

Abstract
We present evidence for a complex regulatory interplay between the initiation of DNA replication and deoxyribonucleotide synthesis. In Escherichia coli, the ATP-bound DnaA protein initiates chromosomal replication. Upon loading of the β-clamp subunit (DnaN) of the replicase, DnaA is inactivated as its intrinsic ATPase activity is stimulated by the protein Hda. The β-subunit acts as a matchmaker between Hda and DnaA. Chain elongation of DNA requires a sufficient supply of deoxyribonucleotides (dNTPs), which are produced by ribonucleotide reductase (RNR). We present evidence suggesting that the molecular switch from ATP-DnaA to ADP-DnaA is a critical step coordinating DNA replication with increased deoxyribonucleotide synthesis. Characterization of dnaA and dnaN mutations that result in a constitutively high expression of RNR reveal this mechanism. We propose that the nucleotide bound state of DnaA regulates the transcription of the genes encoding ribonucleotide reductase (nrdAB). Accordingly, the conversion of ATP-DnaA to ADP-DnaA after initiation and loading of the β-subunit DnaN would allow increased nrdAB expression, and consequently, coordinated RNR synthesis and DNA replication during the cell cycle.
Keywords: β-clamp, deoxyribonucleotides (dNTPs), DnaA, Hda, ribonucleotide reductase (RNR)
Introduction
The initiation of chromosome replication is tightly regulated. In prokaryotes, DnaA plays a key role in promoting and regulating this process, binding directly to the origin of replication (Kornberg and Baker, 1992; Baker and Bell, 1998; Messer, 2002). This binding facilitates DNA strand opening, allowing loading of helicase, primase and DNA polymerase (pol) III holoenzyme (the chromosomal replicase), thus leading to chromosomal replication (Kornberg and Baker, 1992). An early step in replicase assembly is loading of the sliding clamp, a ring-shaped dimer of the β-subunit (DnaN) of pol III. After encircling the DNA strand, β-subunit stabilizes the replicase, enabling the extension of RNA primers and processive replication (Kornberg and Baker, 1992; Baker and Bell, 1998). These steps in DNA replication parallel those in eukaryotic cells (Bell and Dutta, 2002).
In Escherichia coli, DnaA protein forms a stable complex with ATP or ADP, but only the ATP-bound form is active in DNA strand opening during initiation (Sekimizu et al, 1987). After completion of initiation, loading of the β-subunit (DnaN) protein as a sliding clamp onto template promotes the accelerated hydrolysis of active ATP-DnaA to inactive ADP-DnaA, preventing premature reinitiation of chromosome replication, a process termed ‘regulatory inactivation of DnaA (RIDA)' (Katayama et al, 1998). That the DNA-bound form of β-subunit is required for DnaA-ATP hydrolysis ensures timely coupling between DnaA inactivation and the elongation stage of DNA replication. Also involved in this process is the protein, Hda (Kato and Katayama, 2001). The ATPase activity is induced by the interaction of Hda with DnaA, facilitated by β-subunit loaded onto DNA as a sliding clamp (Su'etsugu et al, 2005).
Another essential enzyme required for DNA replication is ribonucleotide reductase (RNR) which catalyzes the reduction of ribonucleotides to deoxyribonucleotides (Thelander and Reichard, 1979). E. coli RNR comprises two nonidentical and coordinately synthesized subunits, R1 and R2 (Hanke and Fuchs, 1983). The activity of the holoenzyme is controlled by feedback regulation through binding of allosteric effectors to R1 (Eliasson et al, 1994). However, the transcriptional control of RNR synthesis is poorly understood.
The amount of RNR protein, and thus, deoxyribonucleotides, is coordinated with the rate of DNA synthesis, which is determined by the initiation events. In E. coli growing with a generation time longer than the time required for chromosome replication, the need for RNR goes from zero to a maximum as DNA initiation occurs. The genes encoding the two RNR subunits, nrdA and nrdB, comprise an operon and are cell cycle regulated. nrd mRNA synthesis increases at approximately the time of DNA initiation (Sun and Fuchs, 1992), similarly to what is observed in eukaryotic organisms (Engstrom et al, 1985). This regulatory mechanism generates a controlled burst of RNR synthesis every time a new replication fork is initiated and ensures the appropriate amount of enzyme under any growth condition.
In addition to its role in initiation of replication, DnaA also acts as a transcriptional regulator (Messer and Weigel, 1997). DnaA autoregulates its own transcription and controls either positively or negatively the expression of several other genes (Messer and Weigel, 1997). The nucleotide-bound state of DnaA is important for its activity as a transcriptional regulator. While ATP-DnaA and ADP-DnaA bind to 9-mer DnaA boxes with equal affinity, ATP-DnaA, but not ADP-DnaA, binds to a 6-mer ATP-DnaA boxes (Speck et al, 1999). Thus, ATP-DnaA represses dnaA transcription much more efficiently than ADP-DnaA (Speck et al, 1999). The nrd promoter region contains two 9-mer DnaA boxes as well as sequences conforming to ATP-DnaA boxes located immediately upstream of the nrdAB putative promoter (Tuggle and Fuchs, 1986; Speck et al, 1999). DnaA binds to these DnaA boxes and regulates transcription of the nrdAB operon (Augustin et al, 1994). Thus, DnaA would be a good candidate for coupling nrdAB expression in a cell cycle-dependent manner to DNA replication initiation.
In previous studies, we isolated suppressor mutations in the dnaAN locus that restored growth to a mutant depleted of the enzymes required for RNR reduction (Ortenberg et al, 2004). Mutations dnaAA345S, dnaAT174P and dnaNG157C result in derepression of the operon (Ortenberg et al, 2004). This regulation appears to act at the transcription level as fusions of the lacZ gene to nrdAB show a similar derepression for β-galactosidase in these mutant backgrounds. In the present studies, we explore the molecular mechanism that links nrdAB expression and DNA replication. We investigate the in vitro and in vivo replication initiation behavior of DnaA proteins from both dnaA mutant strains. We show that the DnaAA345S and DnaAT174P mutant proteins are constitutively inactivated, exhibiting either high intrinsic ATPase activity (DnaAT174P) or a defect in ATP binding combined with higher intrinsic ATPase activity (DnaAA345S). Therefore, neither DnaA mutant protein requires Hda for its conversion to the inactive ADP form. We show that the switch of ATP-DnaA to ADP-DnaA is not only a negative feedback mechanism that prevents premature reinitiation from occurring, but is also the mechanism that allows the coupling of DNA replication initiation with synthesis of dNTPs required during the elongation step of DNA replication.
Results
Defect of ATP binding in DnaAA345S and high intrinsic ATPase activity in DnaAT174P mutant proteins
ATP binding. We speculated that the increase in RNR seen in the dnaA and dnaN mutant strains previously isolated by us (see Introduction) might be due to alterations in the switch between ATP-DnaA and ADP-DnaA, since changes in either DnaA or β-subunit might be expected to influence this process. The mutations might affect ATP binding to DnaA or hydrolysis of DnaA-bound ATP. We purified DnaAA345S and DnaAT174P mutant proteins and wild-type DnaA protein and examined them for ATP binding using a filter retention assay (Sekimizu et al, 1987). This assay revealed that DnaAA345S but not DnaAT174P is defective in ATP binding (Figure 1A).
Figure 1.
