

Abstract
The central dogma of molecular biology inspired by classical work in prokaryotic organisms accounts for only part of the genetic agenda of complex eukaryotes. First, post-transcriptional events lead to the generation of multiple mRNAs, proteins and functions from a single primary transcript, revealing regulatory networks distinct in mechanism and biological function from those controlling RNA transcription. Second, a variety of populous families of small RNAs (small nuclear RNAs, small nucleolar RNAs, microRNAs, siRNAs and shRNAs) assemble on ribonucleoprotein complexes and regulate virtually all aspects of the gene expression pathway, with profound biological consequences. Third, high-throughput methods of genomic analysis reveal that RNAs other than non-protein-coding RNAs (ncRNAs) represent a major component of the transcriptome that may perform novel functions in gene regulation and beyond. Post-transcriptional regulation, small RNAs and ncRNAs provide an expanding picture of the transcriptome that enriches our views of what genes are, how they operate, evolve and are regulated.
Keywords: gene, noncoding, RNA, splicing, transcription
‘… his book was known as the Book of Sand, because neither the Book nor the sand have beginning or end. JL Borges
Introduction
Describing the genome of an organism appears to be a well-defined task, one made feasible today at virtually all levels of biological complexity by spectacular advances in DNA manipulation and sequencing (Hood and Galas, 2003). One key motivation behind genome-sequencing projects is the assumption that the nucleotide sequence of an organism provide a description of the genes, gene products and gene networks that orchestrate programs like those sustaining the metabolic activity of a cell or deploying a body plan. This assumption is largely rooted in the classical view of DNA as the repository of genetic information, proteins as effector molecules and messenger RNAs as transcripts of the DNA that are decoded into protein by the ribosome. Additional functions of RNA, for example, as transfer and ribosomal RNAs in protein translation, have been recognized as important but of limited functional repertoire (Noller, 2005). We will review here recent progress that suggests a significantly more diverse picture for the content and function of the transcriptome, the set of RNAs transcribed from the genome. In contrast with the rather univocal—and today reachable—description of genomes, transcriptomes are entities as diverse as the cell types, developmental stages, environmental conditions and pathological states that the organism harbors or faces. Their full description requires high-throughput technologies that are still under development, with new major categories of transcripts being recently discovered. We will also discuss how these new developments significantly expand—and even challenge—the classical concept of the gene and how post-transcriptional molecular events are becoming key to understand gene regulation in higher eukaryotes. A picture is emerging where the central dogma inspired by prokaryotic molecular biology provides only a peep-hole view of eukaryotic genome function, and where large fractions of what was until recently considered junk DNA are indeed transcribed and may play a role as fundamental for understanding genomes as that of dark matter for understanding cosmological phenomena (Ostriker and Steinhardt, 2003).
Prevalent alternative splicing
In contrast with the gene-dense structure of prokaryotic genomes, where genes of related function are often transcribed as single units that are directly translated, protein-coding genes are dispersed in, and represent only a small fraction of, eukaryotic genomes (Mattick, 2004a). Furthermore, while prokaryotic transcripts can be directly translated, the primary products of transcription of eukaryotic protein-coding genes are subject to extensive processing, including removal of internal sequences—introns—and splicing of the remaining and typically shorter sequences—exons—to generate mature, translatable mRNAs (Black, 2003) (Figure 1). This organization makes gene expression dependent on the spliceosome, the complex enzymatic machinery in charge of intron removal (Nilsen, 2003). However, it also provides opportunities for regulation, because it allows particular sequences to be interpreted as intronic or exonic depending on cell type or environmental conditions (Black, 2003; Matlin et al, 2005). This flexibility in the splicing process allows the generation of multiple mRNAs from a single primary transcript, and affects the majority of higher eukaryotic genes (Johnson et al, 2003).
Figure 1.
