ+ differentiation. (A, B) Decrease in proteasome subunits. Extracts from cells stimulated for the indicated times with LPS were resolved by SDS–PAGE and blots decorated with antibodies to different proteasomal subunits, Ig-μ chains or actin. F, Ficoll-purified live cells (A). The densitometric quantification of the relevant bands is shown in panel B, relative to day 0. Data represent the means of two independent I.29μ+ inductions±s.e.m. (C–F) Decrease in proteasome activity. Proteasomal chymotrypsin-like activity was assessed in extracts from LPS-stimulated I.29μ+ cells and expressed as percentage relative to day 0. Mean of four independent LPS inductions±s.e.m. (C). All three proteasomal activities decrease in a similar way in the total pool of cells (gray bars) and in Ficoll-purified cells (black bars). Chymotrypsin-like (D), trypsin-like (E) and caspase-like (F) activities are expressed as specific activities. Mean of three different determinations from one of the inductions utilized in panel C, ±s.e.m. (D).
As a functional counterpart of these results, the proteasomal chymotryptic activity, normalized per protein content, decreased significantly at days 3 and 4 (Figure 2C). Proteasome activity fell dramatically also on a per cell basis (Supplementary Figure 2), indicating that plasma cells do not increase the synthesis and/or assembly of proteasomes to match Ig production.
Caspase activation can inhibit proteasomal activity by cleaving subunits of the 19S regulatory particle (Sun et al, 2004). The decrease in proteasome activity could thus be a consequence of I.29μ+ cells undergoing apoptosis. To exclude this possibility, we compared lysates of total or live cells. Despite Ficoll centrifugation efficiently eliminated dead cells (less than 3 and 7% dead cells were present at days 3 and 4), neither the three main proteasomal activities (panels D–F) nor the abundance of individual subunits differed significantly (panel A, compare lanes 3, 4 and 3F, 4F), apart from μ-chains being slightly more abundant in the fraction enriched in live cells (A, second panel from top). Therefore, the reduction of proteasome activity is not caused by cell death (see also below).
Accumulation of polyubiquitinated proteins in differentiated I.29μ+
Consistent with the observed decline of proteasomal activity, polyubiquitinated proteins markedly increased in LPS-activated cells, at the expense of the pool of free Ub (Figure 3A).
Figure 3.
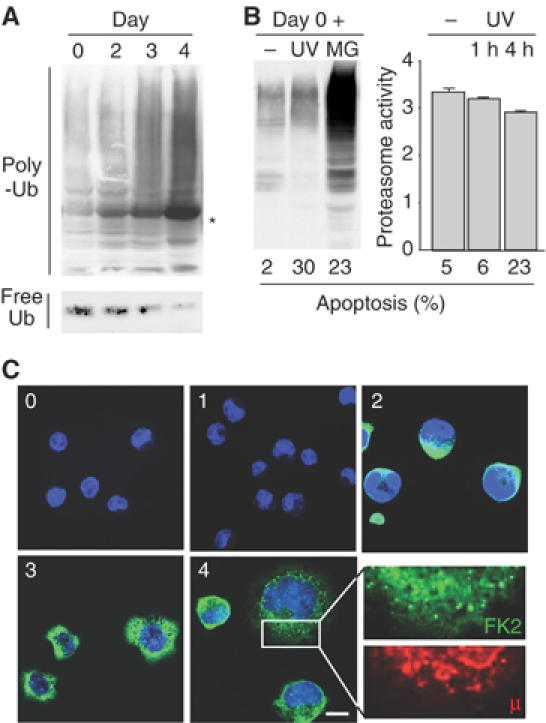
Accumulation of polyubiquitinated proteins in differentiating I.29μ+ cells. (A) Extracts of I.29μ+ cells induced with LPS for the indicated times were blotted with anti-Ub. The band indicated with an asterisk consists of μ-chains that crossreact with secondary antibodies. To detect free Ub, a denser gel was employed (bottom panel). (B) I.29μ+ cells were treated for 4 h with MG132 (MG) or exposed to ultraviolet light for 35 s and cultured for 4 h (UV, lane 6). In the right panel, proteasome activity was measured in unstimulated I.29μ+ cells cultured for 1 or 4 h after exposure to UV. The percentages of apoptotic cells are indicated at the bottom. (C) Immunofluorescent visualization of poly-Ub proteins during I.29μ+ differentiation. Cells were stained with Fk2 antibodies, recognizing poly-Ub proteins, and analyzed by confocal microscopy. Numbers indicate days after LPS stimulation. No overlapping between the Fk2 and anti-μ staining patterns was detected (see a particular at higher magnification in the bottom right panel). Bar: 5 μm.
To further confirm that the accumulation of polyubiquitinated proteins was not a consequence of apoptosis, we exposed I.29μ+ cells to UV rays or MG132 (Figure 3B, left panel). Despite UV treatment induced more apoptosis (30%) than proteasomal inhibition (23%), polyubiquitinated proteins did not increase significantly in UV-treated cells. Only a modest decrease of proteasome activity was detected upon UV treatment (Figure 3B, right panel). Induction of apoptosis by UV failed to increase polyubiquitinated proteins and to decrease proteasome activity also in day 3 LPS-stimulated I.29μ+ (not shown). Thus, apoptosis does not per se induce the accumulation of polyubiquitinated proteins in differentiating I.29μ+ cells.
Accumulation of polyubiquinated proteins was visualized by confocal microscopy with specific antibodies well before overt apoptosis (Figure 3C). Numerous fluorescent dots were detected throughout the cytoplasm after day 3. These structures differed from aggresomes (Kopito and Sitia, 2000) and dendritic cell-induced aggresome-like structures (DALIS) (Lelouard et al, 2002). These structures did not colocalize with Ig-μ, which remained confined to the ER (Figure 3C, lower right panel). However, it should be noted that our anti-μ antibodies preferentially recognize determinants present on folded μ-chains. Reduced μ-chains or fragments thereof would have been stained weakly, if at all.
Taken together, these findings suggest that the fewer proteasomes in differentiating I.29μ+ are insufficient to cope with the protein load, and polyubiquinated proteins accumulate intracellularly.