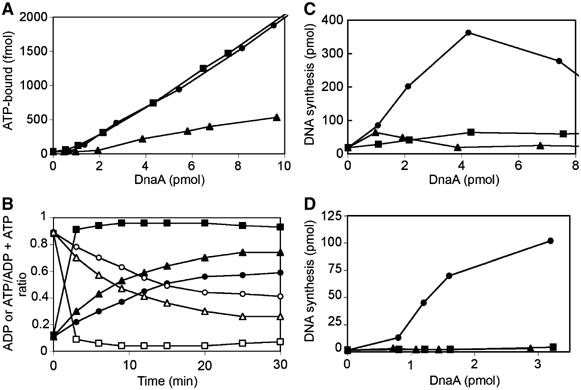
DnaAT174P and DnaAA345S mutant proteins with increased intrinsic ATPase activity and/or defect in ATP binding are unable to initiate replication in vitro. (A) ATP binding. DnaAA345S mutant is defective in its ability to bind ATP. Purified wild-type DnaA (•), mutant DnaA proteins DnaAA345S (▴) and DnaAT174P (▪) were incubated with [α-32P]ATP for 15 min on ice. Samples were filtered through nitrocellulose filters and the remaining protein-bound ATP was measured by liquid scintillation counting. (B) Intrinsic ATPase activity. Purified wild-type DnaA (•), mutant DnaA proteins DnaAA345S (▴) and DnaAT174P (▪) were incubated with [α-32P]ATP for 15 min on ice. The resulting ATP-bound proteins were incubated at 38°C with pBSoriC. Protein-bound ATP (open symbols) and ADP (closed symbols) were captured by filter retention, extracted from the filters, and separated by thin-layer chromatography. The ratio of extracted total bound nucleotide (ATP+ADP) for the last two time points (25 and 30 min) relative to the first two time points (0 and 3 min) for DnaA, DnaAT174P, and DnaAA435S were 0.77, 0.61, and 0.53. (C, D) In vitro replication. DnaAA345S and DnaAT174P are inactive in oriC plasmid replication. Purified wild-type DnaA (•) and mutant DnaA proteins DnaAA345S (▴) and DnaAT174P (▪) were examined for their abilities to support in vitro replication of an oriC plasmid in a system reconstituted with purified protein components (C) and in a crude extract lacking DnaA activity (D).
ATPase activity. DnaAT174P, which binds ATP similarly to wild-type DnaA, has a dramatically increased intrinsic ATPase activity. Bound ATP is totally hydrolyzed in less than 3 min, while, for wild-type DnaA, only half of the bound ATP is hydrolyzed in 15 min (Figure 1B). Detectable increases in the extent and the rate of the low amount of bound ATP hydrolysis are also seen with the DnaAA345S protein (Figure 1B).
Mutations in dnaA and dnaN reduce initiation efficiency
In vitro oriC replication. Since the ATP-bound form of DnaA is thought to initiate chromosomal replication, the two mutant DnaA proteins should be defective for replication initiation, as they exhibit either reduced ATP binding or high intrinsic ATPase activity. In agreement with this presumption, both DnaAA345S and DnaAT174P, in contrast to wild-type DnaA, are inert for in vitro DNA replication from oriC in a system reconstituted with purified proteins (Figure 1C) or when added to crude cell extracts lacking DnaA activity (Figure 1D), behaving similarly to ADP-bound wild-type DnaA protein (data not shown).
Flow cytometry analysis. Despite the in vitro replication initiation deficiency of the DnaAA345S and DnaAT174P proteins, the cells carrying either of the dnaA mutations present only a slight reduction in growth rate (Figure 2A) and form normal-sized colonies on solid media (data not shown). This finding suggests that the mutant DnaA protein can support the required functions of DnaA in vivo. Thus, the lack of initiation activity in vitro is likely not due to the mutations adversely affecting other essential DnaA functions. In order to further assess the in vivo effects of dnaAA345S, dnaAT174P and dnaNG157C mutations on replication initiation, we performed flow cytometry on these strains to determine whether the mutants ‘under-replicate', that is, replicate fewer chromosomes per mass.
Figure 2.
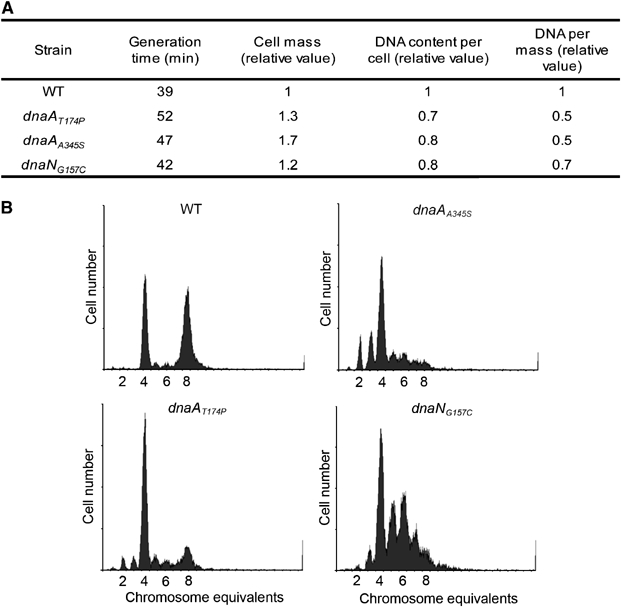
Under-replication in dnaAT174P, dnaAA345S and dnaNG157C mutants. (A) Mutations dnaAT174P, dnaAA345S and dnaNG157C lead to reduced chromosome content. Cells (DHB4, SMG275, SMG217 and SMG219) grown exponentially were stained and analyzed by flow cytometry. Average cell mass and DNA contents were derived from the flow cytometry histograms (see Materials and methods). The average numbers from three experiments are given. (B) Cells growing exponentially in AB glucose-CAA medium with uridine (100 μg/ml) were treated with rifampin and cephalexin to block initiation but allow elongation. Distinct peaks represent the accumulation of cells with integral numbers of chromosomes that reflect the numbers of origins at the time of drug action. Top left, wild-type (MG1655); top right, dnaAA345S (SMG379); bottom left, dnaAT174P (SMG378); bottom right, dnaNG157C (SMG380).
These measurements show a decrease in DNA content per cell and also per mass in the dnaAA345S, dnaAT174P and dnaNG157C mutant cells compared to wild-type cells (Figure 2A). The increased mass/DNA ratio suggests under-replication in the dnaAA345S, dnaAT174P and dnaNG157C mutants, and indicates that these cells have problems initiating replication. To further investigate the replication pattern, exponentially growing cells were treated with rifampicin and cephalexin to inhibit initiation of replication and cell division. Growth was continued for four to five generations to allow ongoing rounds of replication to finish. The number of completed chromosomes after rifampicin–cephalexin treatment reflects the number of origins present at the time the inhibitors were added. In a culture of cells with synchronous initiation, the number of chromosomes is 2, 4, 8, etc. depending on the growth rate, whereas in a culture with asynchronous initiation some of the cells also contain ‘irregular' numbers of origins (Skarstad et al, 1986). Wild-type cells were found to initiate synchronously and at four origins early in the cell cycle such that the older part of the population contained eight origins (Figure 2B). The dnaAA345S, dnaAT174P mutant cells mostly contained four origins (Figure 2B), indicating that initiation of replication still occurs fairly synchronously in these cells, but later in the cell cycle so that few cells contain eight origins. The average number of origins per cell was about six in the wild-type, compared to about four in the dnaAA345S and dnaAT174P mutant strains and about five in the dnaNG157C mutant, indicating that all three mutants initiate less efficiently. The dnaNG157C culture contained a substantial number of five or six chromosome cells in addition to four chromosome cells, indicating that initiation was asynchronous. The reason for an increased asynchrony phenotype in the dnaNG157C cells is not clear.
For all three strains, the decrease, compared to wild type, in both average number of origins per cell and in DNA concentration (Figure 2A), indicates that initiation occurred less efficiently. The result suggests that mutant cells tend to underinitiate, consistent with the results for in vitro replication.
Deletion of hda leads to a strong growth defect in a wild-type strain
Stimulated hydrolysis of DnaA-bound ATP by Hda requires DNA-bound β-subunit, which serves as a matchmaker between DnaA and Hda. The DnaAA345S and DnaAT174P mutant proteins, being constitutively inactivated, are already mainly either in the ADP-bound or nucleotide-free form. Therefore, we predicted that the dnaAA345S and dnaAT174P mutant strains should bypass the requirement for Hda to regulate initiation. In contrast, the wild-type strain should still be affected by a loss of hda.
To test this hypothesis, we transduced the Δhda∷TetR allele into the wild-type strain, DHB4. Only very small transductants appear on rich media plates and only after 2 days of incubation at 37°C, although the frequency with which these colonies appear is normal (data not shown and Camara et al, 2003). Importantly, transductants picked from these plates do not form isolated colonies upon restreaking on fresh medium (Figure 3A). Transductants of the hda mutation into a wild-type DHB4 strain carrying a plasmid with a complementing copy of hda (pSMG19) grew normally in contrast to the strain with an empty vector (data not shown). Thus, in the wild-type strain, the hda deletion confers a severe growth defect.
Figure 3.