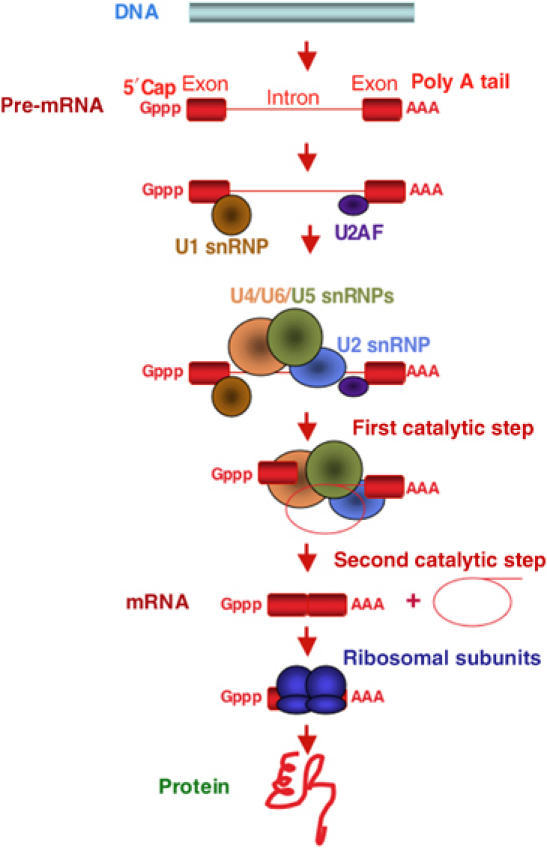
Example of an RNA processing pathway in eukaryotic gene expression. Messenger RNAs are synthesized as precursors containing a 7-methyl-guanidine triphosphate cap at the 5′ end, a polyadenylate tail at the 3′ end and intervening sequences (introns, thin red line) that are eliminated to generate translatable mRNAs where exons (red rectangles) are spliced together. Also represented are some of the components of the spliceosome, the complex that catalyzes intron removal through two consecutive trans-esterification steps. Introns are released in a lariat configuration and are often degraded in the nucleus. Spliceosomal factors include proteins like U2AF and small nuclear RNP particles like U1–U6 snRNPs; many other spliceosomal components are not represented. Most primary transcripts, including rRNAs, tRNAs, snoRNAs and miRNAs, undergo various processing steps catalyzed by protein and/or RNP complexes that eliminate internal or flanking sequences or introduce chemical modifications.
Alternatively spliced mRNAs often encode different proteins with distinct—sometimes opposite—functions (Figure 2), or contain premature stop codons that lead to RNA degradation by the nonsense-mediated decay (NMD) pathway (Stamm et al, 2005). Both its prevalence and its—sometimes extreme—flexibility to generate multiple products make alternative splicing a key contributor to proteomic complexity and an important focus point for gene regulation (Graveley, 2001; Maniatis and Tasic, 2002; Black, 2003; Matlin et al, 2005). Together with, and often coupled to, the use of alternative promoters and alternative sites of 3′ end processing, the use of alternative 5′ and 3′ splice sites and exon inclusion/skipping provide a picture of a gene's output that is best described as a diverse and dynamic set of products and functions (Figure 3), thus blurring the original view ‘one gene, one enzyme' that was at the foundation of molecular genetics (Kapranov et al, 2005). A truly remarkable example is the Drosophila gene Dscam, which can potentially generate more than 38 000 variants that are important both for neuronal wiring and for the generation of diversity in antibody-like molecules that serve in insects' immune defense (Schmucker et al, 2000; Watson et al, 2005). Although it is not yet clear whether the prevalence of alternative splicing correlates with organism complexity (Harrington et al, 2004; Kim et al, 2004), a significant fraction of alternative splicing events appear to be species-specific (Ast, 2004; Nagasaki et al, 2005), suggesting an important role for this process in driving evolutionary change. A remarkable example of this is the exonization of Alu elements, primate-specific mobile DNA sequences that upon insertion in intronic locations can generate transcripts containing new exons (Ast, 2004).
Figure 2.