Stabilization of proteasomal substrates during I.29μ+ differentiation
Our previous observation that Xbp1s accumulates after day 3 of LPS stimulation in I.29μ+ suggested a possible connection between abundant IgM synthesis and UPR induction (van Anken et al, 2003). This could in turn activate apoptosis (Breckenridge et al, 2003). The reduced proteasomal efficiency in LPS-stimulated cells could exaggerate the cascade, stabilizing crucial signaling molecules. First, we determined the steady-state levels of candidate cell death mediator proteins (Figure 4A). These included Chop, a protein involved in ER stress-induced apoptosis in several systems (Breckenridge et al, 2003), and members of the Bcl2 protein family, which play a key role in life–death decisions within the B-lymphocyte lineage (Borner, 2003). Chop increased in the first days of I.29μ+ differentiation, and decreased thereafter, when apoptotic cells were most abundant. In contrast, the apoptotic executor Bax increased significantly after day 3, Bcl2 remaining essentially constant. As a result, the balance of pro- and antiapoptotic factors is shifted towards cell death, concomitantly with the observed onset of apoptosis. To gain further insights into the regulation of UPR components during differentiation, we monitored Xbp1 splicing (Figure 4B, top panel) and the mRNA levels of EDEM (a Xbp1 target gene (Yoshida et al, 2003) and BiP (Figure 4B, bottom panel). The efficiency of Xbp1 splicing peaked at day 1, whereas EDEM and BiP transcripts increased in abundance throughout differentiation. The elevated mRNA levels of EDEM at later stages of differentiation—when Xbp1 splicing decreases—could reflect post-translational stabilization of the Xbp1 protein. Indeed, like its unspliced counterpart (Lee et al, 2003), Xbp1s is degraded by proteasomes (panel C) and its half-life increases during differentiation (Figure 4D, top panel).
Figure 4.
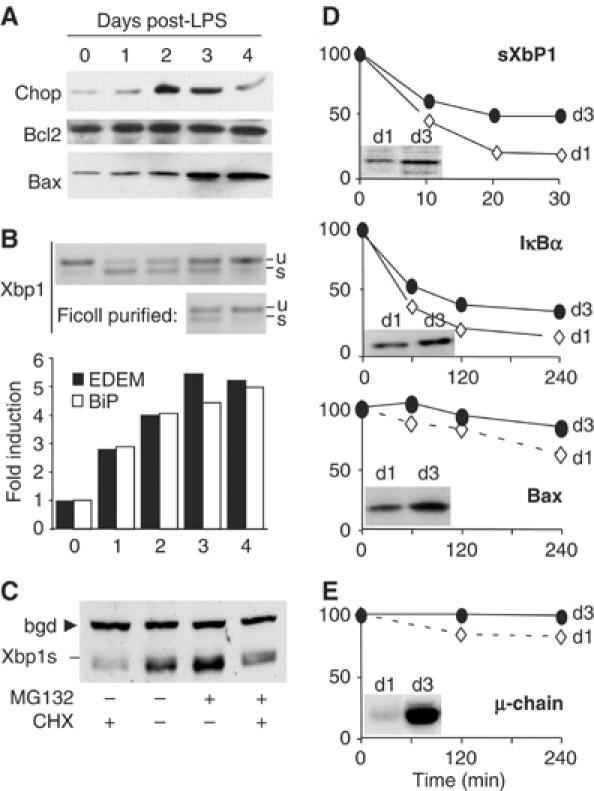
Stabilization of proteasomal substrates in differentiating I.29μ+ cells. (A) Extracts from LPS-stimulated I.29μ+ cells were resolved by SDS–PAGE and blots decorated with antibodies specific for Chop, Bcl2, or Bax, as in Figure 2A. (B) Xbp1 splicing. The RT–PCR products derived from the spliced (s) and unspliced (u) Xbp1 mRNAs are indicated (top panel). Expression of EDEM1 and BiP mRNAs was measured by quantitative real-time RT–PCR analyses, normalized to actin, and expressed as fold change relative to untreated cells (bottom). (C) Xbp1s is degraded by proteasomes. LPS-activated I.29μ+ cells (day 3) were incubated for 20 min with cycloheximide (CHX) and/or MG132, as indicated, lysed and resolved by SDS–PAGE. Blots were decorated with anti-Xbp1. The arrow points to a background stable band (bgd), which served as a loading control. (D) I.29μ+ cells, stimulated for 1 or 3 days with LPS, were incubated with CHX for the indicated times. Aliquots of nuclear (IκBα, Xbp1s) or cytosolic (Bax) extracts were resolved by SDS–PAGE and decorated with specific antibodies, as indicated. Relevant bands were quantified by densitometry, and their intensity expressed as the percentage relative to time 0. Inserts show the steady-state protein levels at days 1 and 3. (E) I.29μ+ cells treated as above were pulsed for 10 min, chased for the indicated times, and immunoprecipitated with anti-μ. Degradation was calculated by the formula: (intracellular+secreted μ at time x): (total μ at time 0) × 100.
Two other proteasome substrates important for B-cell fate, IκBα (Lin et al, 1998) and Bax (Li and Dou, 2000), were stabilized in LPS-stimulated cells (panel D), probably reflecting the decrease in degradative capacity upon differentiation. Ig-μ chains were stabilized as well, as revealed by pulse-chase assays (panel E).
These results indicate that the increased load on the fewer proteasomes causes stabilization of proteins that differ in intracellular localization and intrinsic stability, some of which may facilitate apoptosis.
Increased susceptibility of differentiating I.29μ+ cells to PI
PI are emerging as powerful drugs in MM treatment, but why these tumors are so sensitive is not entirely clear (Hideshima and Anderson, 2002; Goldberg, 2003; Adams, 2004). The above results suggested that the unfavorable load-capacity ratio in Ig-secreting cells could make the latter more prone to enter apoptosis, either basally or following treatment with PI. In this scenario, one would predict that the sensitivity of I.29μ+ cells to PI would increase following LPS stimulation. Therefore, we exposed cells stimulated for 0 or 3 days with LPS to increasing doses of MG132, and determined the fraction of apoptotic cells after 5 h (Figure 5A). Clearly, the sensitivity of LPS-stimulated cells was significantly higher in day 3-stimulated cells. Three additional PI gave similar results (data not shown). To determine whether ER stress could increase the sensitivity to PI, we coincubated cells with tunicamycin (TM), at a dose that did not per se induce apoptosis in the experimental time frame utilized (panel B). TM synergized with MG132 in inducing apoptosis in resting, but not in day 3-stimulated cells (panel A), possibly suggesting that the latter were already experiencing ER stress.