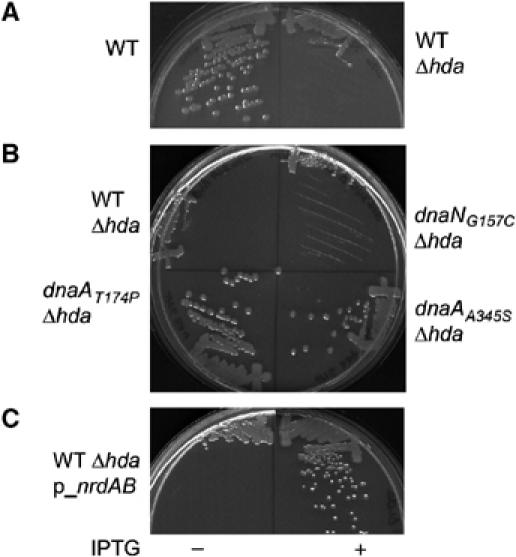
Growth defect due to hda deletion in a wild-type DHB4 strain is suppressed by increased amounts of dNTP. (A) Deletion of hda leads to growth defect in wild-type DHB4. The hda∷TetR allele was transduced into the wild-type DHB4 strain and transductants were selected at 37°C on NZ plates containing tetracycline. Small transductants that appeared after 2 days were restreaked on new NZ plates containing tetracycline (right). (B) Deletion of hda did not lead to growth defect in dnaAT174P and dnaAA345S mutant strains. The hda∷TetR allele was transduced into the wild-type DHB4, dnaAT174P, dnaAA345S and dnaNG157C strains. Transductants were selected at 37°C on NZ plates containing tetracycline and restreaked on identical fresh plates. (C) Overexpression of RNR suppresses the growth defect in the wild-type DHB4. A wild-type DHB4 strain containing a plasmid carrying the nrdAB operon under IPTG control (pSMG7) was transduced with the hda∷TetR allele in presence of IPTG. Transductants were restreaked on NZ plates containing (right) or not (left) 100 μM IPTG.
However, when we transduced the hda deletion into the dnaAA345S or dnaAT174P mutants (in a DHB4 background), the transductants grew to normal colony size after 1 day (Figure 3B); both dnaA mutants eliminate the requirement for Hda. It appears that, since these dnaA mutations result in a shift in DnaA from the ATP-bound to the ADP-bound state, Hda is no longer required for the switch to occur. In contrast, transduction of the hda deletion into the dnaNG157C mutant strain led to the same growth defect as observed with the wild-type strain (Figure 3B), indicating that the dnaNG157C mutant strain is still dependent on Hda. In contrast to the dnaA mutations, the dnaNG157C mutation presumably affects DnaA indirectly by altering the Hda–DnaA interaction, so that Hda is still required for the switch to occur.
Overexpression of ribonucleotide reductase (nrdAB) suppresses the growth defect in the hda deletion strain
Two quite different properties shared by the dnaAA345S and dnaAT174P mutants—elimination of the Hda requirement and the high levels of RNR (Ortenberg et al, 2004)—may provide an explanation for the growth defect of the hda mutant. Deletion of hda, which prevents the conversion of ATP-DnaA to ADP-DnaA, may interfere with the normal necessary derepression of RNR. A lack of dNTP's to complete a round of DNA replication could lead to DNA fork stalling during the elongation step of chromosomal replication and failure to grow. Overexpression of RNR in such a strain would then be expected to restore growth. To test this hypothesis, plasmid pSMG7, which contains the nrdAB operon under IPTG control (Ortenberg et al, 2004), was introduced into the wild-type DHB4 strain. The hda∷TetR allele was then transduced into this strain in the presence of IPTG. Transductants were restreaked on media with or without IPTG. As shown in Figure 3C, transductants overexpressing RNR grow normally, in contrast to transductants that do not overexpress RNR (no IPTG). These results show that high levels of RNR suppress the growth defect in the wild-type strain whose hda has been deleted, supporting the proposal that the growth defect of hda-deleted cells is due to a lack of dNTPs to complete the elongation step of DNA replication.
The correlation between RNR levels and hda was further analyzed by determining the RNR levels in wild-type, dnaAA345S, dnaAT174P and dnaNG157C strains deleted for hda. As deletion of hda leads to a strong growth defect in wild-type and dnaNG157C strains (Figure 3B), we performed the Western blot analysis on cells from the beginning of the streak (cells do not form isolated colonies but PCR analysis of the bacteria from the top of the streak confirm the presence of the hda∷TetR allele). The levels of RNR are high in dnaAA345S, dnaAT174P and dnaNG157C strains carrying wild-type hda in comparison to the wild-type DHB4 strain (Figure 4A), with the cytoplasmic protein thioredoxin serving as a loading control. In the absence of hda, RNR levels are still high in both dnaA mutant strains and no growth defect is observed. In contrast, deletion of hda in the dnaNG157C strain leads to low levels of RNR and a strong growth defect. These results are consistent with our proposal that the growth defect in the hda null mutant is due to a lack of RNR.
Figure 4.
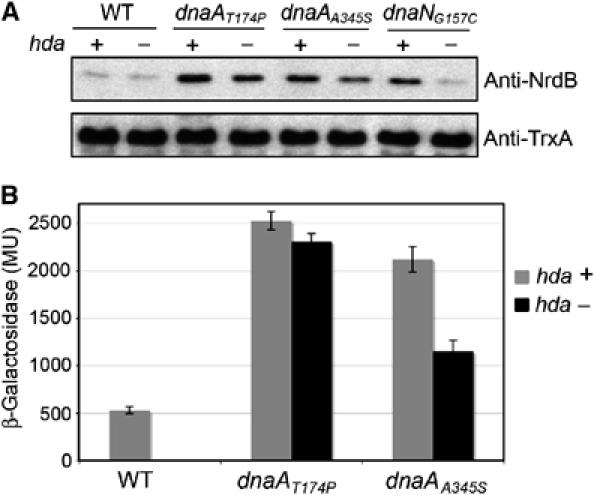
Deletion of hda leads to decreased RNR levels in dnaNG157C but not in dnaAA345S and dnaAT174P mutant strains. (A) Equal amounts of total cellular protein from wild-type DHB4, dnaAA345S, dnaAT174P and dnaNG157C mutant strains harboring (+) or not (−) the hda gene were separated by SDS–15% PAGE. Subunit R2 (nrdB) of the RNR was detected by Western blotting. As loading control, cytoplasmic thioredoxin was also detected by Western blotting. (B) β-Galactosidase activity of the nrdA'–‘lacZ fusion in wild-type DHB4, dnaAA345S and dnaAT174P strains harboring (gray) or not (black) the hda gene.
One attractive explanation for our findings is that Hda not only has a role in preventing the premature reinitiation of DNA replication but has also a role in coordinating the expression of RNR with replication initiation. Interestingly, in the dnaAT174P mutant strain the derepression of nrdAB expression is totally independent of hda, whereas derepression in the dnaAA345S mutant is still partially dependent on hda, as shown by a decrease of RNR levels in the hda deletion strain (Figure 4A). This result is confirmed by assays of β-galactosidase activity of an nrdA'–‘lacZ fusion in dnaAA345S and dnaAT174P strains (Figure 4B). One main difference between the two DnaA mutants is that DnaAT174P has a very high intrinsic ATPase activity which leads to almost instant hydrolysis of ATP, whereas DnaAA345S presents an intermediate phenotype, since it exhibits a diminished ATP binding combined with a slight but significant increase of its ATPase activity. Thus, the DnaAT174P mutant protein is likely to be mainly in the ADP-bound form, whereas in the DnaAA345S mutant the ratio of [ATP−DnaA]/[DnaA total] is lower than in a wild-type strain. The in vivo stability of DnaAA345S and DnaAT174P are comparable to that of wild-type DnaA (data not shown), ruling out the possibility that ndrAB derepression is caused by rapid degradation of DnaAA345S or DnaAT174P. These observations strengthen the notion that the nucleotide-bound state of DnaA plays a central role in the regulation of RNR, with DnaA serving as a transcriptional regulator. However, these results do not allow us to determine whether ATP-DnaA acts as a repressor or if ADP-DnaA acts as an activator.