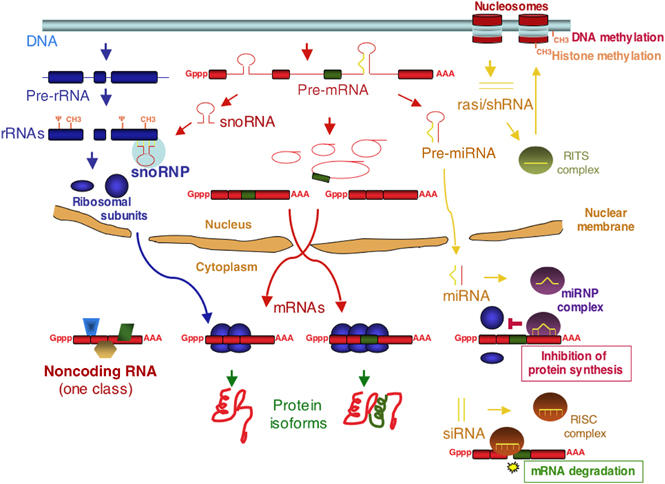
An overview of the eukaryotic transcriptome through examples of its products. Transcription by RNA polymerase I of ribosomal RNA precursors is followed by removal of 5′, 3′ extensions and intervening sequences (blue thin lines) to generate ribosomal RNAs (blue rectangles), which are further modified by pseudouridynilation (Ψ) and ribose methylation (CH3) at specific residues and assemble onto ribosomal subunits (blue circle and ellipse), which are then exported to the cytoplasm, where they mediate protein synthesis. Transcription by RNA polymerase II of mRNA precursors follows the processing pathway depicted in Figure 1. Exonic sequences can also be removed as part of an intron (green rectangle, in the example) to generate alternative mRNAs that can direct the synthesis of distinct protein isoforms. One class of transcripts that do not have extensive coding capacity (ncRNAs) are similarly generated and may play important cellular functions (in the example, by acting as a scaffold for the assembly of functionally connected protein factors, represented as polygons of various shapes). Some introns can generate additional transcripts with important functions in gene regulation. One example are snoRNAs, which assemble onto snoRNP RNP complexes and direct pseudouridynilation and ribose methylation of rRNA and other transcripts. Another example are precursors of miRNAs, which after cleavage by Drosha-type RNases in the nucleus and by Dicer-type enzymes in the cytoplasm, generate 20–28 ds miRNAs that assemble onto RNP complexes that repress translation by binding to 3′ UTRs of mRNAs and/or cause mRNA degradation, depending on the degree of complementarity with their target sequence. Other dsRNAs (e.g. siRNAs) can also trigger mRNA decay through the same mechanism. snoRNAs and miRNA precursors can also be generated from exonic sequences of dedicated transcripts. Small dsRNA fragments generated through bidirectional transcription—shRNAs (often from repetitive DNA—rasiRNAs) and cleavage can also induce transcriptional silencing through histone and DNA methylation.
Figure 3.
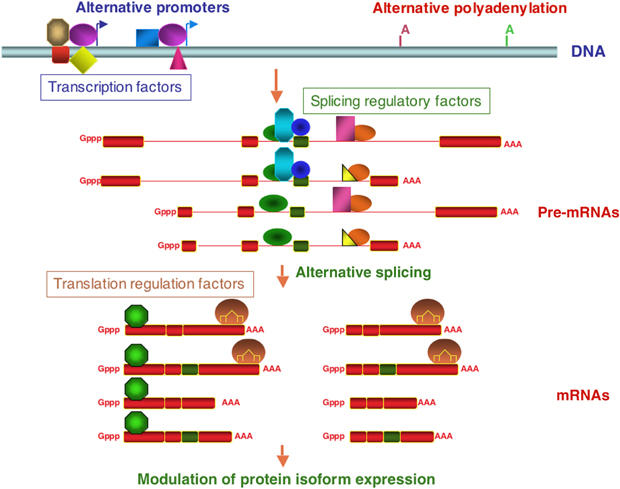
Transcript diversity and combinatorial regulation of gene expression. Multiple transcripts can be generated from the same gene locus through the use of alternative promoters, alternative polyadenylation sites and alternative splicing. The relative use of these signals is determined by the levels of general factors or by factors specific of particular classes of promoters or splice sites (represented by symbols of various shapes and colors), which may be expressed in a tissue-specific fashion. Coupling between these processes can influence the relative levels of mRNA isoforms. For example, the use of particular promoters or polyadenylation sites can affect the association of splicing regulatory factors and consequently the proportion of alternatively spliced transcripts. Similarly, the combination of different 5′-, 3′-ends and exons can determine the binding of factors and influence the fate of the RNA, including translational efficiency, decay rate or localization. In the example, the length of the 5′ and 3′-most exons allows binding of combinations of factors (e.g. an RNA-binding protein and a miRNA complex) that can modulate the levels of expression of the different protein isoforms.
Distinct regulatory programs
Although estimates of the fraction of alternative transcripts that provide distinct/distinguishable biological functions are not yet available, there exist enough examples of functional diversity among mRNA isoforms so as to suggest significant implications for the interpretation of classical genetic results, gene knockout studies and high-throughput data that do not distinguish between alternative transcripts and their diverse associated products.