Figure 5.
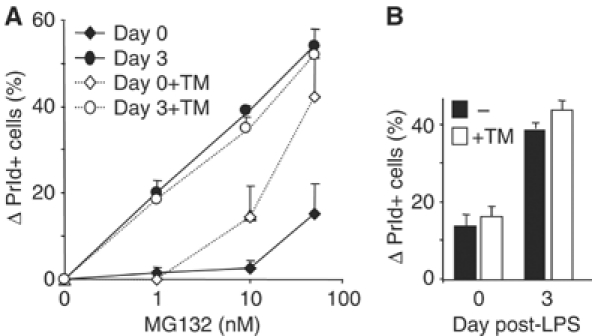
Increased sensitivity to proteasome inhibitors in LPS-stimulated I.29μ+ cells. (A) I.29μ+ cells, untreated or stimulated with LPS for 3 days, were cultured for 5 h in the presence of increasing concentrations of MG132, with or without the simultaneous addition of TM (2.5 μg/ml). The percentage of propidium iodide (PrId) positive cells determined by FACS was plotted after subtracting the value obtained without treatment. Mean±s.d. of three independent experiments. (B) TM alone did not significantly induce apoptosis, implying synergy with MG132 in inducing apoptosis in unstimulated I.29μ+ cells.
Exuberant synthesis of Ig-μ chains makes HeLa cells more sensitive to PI
The above results revealed a correlation between increased Ig-synthesis, decreased proteasomal degradation, ER stress, and apoptosis, both basal and PI-induced. Owing to the complexity of terminal I.29μ+ differentiation (van Anken et al, 2003), and to the intrinsic difficulties encountered in transferring genes into B cells, it was not possible to establish causal links. To circumvent this problem and determine whether a cause–effect relationship exists between excessive proteasomal load and apoptosis, we generated stable HeLa cells expressing secretory μ-chains under an inducible promoter (Tet-Off) and analyzed their sensitivity to PI after induction (Figure 6). At day 2, when μ-chain accumulation is still minimal, no differences were detected (data not shown). At day 4, the percentages of apoptotic cells present before the addition of PI were similar in Tet+ or Tet− cells. However, the synthesis of orphan μ-chains clearly made HeLa cells more sensitive to PI (Figure 6A, left panel). The effects of μ-chain synthesis were particularly evident at intermediate doses. After 6 days of induction, μ-chains caused apoptosis in HeLa cells even in the absence of PI (Figure 6A, right panel). Because of the few HeLa-μs Tet-Off cells undergoing apoptosis, single-cell assays were developed based on the expression of short-lived GFPs rapidly degraded by proteasomes as reporters of proteasomal overload (Dantuma et al, 2000). After 4 days of μ-chain synthesis, more cells became positive if treated with low doses of PI (Figure 6B, left panel). At day 6, a small yet significant percentage of cells were positive even in the absence of PI (Figure 6B, right panel). Therefore, the synthesis of orphan μ-chains increases the sensitivity of HeLa cells to PI in a time- and dose-dependent way, and eventually causes apoptosis, in correlation with proteasomal overload. Activation of μ-chain synthesis did not affect proteasomal activity (Supplementary Figure 3), indicating that synthetic overload is sufficient to sensitize to PI also in the presence of intact proteasome activity.
Figure 6.
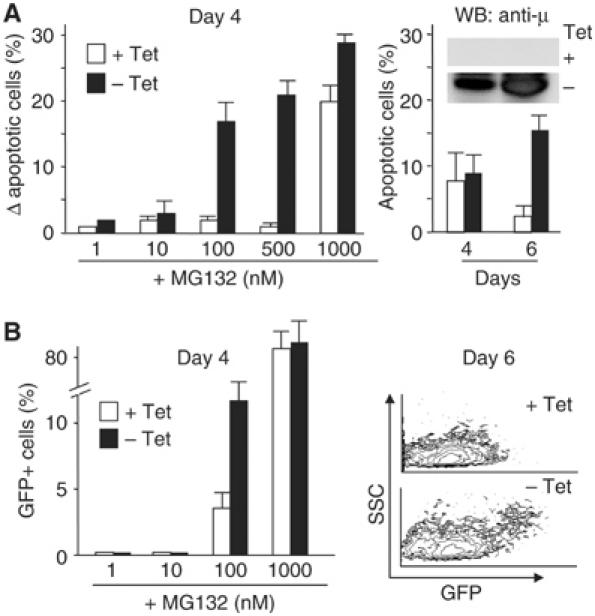
Synthesis of orphan Ig-μ chains sensitizes HeLa cells to proteasome inhibitors and stabilizes a proteasomal reporter. (A) Sensitivity to PI increases in HeLa upon Ig-μ synthesis. HeLa-μs Tet-Off cells were cultured with or without tetracycline (Tet) for 2, 4, or 6 days and exposed to increasing concentrations of MG132 for 24 h before FACS analysis. The left panel shows the percentage of apoptotic cells at day 4, after subtracting the values observed without MG132. The right panel shows the basal levels of apoptosis observed at days 4 and 6. The insert shows the accumulation of μs-chains, as revealed by Western blot analyses. At day 2, when little μ-chains accumulated, there were no significant differences in the number of apoptotic cells between induced and noninduced cells (not shown). Data expressed as mean±s.e.m. of three independent experiments. (B) Accumulation of short-lived GFP upon Ig-μ synthesis. HeLa-μs Tet-Off cells stably expressing short-lived GFPs rapidly degraded by proteasomes were generated by lentiviral transduction. As assessed by FACS (left panel), 4 days after Tet removal, GFP accumulated more readily in response to PI (mean±s.e.m.). After 6 days of intensive Ig-μ synthesis (right panel), some cells displayed spontaneous GFP accumulation by FACS. Axes show side scatter (SSC) on linear scale and GFP fluorescence on log scale.
Normal differentiating B splenocytes downregulate proteasomes and undergo basal and PI-induced apoptosis
To further generalize our observations, we analyzed primary murine B splenocytes. After LPS stimulation, the relative proteasome capacity fell in the last days of differentiation (Figure 7A), when IgM production becomes maximal (see the Ig-L blot in panel B). Polyubiquitinated proteins accumulated in LPS-activated CD19+ splenocytes, peaking at day 3 (panel B). Next, to activate B cells by means other than LPS, we cocultured them with CD3-activated T cells (Banchereau et al, 1994; Johnson-Leger et al, 1998). After an initial rise, proteasomal activity fell dramatically at day 6 (panel C), concomitantly with intracellular accumulation of polyubiquitinated proteins (panel D) and Ig-μ chains (not shown), as revealed by immunofluorescence.