ATP-DnaA may act as a repressor of nrdAB transcription
If ATP-DnaA is a repressor, overexpression of the wild-type DnaA in a dnaAT174P mutant strain (where levels of RNR are abnormally high) might be expected to lead to decreased amounts of RNR, while there may be no effect in a wild-type strain. In contrast, if ADP-DnaA is an activator, overexpression of the DnaAT174P mutant protein (mainly ADP-DnaA) may increase RNR expression in a wild-type strain. An empty plasmid or a plasmid expressing either wild-type DnaA, DnaAA345S or DnaAT174P were introduced into a wild-type and a dnaAT174P mutant strain. Equivalent amounts of cell extracts from these strains were analyzed by SDS–polyacrylamide gel electrophoresis (PAGE) and Western blot analysis was performed using antibodies raised against NrdB (cytoplasmic protein thioredoxin serving as a loading control). As shown in Figure 5A, expression of DnaAT174P in a wild-type strain does not affect the RNR levels when compared to a strain carrying the empty vector. However, overexpression of the wild-type DnaA in the dnaAT174P mutant strain leads to lower amounts of RNR, while the empty vector or the vector overexpressing DnaAT174P has no effect (Figure 5A). Overexpression of DnaAA345S in the dnaAT174P mutant strain leads to partial reduction of RNR levels. With this mutant protein, which exhibits an ATP binding defect, overexpression leads to accumulation of some ATP-bound form and can explain the partial decrease of RNR levels. These results are consistent with the proposal that the ATP-bound form of DnaA acts as a repressor, which regulates RNR expression.
Figure 5.
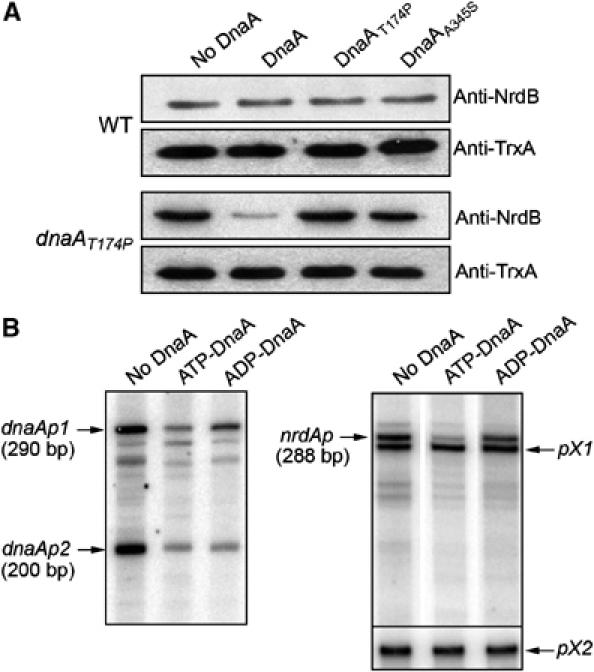
ATP-DnaA represses the nrdAB promoter more effectively than ADP-DnaA. (A) Western blot analysis. Wild-type DHB4 strain (top) or dnaAT174P mutant strain (bottom) containing an empty vector or a plasmid carrying either dnaA, dnaAA345S or dnaAT174P gene under IPTG control were grown in the presence of 100 μM of IPTG to mid exponential growth. Equal amounts of total cellular proteins were separated by SDS–15% PAGE. Subunit R2 (nrdB) of the RNR was detected by Western blotting. As loading control, the cytoplasmic thioredoxin was also detected by Western blotting. (B) In vitro transcription. Run-off in vitro transcripts from a 381 bp FokI–ClaI restriction fragment that contains promoters dnaAp1 and dnaAp2, and from a 608 bp PCR fragment that contains the nrdAB promoter and two additional uncharacterized promoters pX1 and pX2. Purified ATP- or ADP-DnaA wild-type protein (200 nM) was preincubated with DNA template for 5 min, then the RNA synthesis was started by addition of RNA polymerase (66 nM) and NTPs. Samples were separated by 6%-urea polyacrylamide gel electrophoresis. The transcripts dnaAp1, dnaAp2 and nrdAp (290, 200 and 288 bp, respectively) are indicated. Transcription from pX1 and pX2 are used as internal control for the experiments.
In order to obtain further evidence for the role of ATP-DnaA as a repressor of RNR levels, we attempted to measure the effects of ATP-DnaA and ADP-DnaA protein on nrdAB operon expression in an in vitro transcription assay. We used a 608 bp DNA fragment containing the nrdA promoter and we also used, as a positive control, a 381 bp DNA fragment containing both dnaA promoters. The regulation of both dnaA promoters is known to be under repression control of the ATP-bound form of DnaA (Speck et al, 1999). In vitro run-off transcription by RNA polymerase from the fragment containing both dnaA promoters generates two main fragments for the dnaA promoter: two transcripts 290 and 200 bp in length transcribed from dnaAp1 and dnaAp2 promoters, respectively (Figure 5B). The ATP-DnaA protein represses transcription from both dnaA promoters, dnaAp1 and dnaAp2, very efficiently. In contrast, ADP-DnaA protein represses both promoters rather weakly, dnaAp2 better than dnaAp1 (Figure 5B). These results are similar to those previously published by Speck et al (1999) and serve as a positive control to confirm that our preparation of purified DnaA protein behaves as expected. In parallel, in vitro run-off transcription by RNA polymerase from the fragment containing the nrdAB promoter generates an expected transcript of 288 bp in length (Figure 5B). Two additional transcripts of shorter length are also detected on the gel. The promoter of the nrdAB operon has been well characterized by primer extension analysis but no additional transcript has been identified (Tuggle and Fuchs, 1986). However, it is not surprising to find an additional site of transcription initiation in vitro. We used these two additional promoters, named pX1 and pX2 as internal controls for the experiment. As shown on Figure 5B, the transcription from these two promoters are not modified by the addition of the DnaA protein. In comparison, ATP-DnaA represses transcription from the 288 pb fragment of nrdAB promoter, whereas the ADP-DnaA represses the nrdAB promoter rather weakly (Figure 5B), suggesting that the effect of the ATP-DnaA on the nrdAB promoter is specific. Taken together, the in vivo studies, indicating the dominant effects of the wild-type DnaA over the two mutant alleles, and the in vitro transcription results are consistent with DnaA acting as a repressor of NrdAB expression. We did not attempt to assess the activity of the DnaA mutant proteins in the in vitro studies, since the ATP binding defects and/or the increased hydrolysis of ATP of these proteins make it difficult to conceive of how to obtain preparations of the protein that would reflect their in vivo state.
We also have attempted to obtain further in vitro evidence for our proposal that DnaA-ATP acts as a repressor of the nrdAB operon. However, footprinting experiments did not allow us to define a DnaA-binding site. These results may indicate that other protein factors are involved in the transcription regulation of nrdAB.
High levels of RNR lead to increased mutation rate
In E. coli, the major need for deoxyribonucleotide triphosphates is to allow DNA replication. Thus, the required amount of RNR, the sole enzyme that is capable of generating deoxyribonucleotides in E. coli grown aerobically, is directly proportional to the rate of DNA synthesis, which is determined by the rate of DNA initiation. If a strain overexpresses RNR, it may accumulate high levels of dNTPs. Indeed, in yeast and mammalian cells, high levels of dNTP pools are responsible for misincorporation of dNTPs which results in DNA mutagenesis. No studies examining this phenomenon have been carried out in E. coli. To address this issue, we measured the spontaneous mutagenesis frequency rate using the frequency of rifampicin resistance in a wild-type DHB4 strain overexpressing or not RNR (SMG300 and SMG301) under IPTG control. The frequency of rifampicin-resistant cells is increased by 10-fold in the strain overexpressing RNR, in comparison to the strain carrying the empty vector (data not shown). The frequency of rifampicin-resistant cells is also increased by three-fold over the wild type in the dnaAT174P and dnaAA345S mutant strains that exhibit constitutively high RNR levels (data not shown). These results show that high levels of dNTP pools increase the mutagenesis rate in E. coli, suggesting that dNTP pools may have to be regulated to avoid deleterious mutagenic effects on the cells.
Expression of RNR is negatively regulated by the dNTP pools
We asked whether there might be other modes of regulation of RNR. To determine whether the RNR NrdAB (or its products) might regulate its own synthesis, we introduced into the wild-type DHB4 strain carrying the nrdA'–‘lacZ fusion, the plasmid expressing nrdAB under the control of an IPTG-inducible promoter (pSMG12) or an empty plasmid (pSMG11). We then examined the effect of overproduction of RNR on its own promoter by following the expression of the nrdA'–‘lacZ fusion. Overexpression of nrdAB led to a five-fold decrease of nrdA'–‘lacZ fusion expression when compared to the uninduced strain (Figure 6A).