One example of the latter is gene expression profiling using microarrays. Microarrays allow parallel detection of tens of thousands of RNA species that hybridize to a solid support (typically a glass slide) containing complementary probes spotted at defined positions in the array (Ferea and Brown, 1999). Conventional microarray designs use either cDNAs or oligonucleotides that hybridize along the length of a reference transcript. Such designs do not usually provide information about the various transcript isoforms present in an RNA sample. A more systematic approach to determine all the sequences transcribed from a genomic region is achieved by the use of tiling arrays (Kapranov et al, 2002; Bertone et al, 2005). These consist of sets of oligonucleotides that cover whole genomic regions, chromosomes or genomes through partially overlapping probes. Tiling arrays have the potential for high-resolution determination of the regions of a gene that are transcribed, for example, identifying novel exons. The identification of the structure of individual transcripts, however, depends on additional information connecting the various transcript elements, for example, through the use of strand-specific RACE (Cheng et al, 2005; Kapranov et al, 2005) or probes directed against exon–exon splice junctions (Matlin et al, 2005). Efforts to detect alternatively spliced isoforms using splicing-sensitive microarrays are starting to provide valuable clues about global changes in alternative splicing and its regulation, from discovery of new alternative splicing events to clustering of events according to cell type or disease stage, to the discovery of regulatory networks orchestrating multiple aspects of a biological process like synaptic transmission (Yeakley et al, 2002; Johnson et al, 2003; Pan et al, 2004; Blanchette et al, 2005; Relògio et al, 2005; Ule et al, 2005).
These global analyses complement in vitro studies on the detailed molecular mechanisms that regulate particular splice site choices. The picture emerging for how splicing regulation is achieved is one in which multiple sequence elements—not only the rather short and degenerate signals that define exon–intron boundaries—contribute to establish the exonic or intronic fate of a piece of RNA (Black, 2003; Matlin et al, 2005; Singh and Valcárcel, 2005). These auxiliary sequences are frequent in coding exons, and therefore mutational changes (either during evolution or those arising in disease) are constrained by—and may have effects on—both the coding capacity and the appropriate processing of the transcript (Cartegni et al, 2002; Carlini and Genut, 2006; Parmley et al, 2006). Regulatory sequences can engage in alternative RNA secondary structures that influence splice site pairing (Buratti and Baralle, 2004; Graveley, 2005). More often, however, regulatory sequences are recognized by various families of polypeptides, like SR and hnRNP proteins, resulting in ribonucleoprotein (RNP) assemblies where synergistic and antagonistic combinatorial effects have the potential to explain differential splice site recognition by the basic splicing enzymes (Black, 2003; Singh and Valcárcel, 2005). The orchestration of such changes—for example, in a tissue-specific manner—can rely on the tissue-specific expression of key regulators (e.g. sex-specific regulators in Drosophila, or neuron-specific Nova proteins in mammals), but in many cases seems to rely upon cellular codes established by the relative levels of expression of general, ubiquitous factors (Black, 2003). The latter set of mechanisms imply regulatory networks distinct from those governing transcriptional control, with lower numbers and less tissue-specific expression of factors involved in splicing regulation. Indeed, recent data suggest that cell type-specific programs of transcription and splicing are largely nonoverlapping, implying that they provide distinct readouts of a gene's function (Pan et al, 2004; Ule et al, 2005). Understanding the operation of these mechanisms will also require insights into how splicing regulation is coupled to transcription and polyadenylation, its interface with signaling pathways, as well as the development of imaging techniques to visualize the recruitment of factors to nascent transcripts in vivo (Shin and Manley, 2004; Kornblihtt, 2005; Mabon and Misteli, 2005).
Ever smaller RNAs with increasingly important functions
In addition to its role in relying information from the genome for protein synthesis, RNA is well known for its structural versatility, which is the basis of its remarkable catalytic properties (Caprara and Nilsen, 2000; Hollbrook, 2005). The ribosome and possibly the spliceosome are ribozymes where protein components play important roles in correct assembly of the complexes, but where catalysis is mediated by chemical moieties of the RNA (Konarska and Query, 2005; Noller, 2005). Important components of the spliceosome are small nuclear RNAs (snRNA) that recognize splice sites and establish an RNA scaffold that holds together the sequences involved in the splicing reaction (Villa et al, 2002) (Figure 1). Other snRNAs serve in 3′-end formation of particular transcripts and in other RNA-processing events (Beggs and Tollervey, 2005).
Another extensive family comprises small nucleolar RNAs (snoRNAs). They belong to two main classes (C/D box and H/ACA box) which are required for two types of chemical modifications (2′-O-ribose methylation and conversion of uridines to pseudouridines) occurring at specific residues in rRNAs, snRNAs, telomerase and other RNAs (Bachellerie et al, 2002; Vitali et al, 2005) (Figure 2). Each snoRNA guides one type of modification at a specific site within their target RNAs. These modifications—and probably also the interaction of snoRNP complexes themselves—are believed to be important for correct folding and/or function of ribosomal RNAs (Filipowicz and Pogacic, 2002). RNAs other than ribosomal RNAs can also be targets of snoRNAs, modulating, for example, editing or alternative splicing of pre-mRNAs (Bachellerie et al, 2002; Vitali et al, 2005; Kishore and Stamm, 2006).