Figure 7.
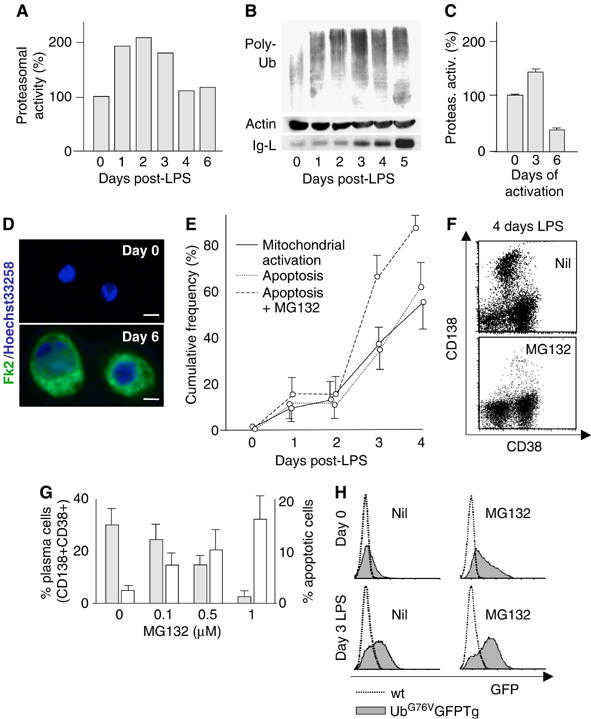
Primary plasma cells accumulate poly-Ub proteins and become sensitive to proteasome inhibitors. (A) Proteasomal activity during LPS-induced primary plasma cell differentiation. Proteasomal chymotrypsin-like activity was assessed in extracts from stimulated primary splenocytes as described in legend to Figure 2, and expressed as % relative to day 0. (B) Accumulation of poly-Ub proteins in differentiating primary immunomagnetically purified CD19+ cells. Extracts from cells stimulated with LPS for the indicated times were blotted with anti-Ub. Ig light chains (Ig-L) were detected in a denser gel (bottom panel). (C) LPS-independent B-cell activation. Unfractionated splenocytes were seeded onto insolubilized anti-CD3 to activate T cells, which in turn cause polyclonal B-cell activation (Banchereau et al, 1994; Johnson-Leger et al, 1998). At the indicated times CD38+ cells were tested for proteasomal chymotryptic activity as in panel A. (D) Cells treated as in (C) were stained with Fk2 antibodies as in legend to Figure 3C. Bar: 5 μm. (E) Occurrence of mitochondrial activation (continuous line) and apoptotic death (dotted lines) during LPS-induced plasma cell differentiation from primary B cells. MG132 (100 nM) was added to cultures from day 0. Measurements performed by FACS analysis upon staining with Annexin V and propidium iodide (apoptotic death) or JC-1 (to detect the loss of transmembrane potential across the outer mitochondrial membrane). Mean±s.d. of three independent experiments. (F) FACS profiles of CD38 and CD138 expression in splenocytes 4 days after intraperitoneal injection with 1 mg LPS. Cells were cultured for 6 h in the absence (top panel) or presence of MG132 (bottom panel). (G) Proportion of plasma cells (filled bars) and prevalence of apoptosis (empty bars) in splenocytes from in vivo LPS-treated mice treated in vitro with increasing doses of MG132 for 6 h. Apoptosis was assessed as the proportion of Annexin V-positive cells. Mean±s.d. (H) Accumulation of a short-lived GFP reporter of the Ub–proteasome system in primary B cells from transgenic mice (Lindsten et al, 2003) after LPS stimulation. CD19+ cells from UbG76VGFP transgenic mice and wild-type littermates were immunomagnetically purified and stimulated in vitro with LPS for 3 days and with MG132 (1.5 μM for 2.5 h) as indicated.
In LPS-activated CD19+ splenocytes, apoptosis increased after day 3 in parallel with mitochondrial damage (panel E), suggesting the involvement of intrinsic pathways. Proteasome inhibition significantly accelerated the death of normal plasma cells, causing apoptosis mainly after day 3 (panel E), again in correlation with the onset of abundant IgM secretion.
To further explore whether proteasomal inhibition is selectively toxic to primary plasma cells, unfractionated white splenocytes were prepared from mice 4 days after LPS injection and exposed ex vivo to increasing doses of MG132 for up to 24 h in vitro. LPS-treated mice accumulated CD138+ plasma cells in their spleen (∼30% after 4 days in these experiments). Administration of PI in vitro caused a dose-dependent and preferential depletion of CD38+ CD138+ plasmablasts and plasma cells in as little as 6 h of treatment (panels F and G). Thus, like their malignant counterparts (Hideshima et al, 2001; Zavrski et al, 2003), normal Ig-secreting cells are extremely sensitive to PI.
Further evidence for proteasomal insufficiency during B-cell differentiation was obtained by analyzing transgenic mice expressing a chimeric uncleavable ubiquitin-GFP (UbG76VGFP) that is rapidly degraded by proteasomes (Dantuma et al, 2000). A short exposure to PI caused a strong accumulation of the reporter in unstimulated CD19+ B cells (panel H, top). After 3 days of LPS stimulation in vitro, most B cells expressed CD138 (not shown) and accumulated the GFP reporter even in the absence of PI (panel H, bottom).
Altogether, these findings demonstrate that also in primary splenocytes, plasma cell differentiation causes loss of proteasomal capacity, accumulation of endogenous proteasomal substrates, and sensitization to PI before spontaneous cell death.
Discussion
The short lifespan of the majority of plasma cells is important to limit antibody responses (Ho et al, 1986). However, some of them home in the bone marrow where they find conditions for longer survival, which maintains protective Ig titers in the blood (Manz et al, 2002). While it is clear that extrinsic factors such as cytokines and contacts with stromal cells play important roles in shaping the environmental niche that promotes survival of long-lived plasma cells, little is known about the mechanisms inducing the death of short-lived ones (Donjerkovic and Scott, 2000; Hase et al, 2002; Kroesen et al, 2003). In particular, the existence of intrinsic factors regulating plasma cell lifespan has been proposed, but never demonstrated. Our findings reveal that in differentiating I.29μ+ and primary spleen B cells, the proteasomal capacity decreases despite the higher demand imposed by the high rates of protein production. The finding that an unfavorable proteasomal load/capacity ratio can predispose cells to apoptosis has important implications for the physiology of the immune system and for different pathological conditions, in primis Ig-secreting tumors.