Figure 6.
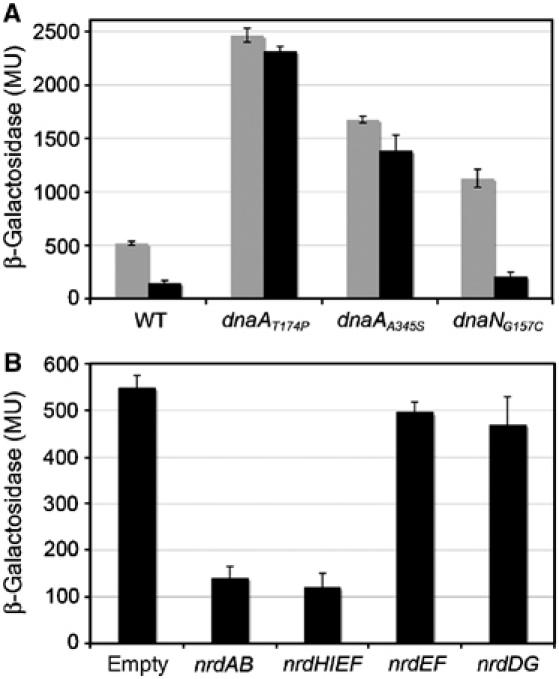
Increased dNTP levels lead to repression of the nrdAB promoter in the wild-type DHB4 and dnaNG157C strains but not in the dnaAA345S and dnaAT174P mutant strains. (A) β-Galactosidase activity, expressed in Miller units, of the nrdA'–‘lacZ fusion of a wild-type strain (DHB4), dnaAA345S, dnaAT174P and dnaNG157C strains containing an empty vector (gray) or a plasmid carrying the nrdAB operon under IPTG control, grown in presence of 100 μM IPTG to mid exponential growth. (B) β-Galactosidase activity, expressed in Miller units, of the nrdA'–‘lacZ fusion of a wild-type strain (DHB4) containing an empty vector (Ctrl) or a plasmid carrying either nrdAB, nrdHIEF, nrdEF or nrdDG under IPTG control grown in the presence of 100 μM IPTG to mid exponential growth.
To determine whether the feedback regulation of RNR on its own synthesis might be operating via the DnaA regulation of RNR, we examined the effects of overproduction of RNR in dnaAA345S and dnaAT174P cells. We found that the dnaA mutations eliminate the feedback regulation by overexpressed RNR, whereas the dnaNG157C mutation does not (Figure 6A). These results are consistent with a model in which negative regulation of nrdAB triggered by overproduction of RNR operates through DnaA. The finding that the feedback mechanism is not eliminated in dnaNG157C cells could be due to the fact that this strain is expressing wild-type DnaA, in contrast to the two dnaA mutant strains. In the dnaA mutant strains, newly formed DnaA rapidly adopts the inactive ADP-bound form or remains nucleotide-free. In contrast, in dnaNG157C cells, newly synthesized DnaA is likely in its active ATP-DnaA form, the conformation of DnaA that is responsible for nrdAB repression.
Overexpression of RNR leads to higher amounts of RNR as well as higher dNTP pools. Therefore, either the dNTPs or the RNR enzyme itself could be responsible for the negative feedback mechanism. To address this question, we artificially overexpressed two other E. coli RNRs, NrdDG, which is active in anaerobiosis, and NrdEF, whose structure is similar to NrdAB, but whose expression is cryptic. In the case of NrdEF, we can distinguish between the regulatory effects of the synthesis of dNTPs and of the enzyme itself, since NrdEF only synthesizes dNTPs if NrdH, a specific reductant of NrdEF is expressed at the same time. Overexpression of NrdEF allows synthesis of the enzyme whereas overexpression of the entire operon nrdHIEF leads to dNTP synthesis as well (unpublished data). Plasmids expressing nrdEF (pSMG13), nrdHIEF (pSMG14) or nrdDG (pSMG15) under the control of an IPTG inducible promoter were introduced into the wild-type strain carrying an nrdA'–‘lacZ fusion. Upon overexpression of nrdHIEF, the activity of the nrdA'–‘lacZ fusion decreased five-fold (Figure 6B). However, expression of nrdEF alone or nrdDG did not lead to repression of the nrdA'–‘lacZ fusion. These results strongly suggest that the level of dNTPs and not the RNR itself is responsible for the negative regulation of RNR expression.
Discussion
A dual function in the cell cycle for the ATP-DnaA to ADP-DnaA switch
The ability to set into motion a program of gene expression in response to major cellular events is critical for cell function (Volkert and Landini, 2001; Zheng et al, 2001; Patten et al, 2004). However, the transcriptional response throughout the cell cycle, notably in response to initiation of DNA replication, has been poorly studied in E. coli in contrast to eukaryotic systems or to the prokaryote Caulobacter crescentus (Lania et al, 1999; Martin et al, 2004). Here we describe a molecular mechanism which would allow coordination of the synthesis of RNR with the onset of DNA replication. Prior work has shown that the inactivation of DnaA via conversion of the ATP-bound to the ADP-bound form is critical for the prevention of premature reinitiation of replication, thus allowing only a single round of replication per cell cycle (Kato and Katayama, 2001; Camara et al, 2003). In the present study, we provide support for a model in which the ATP-DnaA to ADP-DnaA switch occurring with initiation is also required to provide dNTP synthesis for the elongation phase of DNA replication. We suggest that this latter role of DnaA is the more critical of the two for growth. This is indicated by the finding that RNR overexpression suppresses the growth defect observed in an hda deletion strain where the molecular switch is defective; the growth defect is largely due to a lack of dNTP synthesis (Figure 3).
We propose a model to explain how loading of the DNA replicase affects the expression of the genes encoding RNR (nrdAB) (Figure 7). Prior to initiation, DnaA is largely in its ATP-bound form and represses the nrdAB operon, thereby downregulating the pool of dNTPs. However, as a consequence of the initiation of DNA replication, β-subunit is loaded on the replication fork, facilitating Hda-induced hydrolysis of ATP-DnaA to ADP-DnaA. This relieves repression of nrdAB, giving the high levels of RNR required for the burst of DNA synthesis. As ATP-DnaA also represses its own promoter, the repression of dnaA expression is relieved (although somewhat delayed due to sequestration). The levels of ATP in the cells being much higher than ADP levels, the newly synthesized DnaA will be mainly in its ATP-bound form. ATP-DnaA progressively accumulates in the cell prior to the next round of replication (Kurokawa et al, 1999) leading to progressive repression of nrdAB transcription.
Figure 7.
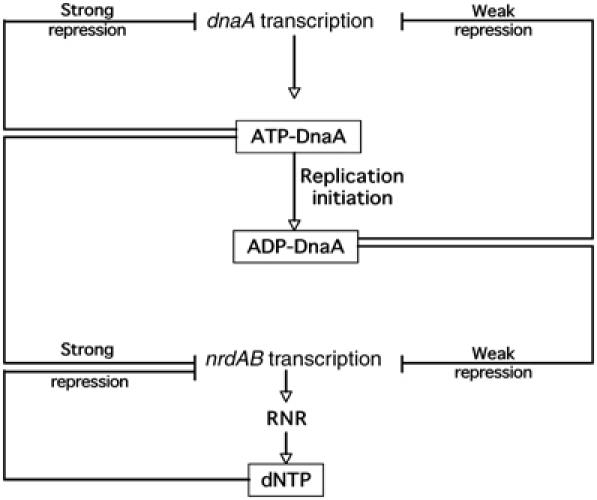
Model for coupling of deoxyribonucleotides (dNTPs) synthesis and DNA replication by the initiation factor DnaA. Newly synthesized DnaA protein is in the ATP-bound form, which is active for DNA replication initiation. ATP-DnaA is also a transcriptional repressor of dnaA and nrdAB genes. At the end of initiation, ATP-DnaA is hydrolyzed to ADP-DnaA in a β-subunit-dependent manner. The fraction of DnaA protein in the cell in the ADP-bound form increases. ADP-DnaA causes only a weak repression of nrdAB operon transcription, which leads to an increase of dNTP synthesis, allowing elongation to occur. ADP-DnaA also causes only weak repression of dnaA leading to newly synthesized DnaA. If DNA replication is slow or does not occur, cells experience a time lag between successive replication cycles. During this lag period, ATP-DnaA accumulates in the cell as RIDA would not be functioning because of the absence of DNA-loaded sliding clamps. Binding of ATP-DnaA to the DnaA boxes of the nrdAB promoter leads to a strong repression of nrdAB transcription and to a cessation of RNR synthesis.