Few would have thought before 1998 that studying pieces of RNAs of 20 nucleotides (nt) in length would be a worthwhile pursuit rendering anything more interesting than pieces of degraded transcripts. This is, however, an intense activity today that provides unexpected insights into a variety of important biological phenomena, from stem cell maintenance to cardiomyocyte differentiation to tumor formation, with applications ranging from large-scale genetic screens to novel therapeutics (Bartel, 2004; Novina and Sharp, 2004; Eckstein, 2005). This paradigm shift started as an additional double-strand (ds) RNA control in an antisense experiment (Fire et al, 1998). dsRNA is processed to 21–28 nt fragments by the RNAse III enzyme Dicer and the fragments (small interfering RNAs (siRNAs)) assemble onto the RNA-Induced Silencing protein Complex (RISC) to direct cleavage of complementary mRNAs, which leads to mRNA degradation and remarkably efficient gene silencing (Hannon and Rossi, 2004; Meister and Tuschl, 2004; Zamore and Haley, 2005) (Figure 2). This is a natural mechanism of defense against viruses—which often have dsRNAs as replicative intermediates—but it seems to have evolved also as an important repressor of the transcriptional activity of heterochromatic regions of the genome, including centromeres and other regions with repetitive sequences and transposons (Bernstein and Allis, 2005; Sontheimer and Carthew, 2005). Repeat-associated siRNAs (rasiRNAs) are generated from dsRNA through overlapping bidirectional transcription or—at least in some organisms—by an RNA-dependent RNA polymerase able to amplify the initial dsRNA signal. Dicer- and RISC-like enzymes and complexes lead to histone and DNA modifications that induce heterochromatin formation and transcriptional repression by poorly understood mechanisms (Verdel and Moazed, 2005) (Figure 2).
MicroRNAs and the orchestration of cellular functions
Dicer, RISC and related activities are also involved in the generation of microRNAs (miRNAs) (Lee et al, 1993; Wienholds and Plasterk, 2005). These are transcribed as precursors (pri-miRNAs) that are processed in the nucleus and cytoplasm to generate RNP complexes containing 21-nt miRNAs that are partially complementary to the 3′ untranslated region (UTR) of mRNAs (Cullen, 2004) (Figures 2 and 3). Binding of RISC–miRNA complexes inhibits translation of the cognate mRNA (Pillai et al, 2005), thus silencing gene expression. Although miRNA-mediated silencing does not necessarily involve cleavage of the target RNA, RNA degradation may also occur (Bagga et al, 2005; Lim et al, 2005) at least in some cases linked to 3′ UTR sequences previously known to mediate mRNA turnover (Jing et al, 2005).
Despite the challenge of finding bona fide miRNAs and miRNA targets based on limited sequence complementarity, computational and tailored cloning efforts are providing a growing list of miRNAs in multicellular organisms (Brennecke and Cohen, 2003; Hüttenhofer et al, 2004; Berezikov et al, 2005; Sontheimer and Carthew, 2005; Xie et al, 2005). Frequently, one miRNA can target multiple mRNAs and one mRNA can be regulated by multiple miRNAs targeting different regions of the 3′ UTR. Conversely, miRNA binding sequences are absent from the 3′ UTR of genes involved in basic cellular processes or of genes coexpressed with particular miRNAs (Stark et al, 2005). These features allow coordinated regulation, combinatorial control and precision and robustness to an increasing number of cell fate decisions and developmental transitions (Bartel and Chen, 2004; Farh et al, 2005; Lim et al, 2005; Wienholds et al, 2005). For example, recent reports have revealed important roles of miRNAs in the development of human cancers. The levels of about 200 miRNAs correlated with lineage and differentiation of tumor cells, and were significantly better criteria to classify poorly differentiated tumors than expression profiling of more than 2000 protein-coding genes, arguing for pivotal roles of miRNA levels in tumor development (Lu et al, 2005). Clusters of miRNAs have the properties of classical oncogenes, and modulate—and are modulated by—the activities of other oncogenes (He et al, 2005). For example, a regulatory network was recently discovered in which increased transcription of a cluster of miRNAs by the proto-oncogene c-MYC results in translational downregulation of the transcription factor E2F1, another important regulator of cell division, which is itself transcriptionally regulated by c-MYC (O'Donnell et al, 2005). Thus, miRNAs could serve in this case as part of a safety mechanism that adjusts the levels of expression of a key regulator of cell cycle progression.