Proteasomes in the physiology of plasma cell differentiation
The finding that Xbp1 is essential for plasma cell development (Reimold et al, 2001; Iwakoshi et al, 2003a) suggested that exuberant Ig synthesis causes ER stress, thus inducing ER biogenesis and eventually apoptosis via UPR-related pathways (Ma and Hendershot, 2003; Sitia and Braakman, 2003; Brewer and Hendershot, 2005). While confirming a correlation between massive Ig production and plasma cell death (Figure 1), our findings reveal unexpected aspects. Chop—an UPR-induced protein known to mediate apoptosis—decreased in abundance after day 3 of LPS stimulation. Also, the efficiency of Xbp1 splicing decreased in the last days of differentiation. The concomitant accumulation of Xbp1s may be due in part to its increased stability, which in turn reflects reduced proteasomal capacity (Figure 4). Delayed Xbp1s degradation could allow B cells to rely on the Xbp1 pathway without activating Perk and consequently inhibit translation, which would be detrimental to their synthetic goal. The reports that Xbp1s stabilizes certain ER-targeted proteins (Shaffer et al, 2004) and stimulates μ-chain translation (Tirosh et al, 2005) suggest the existence of complex regulatory networks that likely allow B cells to customize the UPR to fulfill their physiological requirements. Therefore, both the Perk and the Ire1α pathways seem to be tuned down before LPS-stimulated I.29μ+ cells undergo apoptosis. A decreased protection by the UPR might actually promote Chop-independent cell death. What then triggers apoptosis?
As an efficient ERAD is important for folding and secretion capacity (Eriksson et al, 2004), it was surprising to observe that the enzymatic activities and the relative abundance of proteasomes decreased during differentiation of both I.29μ+ lymphoma cells and primary splenocytes. Proteasomes decreased also on a per cell basis, revealing that plasma cell differentiation entails decreased proteasome biogenesis. Intriguingly, only two catalytic subunits (X and Y) were downregulated, the third one (Z) remaining essentially unchanged. This implies specific mechanisms for regulating the expression of proteasomal subunits, in line with recent observations on PA28 α and β (Ossendorp et al, 2005). Proteasomal catalytic subunits are synthesized as inactive precursors, which are cleaved upon assembly into functional proteasomes (Rock and Goldberg, 1999). The cleaved Z-subunits present in LPS-stimulated I.29μ+ cells could be part of particles containing multiple copies of this subunit.
The decreased X and Y levels were not compensated by increased synthesis of immunoproteasomes. In fact, two IFNγ-inducible subunits (LMP2 and MECL-1) slightly decreased, albeit to a lesser extent than X and Y. Paradoxically, therefore, when the need for proteasomes increases, their abundance becomes limiting. This could represent a distinctive feature of short-lived plasma cells, as dendritic cells upregulate proteasomal subunits in response to LPS (Macagno et al, 2001). No changes in proteasome activity were induced by LPS in a monocytic line (Supplementary Figure 3), implying that TLR4 triggering per se is not sufficient to cause proteasome downregulation. Likewise, decreased proteasome activity and accumulation of polyubiquitinated proteins were observed also when primary splenocytes were cocultured with CD3-activated T cells (Figure 7C and D). Whilst polyubiquitinated proteins accumulated in activated B cells, the pool of free Ub decreased. Either the latter or the relative number of proteasomes could limit degradation. As a consequence, endogenous proteasomal substrates were stabilized in differentiating I.29μ+ cells, and a reporter of the Ub–proteasome pathway (Lindsten et al, 2003) accumulated in LPS-activated GFPG76V-transgenic splenocytes (Figure 7F). Altogether, these data confirm that proteasome insufficiency is a feature of plasma cell differentiation, independently from how B cell are activated.
Proteasome decrease and apoptosis: chicken not egg
Although activated caspases may cleave 19S regulator particle subunits during apoptosis (Sun et al, 2004), exposure to UV light did not result in significant accumulation of polyubiquitinated proteins in apoptotic I.29μ+. The detection of live cells with abundant polyubiquitinated proteins (Figures 4C and 7B) further confirms that in differentiating B cells proteasomal insufficiency precedes apoptosis. In the late stages of differentiation, activated caspases could further decrease proteasomal capacity by cleaving certain 19S subunits, possibly leading to an amplification circuit (Sun et al, 2004).
Linking protein production to cell death
A clear cause–effect relationship was established using HeLa cells harboring inducible Ig-μ chains. Overexpression of Ig-μ increased the sensitivity to PI, and resulted in spontaneous apoptosis, similar to what was observed in B cells. Proteasome activity did not decrease in this model, perhaps explaining why the effects were less marked than in activated B cells, where increased load is accompanied by a reduced capacity.
The stabilization of endogenous proteasomal substrates might meet certain functional requirements of plasma cells. For instance, the increased stability of μs chains may favor IgM polymerization, whereas accumulation of death factors may predispose to the apoptotic program. IκBα stabilization would decrease NF-κB activity and lower the apoptotic threshold. Likewise, the stabilization of Bax and Bim, two proteasome substrates known to control B-cell lifespan (Marsden and Strasser, 2003), may—at least in part—explain their relative increase with respect to Bcl2, until the death threshold is reached. In this scenario, plasma cell death would be linked to Ig production, thus contributing to end humoral responses. At the very high rate of Ig production that characterizes the late phases of differentiation, even a small percent of defective degradation would lead to accumulation. Combined with the progressive decrease of proteasomal capacity, this could allow cells to keep track of the amount of Ig secreted.