The following findings provide the basis for our model: (i) three mutations that shift the state of DnaA from mostly ATP-bound to ADP-bound form cause derepression of RNR. Two dnaA mutations affect DnaA directly, while the dnaN mutation affects DnaA indirectly presumably through its interaction with Hda, (ii) ATP-bound DnaA represses nrdAB transcription more effectively than ADP-bound DnaA, and the dnaA mutants are recessive to wild-type dnaA with respect to RNR synthesis, (iii) overexpression of nrdAB suppresses the growth defect of an hda deletion strain in which the switch from ATP- to ADP-DnaA cannot occur, (iv) overexpression of nrdAB leads to high mutation rates, (v) high levels of dNTPs strongly repress nrdAB transcription in a DnaA-dependent manner.
Excess dNTP's trigger repression of nrdAB transcription by a newly identified regulatory mechanism
We also uncovered an additional regulatory mechanism for RNR synthesis in which overproduction of RNR leads to repression of nrdAB transcription (Figure 6). Why does E. coli exhibit such complicated regulation of dNTP synthesis and not simply maintain excess dNTPs throughout the cell cycle. It could be simply that overproduction of dNTP is wasteful. However, in Saccharomyces cerevisiae, an excess of dNTPs in cells results in an increased mutation rate (Kunz and Kohalmi, 1991; Chabes et al, 2003). Our results show that this is also the case in E. coli.
This regulation is likely mediated through the accumulation of dNTP's in the cell, and not by NrdAB itself, as indicated by the finding that when dNTP's are produced from a different RNR (NrdEF), repression still takes place (Figure 6A and B). The findings that this regulation is lost in the dnaAA345S and dnaAT174P mutants, but not in the dnaNG157C mutant, suggest that DnaA is the transcriptional factor involved in this regulation (Figure 6A). The effect on DnaA could be due to binding of a dNTP, consistent with previous findings that DnaA binds dATP (Sekimizu et al, 1987). Since overexpression of RNR leads to abnormally high dNTPs, one could imagine that direct binding of dATP to DnaA leads DnaA to adopt a conformation similar to ATP-bound DnaA. DnaA in this conformation could be responsible for nrdAB repression by binding the ATP-DnaA-binding sites located in the nrdAB promoter. It appears that the utilization of initiation factor DnaA to tightly coordinate production of dNTP and replication allows the cell to precisely regulate the amount of dNTP in both a positive and negative manner so that enough dNTPs are synthesized, but no more.
Coordination of RNR synthesis and DNA replication in other organisms
Notably, regulation of DNA replication initiation is mechanistically similar in all organisms, using a nucleotide switch of the initiator protein. For example, in S. cerevisiae, considerable evidence supports a model in which ORC, an ATP-binding protein and the functional equivalent of DnaA, is inactivated for initiation of DNA replication by ATP hydrolysis soon after initiation (Klemm et al, 1997; Bell and Dutta, 2002). Comparison of the ATP regulation of these factors suggests that ATP binding and hydrolysis act as a molecular switch that couples key events during initiation of replication in prokaryotic and eukaryotic cells.
While it is currently not known, it is tempting to speculate that transcriptional activation of RNR genes in eukaryotic cells during the cell cycle (Engstrom et al, 1985) is also dependent on the nucleotide-state of ORC.
The power of genetics for discovery of novel regulatory networks
The findings presented here were made possible by the characterization of dnaA and dnaN mutants detected as suppressor mutations that restored sufficient RNR activity to a strain missing the glutaredoxin and thioredoxin reductants of the enzyme. The present work reveals additional and unexpected complexity in the cell cycle regulatory mechanisms leading to DNA replication. Our results show how analysis of suppressors of mutants affecting central physiological pathways can lead to the revelation of intricately evolved networks for such pathways even in such a well-characterized organism as E. coli. In addition, the dnaA mutations can be quite useful in further studies as they represent the first viable mutations in dnaA that cause rapid accumulation of ADP-DnaA and confer an DnaA ATP-binding defect and/or constitutive ATPase activity.
Materials and methods
Bacterial strains and growth conditions
The bacterial strains and plasmids used in this study are listed in Table I. Strains were grown routinely in NZ medium (Guzman et al, 1992) or AB glucose-CAA medium (Bach and Skarstad, 2004) at 37°C. For growth of MG1655 strains, 100 μg/ml uridine was added. Antibiotic selection was maintained at the following concentrations: ampicillin, 200 μg/ml (plasmid) or 25 μg/ml (chromosome); chloramphenicol, 10 μg/ml; kanamycin, 40 μg/ml.
Table 1.
Strains and plasmids used in this work
| Strains/plasmids | Relevant genotype | Reference |
|---|---|---|
| Plasmids | ||
| pDSW204 | ori pBR, ampR, IPTG inducible | Weiss et al (1999) |
| pDSW206 | ori pBR, ampR, IPTG inducible | Weiss et al (1999) |
| pBAD18kan | ori pBR, kanR, arabinose inducible | Guzman et al (1995) |
| pBAD43 | ori pSC101, spcR, arabinose inducible | Guzman et al (1995) |
| pSMG7 | pDSW204-nrdAB | Ortenberg et al (2004) |
| pSMG8 | pDSW204-nrdEF | This work |
| pSMG9 | pDSW204-nrdHIEF | This work |
| pSMG10 | pDSW204-nrdDG | This work |
| pSMG11 | pDSW204kan | This work |
| pSMG12 | pDSW204kan-nrdAB | This work |
| pSMG13 | pDSW204kan-nrdEF | This work |
| pSMG14 | pDSW204kan-nrdHIEF | This work |
| pSMG15 | pDSW204kan-nrdDG | This work |
| pSMG16 | pDSW206-dnaA | This work |
| pSMG17 | pDSW206-dnaAA345S | This work |
| pSMG18 | pDSW206-dnaAT174P | This work |
| pSMG19 | pBAD43-hda | This work |
| pZL411 | pET19b-His-dnaA | Li and Crooke (1999) |
| pHisDnaAA345S | pET19b-His-dnaAA345S | This work |
| pHisDnaAT174P | pET19b-His-dnaAT174P | This work |
| pTB101 | pBSoriC | Baker and Kornberg (1988) |
| M13mpRE85 | Fuller et al (1981) | |
| Strains | ||
| DHB4a | Δ(ara–leu)7697 araD139 ΔlacX74 galE galK rpsL phoR Δ(phoA)PvuII ΔmalF3 thi contains a mutation in dnaA leading to a P to S change in DnaA amino acid 18 relative to other wild-type strains (dnaAP18S) | Boyd et al (1987) |
| SMG218 | DHB4 dnaA…MiniTn10CmR | This work |
| SMG217 | DHB4 dnaAA345S…MiniTn10CmR | This work |
| SMG219 | DHB4 dnaNG157C…MiniTn10CmR | This work |
| SMG275 | DHB4 dnaAT174P…MiniTn10CmR | This work |
| MG1655 | Wild type | Jensen (1993) |
| SMG378 | MG1655 dnaAT174P… MiniTn10CmR | This work |
| SMG379 | MG1655 dnaAA345S…MiniTn10CmR | This work |
| SMG380 | MG1655 dnaNG157C…MiniTn10CmR | This work |
| SMG329 | DHB4 Δ(λattL-lom)∷bla nrdA'–'lacZ | This work |
| SMG334 | SMG329 dnaAA345S…MiniTn10CmR | This work |
| SMG337 | SMG329 dnaAT174P…MiniTn10CmR | This work |
| SMG340 | SMG329 dnaNG157C…MiniTn10CmR | This work |
| SMG300 | DHB4 pDSW204 | This work |
| SMG301 | DHB4 pDSW204-nrdAB | This work |
| SMG318 | SMG301 Δhda∷tetR | This work |
| BLR(DE3)pLysS | F− ompT hsdSB(rB−mB−) gal dcm Δ(srl-recA)306∷Tn10(TetR) λ(DE3) [pLysS(CamR)] | Novagen |
| aVariant of DHB4 that had been cured of the F′ plasmid was used throughout this study. | ||
Induction of genes under the control of the trc promoter was accomplished by addition of 0–100 μM isopropyl β-D-thiogalactopyranoside (IPTG).