Another interesting aspect of the genomic organization of many snoRNAs and one-third of miRNAs is that they are transcribed as intronic sequences within pre-mRNAs (Bachellerie et al, 2002; Seitz et al, 2003) (Figure 2). In some cases the mRNA does not code for a protein, and therefore the gene's output are snoRNAs or miRNAs generated from introns, which leads us to the next frontier.
Dark matter and noncoding RNAs
Only about 2% of the human genome corresponds to mature mRNA sequences (Lander et al, 2001). Even considering the corresponding introns, the primary transcripts of protein-coding genes represent around 30% of genomic sequence, with ribosomal and small RNAs adding another small fraction. Recent data from phylogenetic analyses and various high-throughput approaches, however, challenge the picture of higher eukaryotic genes as jewel planets on a much larger void of possibly junk DNA (Bejerano et al, 2004a, 2004b; Siepel et al, 2005), and suggest that a large part of the genome—possibly most of it—is indeed transcribed (Cheng et al, 2005; Kapranov et al, 2005). These technologies detect at least as many RNAs that do not have significant coding capacity (non-protein-coding RNAs (ncRNAs)) as mRNAs, explaining at least part of the discrepancy between the number of transcripts determined by random sequence of cDNA libraries and the number of genes predicted computationally on the basis of their coding potential (Suzuki and Hayashizaki, 2004; Claverie, 2005). For example, massive cDNA sequencing efforts have documented more than 180 000 distinct mouse transcripts from 22 000 estimated genes (Carninci et al, 2005). In addition to revealing more than 16 000 potential novel protein-coding genes, the lion's share of this expanded transcript collection corresponds to ncRNAs. Another surprising observation was that more than 70% of the transcripts overlap with an RNA synthesized from the opposite DNA strand, with frequent pairs of coding and noncoding RNAs adopting this configuration. Very similar conclusions were drawn from results using tiling arrays covering several human chromosomes. For example, combining information on transcription factor binding sites with expression data from tiling arrays of human chromosomes 21 and 22, comparable numbers of protein-coding and noncoding transcripts were identified as regulated by well-known transcription factors (Cawley et al, 2004). Intriguingly, mRNA/ncRNA pairs were often generated from overlapping or neighboring genomic regions, and were often found to be coregulated.
High-resolution tiling arrays detect further complexity in the form of a high proportion of nonpolyadenylated RNAs, and also show that half of all transcribed sequences were present only in the nucleus (Cheng et al, 2005). These features (lack of polyA tails, exclusion from the cytoplasm) are foreign to most sources of annotated transcripts, and therefore open the door to a largely unexplored territory of the transcriptome and of gene regulation.
Dark matter, dark energy?
It is currently unclear to what extent these ncRNAs play relevant biological roles or are spurious products of transcription (junk RNA), or even if a fraction correspond to experimental artifacts of the methods of detection (Hüttenhofer et al, 2005; Johnson et al, 2005). One first consideration is that even if the product of transcription does not have a function, the act of transcription of the corresponding region of the genome may open up chromatin structure and influence expression of other genes, DNA replication, recombination, etc. Second, an increasing number of independently validated ncRNAs show dynamic changes in expression that suggest their involvement in biological responses (Ravasi et al, 2006). Third, enough examples exist of ncRNAs with important functions to warrant further scrutiny. Now classical examples include ncRNAs important to assemble chromatin remodeling complexes that modulate the transcriptional activity of whole chromosomes in Drosophila and mammals to allow X-chromosome dosage compensation in different sexes (Bernstein and Allis, 2005; Lucchesi et al, 2005). Another example of such scaffolding activity has been recently provided by a tissue-culture screen in which the effect of knocking down the expression of 512 ncRNAs on 12 cellular assays was analyzed, rendering eight RNAs with effects on nuclear trafficking, cell viability or Hedgehog signaling (Willingham et al, 2005). For example, the NRON transcript was found to nucleate the assembly of nuclear trafficking factors with important consequences for nucleocytoplasmic transport. It would have been interesting to determine what fraction of a random collection of 512 protein-coding genes would score in the same assays.