Generality of the load versus capacity model
The findings obtained with primary B cells indicate that PI hypersensitivity is not a specific feature of myelomas, but is inherent to the processes of plasma cell differentiation and exuberant Ig synthesis, as sensitization can be observed during B to plasma cell differentiation (Figure 7), and artificially reproduced in nonlymphoid cells by overexpressing orphan μ-chains (Figure 6). This has important implications related to the potential utilization of PI in the treatment of inflammatory disorders, especially those related to excessive antibody production. Indeed, epoxomicin inhibits inflammation, and the mechanisms herein described could potentiate NF-κB deregulation (Meng et al, 1999; Goldberg and Rock, 2002). With respect to cancer treatment, our findings with I.29μ+ cells suggest that certain lymphomas or leukemias can become sensitive to PI if properly induced to differentiate (Figure 5). In many tumors, factors that induce differentiation facilitate or induce apoptosis (Kasibhatla and Tseng, 2003). Our results suggest that apoptotic sensitivity could be increased by inducing protein synthesis and misfolding. This may sound heretic when degenerative disorders owing to proteotoxicity represent a major social concern. Yet, the observation that TM and PI synergize in I.29μ+ cells provides a proof of principle for utilizing stress against cancer. In the context of plasma cell tumors, the secreting potential of myeloma cells has been proposed as a prognostic factor for MM (Symeonidis et al, 2002). Further studies are needed to determine whether PI sensitivity correlates with Ig production and to investigate the features of cytotoxic proteins, with respect to topology, synthesis, folding, or degradation rates.
In principle, the intrinsic ‘self-pollUbtion' mechanism described here could operate in other secretory cells in conjunction with diverse extrinsic factors. The latter play a major role in controlling plasma cell lifespan. By providing appropriate soluble factors and costimulatory molecules, germinal centers allow plasmacyte differentiation and survival (Le Bon et al, 2001; Manz et al, 2002; Poeck et al, 2004). Long-lived plasma cells home in the bone marrow, where they find optimal survival conditions, but die when cultured in vitro. Based on our findings, one could speculate that long-lived plasma cells reduce their unbalance perhaps by decreasing Ig synthesis, or deploying more degradative capacity, for example, by increasing proteasome biogenesis. Comparing the load-capacity ratios in long- and short-lived plasma cells may shed light into the role of extrinsic and intrinsic life-limiting mechanisms.
In conclusion, we propose that the progressive impairment of proteasomal capacity is one of the mechanisms that contribute in limiting plasma cell lifespan, linking it to antibody production. Identifying the factors that control proteasome synthesis and activity might allow one to manipulate protein production and cell lifespan with obvious implications for biotechnology and cancer therapy.
Materials and methods
Cell cultures and spleen B-cell purification
I.29μ+ cells were activated with LPS as described (van Anken et al, 2003). Primary plasma cells were generated from C57Bl/6 mice via three alternative approaches: (1) mice were injected intraperitoneally with 1 mg LPS; after 1–5 days spleen white cells were harvested and assayed for CD38 and CD138 expression and apoptosis by FACS; (2) CD19+ splenocytes from normal or transgenic GFPG76V mice (Lindsten et al, 2003) were prepared by immunomagnetic selection (Miltenyi) and stimulated with LPS in vitro; and (3) unfractionated splenocytes were seeded in wells previously coated with anti-mouse CD3 (BD Pharmingen), harvested after 0, 3, or 6 days, and assayed for B-cell content and activation by FACS, proteasomal activity, and immunofluorescent staining for poly-Ub proteins.
HeLa-μs Tet-Off cells were generated by trasfecting Tet-Off cells (Clontech Laboratories Inc.) with pTRE-μ1 and pTK-Hyg. Hygromycin-resistant clones were screened by immunofluorescence after 72 h in the absence of Tet, selected and maintained in medium supplemented with tetracycline. To generate HeLa-μs Tet-Off cells stably expressing the Ub-GFP reporter, lentiviral transduction was adopted (see below and Supplementary data for details).
Flow-cytometric analyses of apoptosis
Cells were washed with PBS and stained with Annexin V–FITC (1 μg/ml) and propidium iodide (2.5 μg/ml). The loss of mitochondrial transmembrane potential across the outer mitochondrial membrane was assessed with JC-1 dye, as described (Bedner et al, 1999). Flow cytometry data were obtained with FACScalibur (BD Biosciences) and analyzed using the Cellquest software (BD Biosciences).
Antibodies and immunocytochemistry
I.29μ+ cells were harvested and coated on 1% Alcian blue-treated coverslips for 5 min at 37°C, fixed with 3% paraformaldehyde in PBS for 10 min at RT and permeabilized with 0.5 mg/ml saponin in PBS containing 5% FCS and 10 mM glycine. Primary splenocytes and plasma cells were seeded on poly-L-lysine-coated slides, before fixation and permeabilization. Cells were stained with monoclonal antibody Fk2 (Biomol, Exeter, UK) for 1 h, rinsed in PBS followed by staining with Alexa Fluor 488 goat anti-mouse IgG1 antibody and TO-PRO-3 (Molecular Probes Inc., Eugene, OR, USA). Coverslips were observed on a Zeiss LSM 510 confocal microscope.
Proteasome activity assays and immunoblot analyses
Details are provided in Supplementary data.
RT–PCR, vector production, and infection
Details on the protocols and primers utilized for RT–PCR and on the lentiviral vectors encoding unstable Ub-R-GFP, an N-end rule substrate, and UbG76V-GFP, containing a mutated uncleavable Ub moiety (Dantuma et al, 2000), are given in Supplementary material. HeLa-μs Tet-Off cells were transduced using different dilutions of the vector. Cells expressing optimal amounts of the reporter were sorted by FACS upon reversible proteasomal inhibition.
Supplementary Material
Supplementary Materials and Methods
Supplementary Figures
Acknowledgments
We thank Ineke Braakman, Nico Dantuma, Fred Goldberg, Nadia Montani, Kazu Mori, Luigi Naldini, Silvia Nerini-Molteni, Michael Neuberger, Luca Rampoldi, David Ron, Francesca Santoni de Sio, Luisa Schiaffonati, Caterina Valetti, Eelko van Anken, and Larry Wrabetz for reagents, suggestions, and discussions, and Fabrizio Benedicenti and Claudio Fagioli for technical help. This work was supported through grants from the Associazione Italiana per la Ricerca sul Cancro (AIRC), Ministero della Sanità, MIUR (CoFin and Center of Excellence in Physiopathology of Cell Differentiation), and Telethon. AM was recipient of a fellowships from the European Community (CEE52106). We apologize with our colleagues whose pioneering work could not be cited owing to space limitations.