Plasmid construction
The nrdEF, nrdHIEF and nrdDG coding sequences were amplified by PCR using primers that introduce an EcoRI and a HindIII restriction site. The product was digested and ligated into the same sites of the pDSW204 (Weiss et al, 1999) to create plasmid pSMG8, pSMG9 and pSMG10. The DNA fragment containing the kanamycin-resistance gene and its promoter region was extracted from the pBAD18kan (Guzman et al, 1995) by digestion with BsaI and AhdI. This product was ligated into the same sites of pDSW204, pSMG7, pSMG8, pSMG9 and pSMG10 to create plasmids pSMG11, pSMG12, pSMG13, pSMG14 and pSMG15.
The dnaA-coding sequence was amplified by PCR using DHB4, SMG275 and SMG217 chromosomal DNA as template and primers that introduce an NcoI and a HindIII restriction site. The products were digested and ligated into the same sites of pDSW206 plasmid (Weiss et al, 1999). The resulting plasmids, pSMG16, pSMG17 and pSMG18, contain, respectively, dnaA, dnaAT174P and dnaAA345S gene with their own Shine Delgarno sequence under control of trc promoter.
The dnaA-coding sequence was amplified by PCR using SMG217 and SMG275 chromosomal DNA as template with primers that introduce an NdeI and a BamHI restriction site. The digested products were ligated into the same sites of pZL411 plasmid to replace the wild-type dnaA (Li and Crooke, 1999). The resulting plasmids were designated pHisDnaAA345S and pHisDnaAT174P, respectively.
Genetic and molecular biology procedures
Standard techniques were used for cloning and analysis of DNA, PCR, electroporation, transformation and P1 transduction (Sambrook et al, 1989; Miller, 1992).
Construction of strains containing a nrdA'–‘lacZ fusion
To obtain a translational fusion of the N-terminal part of ribonucleotide reductase α-subunit, R1 (encoded by nrdA gene), with β-galactosidase, a DNA fragment was amplified by PCR using primers that hybridize to a position 778 nucleotides upstream of the translation start site of the nrdA gene and contain a HindIII site and a primer that anneals within the nrdA-coding region and contains a BamHI site. The digested product was ligated into the same sites of pNG102 plasmid (Ritz et al, 2000) to give plasmid pSMG5. The construct expresses a hybrid protein with the first 13 amino acids of R1 subunit fused to β-galactosidase. The fusion was then integrated into the chromosome of DHB4 by using specialized transduction with lambda phage InCh (Boyd et al, 2000) to give SMG329. β-Galactosidase activity assays in liquid media were performed as described (Miller, 1992).
Analytical procedure
Proteins were resolved by SDS–PAGE and stained with Coomassie brilliant blue. For Western blotting, proteins were transferred to a nitrocellulose membrane after electrophoresis; the ECL-Western blotting system (Amersham Pharmacia) was used for detection.
Purification of His-tagged DnaA
Histidine-tagged wild-type and mutant DnaAA345S and DnaAT174P proteins were expressed, respectively, from pZL411, pHisDnaAA345S, and pHisDnaAT174P plasmids in strain BLR(DE3)pLysS and purification was performed as previously described (Li and Crooke, 1999).
ATP binding
ATP binding was assessed as previously described (Sekimizu et al, 1987). Briefly, histidine-tagged DnaA proteins were incubated with 1 μM [α-32P]ATP at 0°C for 15 min. Samples were filtered through nitrocellulose filters and retained nucleotides measured by liquid scintillation counting.
DnaA intrinsic ATPase activity
ATP hydrolysis was assessed as previously described (Li and Crooke, 1999). Briefly, histidine-tagged DnaA proteins were incubated with 1 μM [α-32P]ATP at 0°C for 15 min. The resulting ATP-DnaA was incubated at 38°C with pBSoriC (0.1 μg) for various times. Samples were filtered through nitrocellulose membranes. DnaA-bound ATP and ADP were extracted from the filters with 1 M formic acid and spotted onto thin layer PEI Cellulose plates. ADP and ATP were separated and radioactivity was measured using a Molecular Dynamics Storm 840 imaging system.
DNA replication in a crude cell-free extract
In vitro replication of oriC-plasmid M13mpRE85 in a crude enzyme fraction was performed as described (Fuller et al, 1981; Crooke, 1995). Histidine-tagged DnaA protein was incubated at 30°C for 20 min with a crude DnaA-free protein fraction prepared from WM433 (dnaA204(Ts)) strain and with a DNA synthesis reaction containing 0.1 mM each dATP, dGTP, dCTP and [α-32P]TTP and 200 ng M13mpRE85 replicative form I DNA. Nucleotides incorporated into acid-insoluble material were collected on GF/C filters and measured by liquid scintillation counting.
DNA replication with defined components
In vitro replication of pBSoriC, dependent on purified replication proteins, was performed as described (Crooke, 1995). Histidine-tagged DnaA protein was mixed at 0°C with 0.1 mM each of dATP, dGTP, dCTP and [α-32P]TTP and 200 ng pBSoriC; 450 ng SSB; 10 ng HU; 400 ng gyrase A subunit; 180 ng of gyrase B subunit; 150 ng DnaB/DnaC complex; 12 ng DnaG primase; 500 ng DNA polymerase III*; 25 ng of DNA polymerase III holoenzyme β-subunit and the indicated amounts of DnaA proteins. The mixtures were incubated at 30°C for 20 min. Nucleotides incorporated into acid-insoluble material were collected on GF/C filters and measured by liquid scintillation counting.
Flow cytometry analysis
Flow cytometry analysis was performed as described (Torheim et al, 2000). The average DNA content per cell was determined as average Hoechst fluorescence per cell. The average DNA per mass was found by dividing average DNA content by average cell mass.
In vitro transcription
The in vitro transcription assay was performed as described (Speck et al, 1999). A 608 bp fragment amplified by PCR using DHB4 chromosomal DNA as template containing the nrdA promoter as well as a 381 bp FokI–ClaI restriction fragment that contains both dnaA promoters, dnaAp1 and dnaAp2 (Speck et al, 1999), were used as a linear template for single-round run-off transcription assays. These were carried out at 37°C in transcription buffer pp60 (50 mM HEPES–KOH (pH 7.6 at 1 M), 2.5 mM magnesium acetate, 20% (v/v) glycerol, 0.007% (v/v) Triton X-100, 0.3 mM EDTA and 7 mM DTT) in a total volume of 20 μl. The DNA template (10 ng) was preincubated for 5 min with 200 ng of DnaA protein. Subsequently, RNA polymerase was added to a concentration of 66 nM, followed by another 5 min incubation. The transcription reactions were initiated by addition of [α-32P]UTP (7.5 μCi, 3000 Ci/mmol), unlabelled UTP (60 μM), ATP (200 μM), CTP and GTP (each at 400 μM), and heparin (200 μg/ml). Mixtures were incubated for 5 min and then the reaction was terminated by addition of stop solution (50 mM EDTA containing 300 μg/ml of yeast tRNA). After the reaction, RNA was precipitated with ethanol and resuspended in 6 μl of loading buffer (95% (v/v) formamide, 20 mM EDTA, 0.05% (w/v) bromophenol blue, 0.05% (w/v) xylene cyanol). After incubation at 96°C for 5 min, the samples were loaded and electrophoresed on a 6% (w/v) polyacrylamide sequencing gels. Gels were visualized by phosphorimager.
Acknowledgments
We thank Bryce Nickels at Harvard Medical School for helping with in vitro transcription assays; Mali Strand Ellefsen and Anne Wahl at the Institute for Cancer Research for the Flow Cytometry Core Facility and for excellent technical assistance, respectively. This work was supported by fellowships from Human Frontier Science Program (HFSP) (to SG), by National Institutes of Health Grants GM41883 and GM55090 (to JB) and GM49700 (to EC) and by the Norwegian Research Council functional genomics programme (FUGE) (to KS).