Various additional functions can be postulated for the vast continent of ncRNAs. Transcripts antisense of a coding mRNA can obviously modulate expression by forming sense–antisense pairs that prevent translation or act as sources of dsRNA that can be processed to generate siRNA species that affect chromatin structure, mRNA stability or translatability (Katayama et al, 2005) (Figure 2). More frequently, however, sense/antisense pairs do not show a reciprocal expression pattern, suggesting that their frequent coregulation signifies coordinated expression of pairs of protein-coding and noncoding transcripts, perhaps with synergistic functions (Cawley et al, 2004; Mattick, 2004a, 2004b). Based on the known functions of other large categories of ncRNAs in various organisms, it is conceivable that some of the newly discovered transcripts act as allosteric effectors influencing the function or catalytic activities of proteins (Kuwabara et al, 2004; Storz et al, 2005), or act themselves as sensors of the concentrations of metabolites, as is the case of riboswitches in bacteria (Nudler and Mironov, 2004; Tucker and Breaker, 2005). snRNAs, snoRNAs and miRNAs share a number of features, including their synthesis as precursors requiring processing, their stepwise assembly into RNP complexes and the use of base-pairing complementarity to identify their targets. The common thread is the delivery of complexes with enzymatic or regulatory functions on specific RNA or DNA sequences, features that may be shared by other classes of ncRNAs. Finally, given the structural versatility and catalytic properties of RNAs—illustrated by naturally occurring ribozymes like the ribosome or self-spliced introns, and by the wide spectrum of reactions catalyzed by artificially selected RNA molecules (Fedor and Williamson, 2005)—it is not inconceivable that a fraction of ncRNAs contribute enzymatic functions.
One caveat for considering ncRNAs as the genome's second major depository of functional information is why genetic screens have not provided as many hits in ncRNA genes as in coding transcripts. Several explanations can be proposed for this discrepancy. A first consideration is target size, which may lower the likelihood of mutational events in certain small RNAs. Second, we do not know the nature of the functional determinants in these transcripts. Single-nucleotide substitutions or small deletions or insertions anywhere in a coding region can have dramatic effects on the protein-coding potential of an mRNA. Such mutations may be easier to accommodate to maintain functional ncRNAs, consistent with the limited phylogenetic conservation of some ncRNAs with well-known functions (Pang et al, 2006). Third, as some miRNAs appear to regulate multiple targets, it may be difficult to isolate viable mutants with specific phenotypes from extensively networked genetic elements. Finally, a cultural bias may have operated to discard mutants not associated with identifiable genes, a tendency that is likely to be reversed with the increasing awareness of the nature and properties of ncRNA genes.
While the number of protein-coding genes in a genome does not have a clear correlation with the complexity of the organism, the amount of noncoding DNA appears to increase steadily after the transition to multicellularity. It has been argued that ncRNAs (including regulatory small RNAs like miRNAs) may play key roles in orchestrating increasingly more sophisticated regulatory networks that sustain higher levels of complexity (Mattick, 2004a, 2004b; Mattick and Makunin, 2005). If this were true, and similar to cosmologists' postulate for the existence of some form of dark energy that sustains an ever-increasing expansion rate of the universe (Springel et al, 2005), the once considered junk of the genome could be the energy behind the expanding complexity of multicellular organisms.
In addition, it seems likely that the evolution of complexity is not based only on increased numbers of protein-coding genes but on increasingly sophisticated gene networks (Mattick, 2004a, 2004b). An overriding principle is the modularity of regulatory sequences. These allow precise control of promoter activity based upon a specific constellation of transcription factors, control of the proportion between alternatively spliced isoforms upon varying ratios of splicing regulators, control of the efficiency of translation upon the combination of available translation factors—including miRNAs, etc. It is thus easy to envision how a set of genes jointly regulated by a particular set of transcription factors can be differentially expressed by alternative splicing or translational control (Figure 3). These various layers of regulation multiply the combinatorial possibilities at each step of the gene expression pathway, leading to the exquisite finetuning of gene expression in time and space required to establish complex body plans, metabolic pathways and behaviors (Smith and Valcárcel, 2000; Hood and Galas, 2003) through the establishment of dedicated gene networks. An illustrative example is a double-negative feedback loop between two classes of miRNAs and two transcription factors that allows the establishment of left/right asymmetry in taste receptor neurons in Caenorhabditis. elegans (Johnston et al, 2005).
Book of sand
The argentinian writer JL Borges conceived an ‘infinite book' where new pages constantly appear as the reader attempts to open the book between the cover and the first page. The pages of the book of the genome also seem to multiply themselves as we open it. The discovery of the ‘one gene, one enzyme' concept that once unified genetics and biochemistry was followed by the discovery that single genes can express numbers of isoforms that exceed the number of genes in the organism. The emergence of small RNAs as essential actors in RNA processing ushered the discovery of new expanding categories of genetic elements—for example, miRNAs—with remarkable networking properties. Even the once considered empty pages of the genome now appear to be responsible for more than half of the transcriptional output of complex genomes, largely unexplored in identity and function.