References
- Adams J (2004) The proteasome: a suitable antineoplastic target. Nat Rev Cancer 4: 349–360 [DOI] [PubMed] [Google Scholar]
- Banchereau J, Bazan F, Blanchard D, Briere F, Galizzi JP, van Kooten C, Liu YJ, Rousset F, Saeland S (1994) The CD40 antigen and its ligand. Annu Rev Immunol 12: 881–922 [DOI] [PubMed] [Google Scholar]
- Bedner E, Li X, Gorczyca W, Melamed MR, Darzynkiewicz Z (1999) Analysis of apoptosis by laser scanning cytometry. Cytometry 35: 181–195 [DOI] [PubMed] [Google Scholar]
- Borner C (2003) The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions. Mol Immunol 39: 615–647 [DOI] [PubMed] [Google Scholar]
- Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC (2003) Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 22: 8608–8618 [DOI] [PubMed] [Google Scholar]
- Brewer JW, Hendershot LM (2005) Building an antibody factory: a job for the unfolded protein response. Nat Immunol 6: 23–29 [DOI] [PubMed] [Google Scholar]
- Calame KL, Lin KI, Tunyaplin C (2003) Regulatory mechanisms that determine the development and function of plasma cells. Annu Rev Immunol 21: 205–230 [DOI] [PubMed] [Google Scholar]
- Cals MM, Guenzi S, Carelli S, Simmen T, Sparvoli A, Sitia R (1996) IgM polymerization inhibits the Golgi-mediated processing of the mu-chain carboxy-terminal glycans. Mol Immunol 33: 15–24 [DOI] [PubMed] [Google Scholar]
- Cascio P, Hilton C, Kisselev AF, Rock KL, Goldberg AL (2001) 26S proteasomes and immunoproteasomes produce mainly N-extended versions of an antigenic peptide. EMBO J 20: 2357–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG (2000) Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol 18: 538–543 [DOI] [PubMed] [Google Scholar]
- Donjerkovic D, Scott DW (2000) Activation-induced cell death in B lymphocytes. Cell Res 10: 179–192 [DOI] [PubMed] [Google Scholar]
- Eriksson KK, Vago R, Calanca V, Galli C, Paganetti P, Molinari M (2004) EDEM contributes to maintenance of protein folding efficiency and secretory capacity. J Biol Chem 279: 44600–44605 [DOI] [PubMed] [Google Scholar]
- Fagioli C, Mezghrani A, Sitia R (2001) Reduction of interchain disulfide bonds precedes the dislocation of Ig-mu chains from the endoplasmic reticulum to the cytosol for proteasomal degradation. J Biol Chem 276: 40962–40967 [DOI] [PubMed] [Google Scholar]
- Fagioli C, Sitia R (2001) Glycoprotein quality control in the endoplasmic reticulum. Mannose trimming by endoplasmic reticulum mannosidase I times the proteasomal degradation of unassembled immunoglobulin subunits. J Biol Chem 276: 12885–12892 [DOI] [PubMed] [Google Scholar]
- Fra AM, Fagioli C, Finazzi D, Sitia R, Alberini CM (1993) Quality control of ER synthesized proteins: an exposed thiol group as a three-way switch mediating assembly, retention and degradation. EMBO J 12: 4755–4761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AL (2003) Protein degradation and protection against misfolded or damaged proteins. Nature 426: 895–899 [DOI] [PubMed] [Google Scholar]
- Goldberg AL, Rock K (2002) Not just research tools—proteasome inhibitors offer therapeutic promise. Nat Med 8: 338–340 [DOI] [PubMed] [Google Scholar]
- Hase H, Kanno Y, Kojima H, Morimoto C, Okumura K, Kobata T (2002) CD27 and CD40 inhibit p53-independent mitochondrial pathways in apoptosis of B cells induced by B cell receptor ligation. J Biol Chem 277: 46950–46958 [DOI] [PubMed] [Google Scholar]
- Hideshima T, Anderson KC (2002) Molecular mechanisms of novel therapeutic approaches for multiple myeloma. Nat Rev Cancer 2: 927–937 [DOI] [PubMed] [Google Scholar]
- Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, Anderson KC (2001) The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res 61: 3071–3076 [PubMed] [Google Scholar]
- Hideshima T, Richardson PG, Anderson KC (2003) Targeting proteasome inhibition in hematologic malignancies. Rev Clin Exp Hematol 7: 191–204 [PubMed] [Google Scholar]
- Ho F, Lortan JE, MacLennan IC, Khan M (1986) Distinct short-lived and long-lived antibody-producing cell populations. Eur J Immunol 16: 1297–1301 [DOI] [PubMed] [Google Scholar]
- Hoyer BF, Moser K, Hauser AE, Peddinghaus A, Voigt C, Eilat D, Radbruch A, Hiepe F, Manz RA (2004) Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J Exp Med 199: 1577–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakoshi NN, Lee AH, Glimcher LH (2003a) The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol Rev 194: 29–38 [DOI] [PubMed] [Google Scholar]
- Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH (2003b) Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol 4: 321–329 [DOI] [PubMed] [Google Scholar]
- Johnson-Leger C, Christenson JR, Holman M, Klaus GG (1998) Evidence for a critical role for IL-2 in CD40-mediated activation of naive B cells by primary CD4T cells. J Immunol 161: 4618–4626 [PubMed] [Google Scholar]
- Kasibhatla S, Tseng B (2003) Why target apoptosis in cancer treatment? Mol Cancer Ther 2: 573–580 [PubMed] [Google Scholar]
- Kopito RR, Sitia R (2000) Aggresomes and Russell bodies. EMBO Rep 1: 225–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroesen BJ, Jacobs S, Pettus BJ, Sietsma H, Kok JW, Hannun YA, de Leij LF (2003) BcR-induced apoptosis involves differential regulation of C16 and C24-ceramide formation and sphingolipid-dependent activation of the proteasome. J Biol Chem 278: 14723–14731 [DOI] [PubMed] [Google Scholar]
- Kuehl WM, Bergsagel PL (2002) Multiple myeloma: evolving genetic events and host interactions. Nat Rev Cancer 2: 175–187 [DOI] [PubMed] [Google Scholar]
- Le Bon A, Schiavoni G, D'Agostino G, Gresser I, Belardelli F, Tough DF (2001) Type i interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 14: 461–470 [DOI] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH (2003) Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci USA 100: 9946–9951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelouard H, Gatti E, Cappello F, Gresser O, Camosseto V, Pierre P (2002) Transient aggregation of ubiquitinated proteins during dendritic cell maturation. Nature 417: 177–182 [DOI] [PubMed] [Google Scholar]