References
- Augustin LB, Jacobson BA, Fuchs JA (1994) Escherichia coli Fis and DnaA proteins bind specifically to the nrd promoter region and affect expression of an nrd-lac fusion. J Bacteriol 176: 378–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach T, Skarstad K (2004) Re-replication from non-sequesterable origins generates three-nucleoid cells which divide asymmetrically. Mol Microbiol 51: 1589–1600 [DOI] [PubMed] [Google Scholar]
- Baker TA, Bell SP (1998) Polymerases and the replisome: machines within machines. Cell 92: 295–305 [DOI] [PubMed] [Google Scholar]
- Baker TA, Kornberg A (1988) Transcriptional activation of initiation of replication from the E. coli chromosomal origin: an RNA–DNA hybrid near oriC. Cell 55: 113–123 [DOI] [PubMed] [Google Scholar]
- Bell SP, Dutta A (2002) DNA replication in eukaryotic cells. Annu Rev Biochem 71: 333–374 [DOI] [PubMed] [Google Scholar]
- Boyd D, Manoil C, Beckuith J (1987) Determinants of membrane protein topology. Proc Natl Acad Sci USA 84: 8525–8529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd D, Weiss DS, Chen JC, Beckwith J (2000) Towards single-copy gene expression systems making gene cloning physiologically relevant: lambda InCh, a simple Escherichia coli plasmid-chromosome shuttle system. J Bacteriol 182: 842–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camara JE, Skarstad K, Crooke E (2003) Controlled initiation of chromosomal replication in Escherichia coli requires functional Hda protein. J Bacteriol 185: 3244–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabes A, Georgieva B, Domkin V, Zhao X, Rothstein R, Thelander L (2003) Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell 112: 391–401 [DOI] [PubMed] [Google Scholar]
- Crooke E (1995) DNA synthesis initiated at oriC: in vitro replication reactions. Methods Enzymol 262: 500–506 [DOI] [PubMed] [Google Scholar]
- Eliasson R, Pontis E, Sun X, Reichard P (1994) Allosteric control of the substrate specificity of the anaerobic ribonucleotide reductase from Escherichia coli. J Biol Chem 269: 26052–26057 [PubMed] [Google Scholar]
- Engstrom Y, Eriksson S, Jildevik I, Skog S, Thelander L, Tribukait B (1985) Cell cycle-dependent expression of mammalian ribonucleotide reductase. Differential regulation of the two subunits. J Biol Chem 260: 9114–9116 [PubMed] [Google Scholar]
- Fuller RS, Kaguni JM, Kornberg A (1981) Enzymatic replication of the origin of the Escherichia coli chromosome. Proc Natl Acad Sci USA 78: 7370–7374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman LM, Barondess JJ, Beckwith J (1992) FtsL, an essential cytoplasmic membrane protein involved in cell division in Escherichia coli. J Bacteriol 174: 7716–7728 [PMC free article] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson MJ, Beckwith J (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose pBAD promoter. J Bacteriol 177: 4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke PD, Fuchs JA (1983) Characterization of the mRNA coding for ribonucleoside diphosphate reductase in Escherichia coli. J Bacteriol 156: 1192–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K (1993) The Escherichia coli K-12 ‘wild types' W3110 and MG1655 have an rph frameshift mutation that leads to pyrimidine starvation due to low pyrE expression levels. J Bacteriol 175: 3401–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama T, Kubota T, Kurokawa K, Crooke E, Sekimizu K (1998) The initiator function of DnaA protein is negatively regulated by the sliding clamp of the E. coli chromosomal replicase. Cell 94: 61–71 [DOI] [PubMed] [Google Scholar]
- Kato J, Katayama T (2001) Hda, a novel DnaA-related protein, regulates the replication cycle in Escherichia coli. EMBO J 20: 4253–4262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm RD, Austin RJ, Bell SP (1997) Coordinate binding of ATP and origin DNA regulates the ATPase activity of the origin recognition complex. Cell 88: 493–502 [DOI] [PubMed] [Google Scholar]
- Kornberg A, Baker TA (1992) DNA Replication. New York, NY: WH Freeman and Company, NY [Google Scholar]
- Kunz BA, Kohalmi SE (1991) Modulation of mutagenesis by deoxyribonucleotide levels. Annu Rev Genet 25: 339–359 [DOI] [PubMed] [Google Scholar]
- Kurokawa K, Nishida S, Emoto A, Sekimizu K, Katayama T (1999) Replication cycle-coordinated change of the adenine nucleotide-bound forms of DnaA protein in Escherichia coli. EMBO J 18: 6642–6652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lania L, Majello B, Napolitano G (1999) Transcriptional control by cell-cycle regulators: a review. J Cell Physiol 179: 134–141 [DOI] [PubMed] [Google Scholar]
- Li Z, Crooke E (1999) Functional analysis of affinity-purified polyhistidine-tagged DnaA protein. Protein Express Purif 17: 41–48 [DOI] [PubMed] [Google Scholar]
- Martin ME, Trimble MJ, Brun YV (2004) Cell cycle-dependent abundance, stability and localization of FtsA and FtsQ in Caulobacter crescentus. Mol Microbiol 54: 60–74 [DOI] [PubMed] [Google Scholar]
- Messer W (2002) The bacterial replication initiator DnaA. DnaA and oriC the bacterial mode to initiate DNA replication. FEMS Microbiol Rev 26: 355–374 [DOI] [PubMed] [Google Scholar]
- Messer W, Weigel C (1997) DnaA initiator—also a transcription factor. Mol Microbiol 24: 1–6 [DOI] [PubMed] [Google Scholar]
- Miller JH (1992) A Short Course in Bacterial Genetics: a Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Plainview, NY: Cold Spring Harbor Lab. Press [Google Scholar]
- Ortenberg R, Gon S, Porat A, Beckwith J (2004) Interactions of glutaredoxins, ribonucleotide reductase, and components of the DNA replication system of Escherichia coli. Proc Natl Acad Sci USA 101: 7439–7444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patten CL, Kirchhof MG, Schertzberg MR, Morton RA, Schellhorn HE (2004) Microarray analysis of RpoS-mediated gene expression in Escherichia coli K-12. Mol Genet Genomics 272: 580–591 [DOI] [PubMed] [Google Scholar]
- Ritz D, Patel H, Doan B, Zheng M, Aslund F, Storz G, Beckwith J (2000) Thioredoxin 2 is involved in the oxidative stress response in Escherichia coli. J Biol Chem 275: 2505–2512 [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Press [Google Scholar]
- Sekimizu K, Bramhill D, Kornberg A (1987) ATP activates dnaA protein in initiating replication of plasmids bearing the origin of the E. coli chromosome. Cell 50: 259–265 [DOI] [PubMed] [Google Scholar]
- Skarstad K, Boye E, Steen HB (1986) Timing of initiation of chromosome replication in individual Escherichia coli cells. EMBO J 5: 1711–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck C, Weigel C, Messer W (1999) ATP- and ADP-dnaA protein, a molecular switch in gene regulation. EMBO J 18: 6169–6176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su'etsugu M, Shimuta TR, Ishida T, Kawakami H, Katayama T (2005) Protein associations in DnaA-ATP hydrolysis mediated by the Hda-replicase clamp complex. J Biol Chem 280: 6528–6536 [DOI] [PubMed] [Google Scholar]
- Sun L, Fuchs JA (1992) Escherichia coli ribonucleotide reductase expression is cell cycle regulated. Mol Biol Cell 3: 1095–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelander L, Reichard P (1979) Reduction of ribonucleotides. Annu Rev Biochem 48: 133–158 [DOI] [PubMed] [Google Scholar]
- Torheim NK, Boye E, Lobner-Olesen A, Stokke T, Skarstad K (2000) The Escherichia coli SeqA protein destabilizes mutant DnaA204 protein. Mol Microbiol 37: 629–638 [DOI] [PubMed] [Google Scholar]
- Tuggle CK, Fuchs JA (1986) Regulation of the operon encoding ribonucleotide reductase in Escherichia coli: evidence for both positive and negative control. EMBO J 5: 1077–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkert MR, Landini P (2001) Transcriptional responses to DNA damage. Curr Opin Microbiol 4: 178–185 [DOI] [PubMed] [Google Scholar]
- Weiss DS, Chen JC, Ghigo JM, Boyd D, Beckwith J (1999) Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J Bacteriol 181: 508–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Wang X, Templeton LJ, Smulski DR, LaRossa RA, Storz G (2001) DNA microarray-mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J Bacteriol 183: 4562–4570 [DOI] [PMC free article] [PubMed] [Google Scholar]