- Li B, Dou QP (2000) Bax degradation by the ubiquitin/proteasome-dependent pathway: involvement in tumor survival and progression. Proc Natl Acad Sci USA 97: 3850–3855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, DeMartino GN, Greene WC (1998) Cotranslational biogenesis of NF-kappaB p50 by the 26S proteasome. Cell 92: 819–828 [DOI] [PubMed] [Google Scholar]
- Lindsten K, Menendez-Benito V, Masucci MG, Dantuma NP (2003) A transgenic mouse model of the ubiquitin/proteasome system. Nat Biotechnol 21: 897–902 [DOI] [PubMed] [Google Scholar]
- Ma Y, Hendershot LM (2003) The stressful road to antibody secretion. Nat Immunol 4: 310–311 [DOI] [PubMed] [Google Scholar]
- Macagno A, Kuehn L, de Giuli R, Groettrup M (2001) Pronounced up-regulation of the PA28alpha/beta proteasome regulator but little increase in the steady-state content of immunoproteasome during dendritic cell maturation. Eur J Immunol 31: 3271–3280 [DOI] [PubMed] [Google Scholar]
- Mancini R, Fagioli C, Fra AM, Maggioni C, Sitia R (2000) Degradation of unassembled soluble Ig subunits by cytosolic proteasomes: evidence that retrotranslocation and degradation are coupled events. FASEB J 14: 769–778 [DOI] [PubMed] [Google Scholar]
- Manz RA, Arce S, Cassese G, Hauser AE, Hiepe F, Radbruch A (2002) Humoral immunity and long-lived plasma cells. Curr Opin Immunol 14: 517–521 [DOI] [PubMed] [Google Scholar]
- Marsden VS, Strasser A (2003) Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu Rev Immunol 21: 71–105 [DOI] [PubMed] [Google Scholar]
- Meiners S, Heyken D, Weller A, Ludwig A, Stangl K, Kloetzel PM, Kruger E (2003) Inhibition of proteasome activity induces concerted expression of proteasome genes and de novo formation of mammalian proteasomes. J Biol Chem 278: 21517–21525 [DOI] [PubMed] [Google Scholar]
- Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM (1999) Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc Natl Acad Sci USA 96: 10403–10408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossendorp F, Fu N, Camps M, Granucci F, Gobin SJ, van den Elsen PJ, Schuurhuis D, Adema GJ, Lipford GB, Chiba T, Sijts A, Kloetzel PM, Ricciardi-Castagnoli P, Melief CJ (2005) Differential expression regulation of the alpha and beta subunits of the PA28 proteasome activator in mature dendritic cells. J Immunol 174: 7815–7822 [DOI] [PubMed] [Google Scholar]
- Poeck H, Wagner M, Battiany J, Rothenfusser S, Wellisch D, Hornung V, Jahrsdorfer B, Giese T, Endres S, Hartmann G (2004) Plasmacytoid dendritic cells, antigen, and CpG-C license human B cells for plasma cell differentiation and immunoglobulin production in the absence of T-cell help. Blood 103: 3058–3064 [DOI] [PubMed] [Google Scholar]
- Rabinovich E, Kerem A, Frohlich KU, Diamant N, Bar-Nun S (2002) AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol 22: 626–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH (2001) Plasma cell differentiation requires the transcription factor XBP-1. Nature 412: 300–307 [DOI] [PubMed] [Google Scholar]
- Rock KL, Goldberg AL (1999) Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu Rev Immunol 17: 739–779 [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, Hurt EM, Petroulakis E, Sonenberg N, Yewdell JW, Calame K, Glimcher LH, Staudt LM (2004) XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 21: 81–93 [DOI] [PubMed] [Google Scholar]
- Sitia R, Braakman I (2003) Quality control in the endoplasmic reticulum protein factory. Nature 426: 891–894 [DOI] [PubMed] [Google Scholar]
- Sitia R, Neuberger M, Alberini C, Bet P, Fra A, Valetti C, Williams G, Milstein C (1990) Developmental regulation of IgM secretion: the role of the carboxy-terminal cysteine. Cell 60: 781–790 [DOI] [PubMed] [Google Scholar]
- Sitia R, Neuberger MS, Milstein C (1987) Regulation of membrane IgM expression in secretory B cells: translational and post-translational events. EMBO J 6: 3969–3977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriburi R, Jackowski S, Mori K, Brewer JW (2004) XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol 167: 35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XM, Butterworth M, MacFarlane M, Dubiel W, Ciechanover A, Cohen GM (2004) Caspase activation inhibits proteasome function during apoptosis. Mol Cell 14: 81–93 [DOI] [PubMed] [Google Scholar]
- Symeonidis A, Kouraklis-Symeonidis A, Grouzi E, Zolota V, Melachrinou M, Kourea K, Fragopanagou E, Giannakoulasa N, Seimeni U, Tiniakou M, Matsouka P, Zoumbos N (2002) Determination of plasma cell secreting potential as an index of maturity of myelomatous cells and a strong prognostic factor. Leukemia Lymphoma 43: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Tirosh B, Iwakoshi NN, Glimcher LH, Ploegh HL (2005) XBP-1 specifically promotes IgM synthesis and secretion, but is dispensable for degradation of glycoproteins in primary B cells. J Exp Med 202: 505–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Anken E, Romijn EP, Maggioni C, Mezghrani A, Sitia R, Braakman I, Heck AJ (2003) Sequential waves of functionally related proteins are expressed when B cells prepare for antibody secretion. Immunity 18: 243–253 [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Hosokawa N, Kaufman RJ, Nagata K, Mori K (2003) A time-dependent phase shift in the mammalian unfolded protein response. Dev Cell 4: 265–271 [DOI] [PubMed] [Google Scholar]
- Zavrski I, Naujokat C, Niemoller K, Jakob C, Heider U, Langelotz C, Fleissner C, Eucker J, Possinger K, Sezer O (2003) Proteasome inhibitors induce growth inhibition and apoptosis in myeloma cell lines and in human bone marrow myeloma cells irrespective of chromosome 13 deletion. J Cancer Res Clin Oncol 129: 383–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials and Methods
Supplementary Figures