It would probably be unwise to think that this is all the complexity that remains to be discovered. Hybrid transcripts containing pieces of independent transcriptional units (generated for example by read-through transcription or trans-splicing events) are common in some organisms and can occur in higher eukaryotes as well (Hastings, 2005; Takahara et al, 2005). Detection of transcripts antisense to processed mRNAs suggests the possibility that RNA-mediated RNA synthesis produces RNA species that cannot be generated by transcription (Cheng et al, 2005). Such activities may amplify the products of transcription and also mediate transcriptional silencing, as observed in plants (Vaucheret, 2005). Certain signaling pathways (e.g. the unfolded protein response and other forms of stress) generate novel transcripts through processes that integrate gene expression and subcellular dynamics (Patil and Walter, 2001; Prasanth et al, 2005). Finally, RNA editing—the ultimate nightmare for gene annotation aficionados—is known to play a pivotal role in important biological decisions, and analysis of nucleotide products of editing reactions suggests a widespread impact of the process, particularly in the nervous system (Bass, 2002; Eisenberg et al, 2005; Stuart et al, 2005). Far from discouraging efforts to interpret the genome, the existing and foreseeable expanding complexity may prove key to understand the frame and logic of its function.
A list of web resources for analyses of transcript heterogeneity is given in Supplementary Table I.
Supplementary Material
Supplementary data
Supplementary Table I
Acknowledgments
We thank Tom Gingeras, Roderic Guigó, Alexander Hüttenhofer and John Mattick for comments on the manuscript. LMS was supported by a Praxis Program fellowship from the Portuguese government.
References
- Ast G (2004) How did alternative splicing evolve? Nat Rev Genet 5: 773–782 [DOI] [PubMed] [Google Scholar]
- Bachellerie JP, Cavaille J, Huttenhofer A (2002) The expanding snoRNA world. Biochimie 84: 775–790 [DOI] [PubMed] [Google Scholar]
- Bagga S, Bracht J, Hunter S, Massirer K, Holtz J, Eachus R, Pasquinelli AE (2005) Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell 122: 553–563 [DOI] [PubMed] [Google Scholar]
- Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297 [DOI] [PubMed] [Google Scholar]
- Bartel DP, Chen CZ (2004) Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet 5: 396–400 [DOI] [PubMed] [Google Scholar]
- Bass BL (2002) RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 71: 817–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs JD, Tollervey D (2005) Crosstalk between RNA metabolic pathways: an RNOMICS approach. Nat Rev Mol Cell Biol 6: 423–429 [DOI] [PubMed] [Google Scholar]
- Bejerano G, Haussler D, Blanchette M (2004a) Into the heart of darkness: large-scale clustering of human non-coding DNA. Bioinformatics 20 (Suppl 1): I40–I48 [DOI] [PubMed] [Google Scholar]
- Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D (2004b) Ultraconserved elements in the human genome. Science 304: 1321–1325 [DOI] [PubMed] [Google Scholar]
- Berezikov E, Guryev V, van de Belt J, Wienholds E, Plasterk RH, Cuppen E (2005) Phylogenetic shadowing and computational identification of human microRNA genes. Cell 120: 21–24 [DOI] [PubMed] [Google Scholar]
- Bernstein E, Allis CD (2005) RNA meets chromatin. Genes Dev 19: 1635–1655 [DOI] [PubMed] [Google Scholar]
- Bertone P, Gerstein M, Snyder M (2005) Applications of DNA tiling arrays to experimental genome annotation and regulatory pathway discovery. Chromosome Res 13: 259–274 [DOI] [PubMed] [Google Scholar]
- Black DL (2003) Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72: 291–336 [DOI] [PubMed] [Google Scholar]
- Blanchette M, Green RE, Brenner SE, Rio DC (2005) Global analysis of positive and negative pre-mRNA splicing regulators in Drosophila. Genes Dev 19: 1306–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Cohen SM (2003) Towards a complete description of the microRNA complement of animal genomes. Genome Biol 4: 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Baralle FE (2004) Influence of RNA secondary structure on the pre-mRNA splicing process. Mol Cell Biol 24: 10505–10514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprara MG, Nilsen TW (2000) RNA: versatility in form and function. Nat Struct Biol 7: 831–833 [DOI] [PubMed] [Google Scholar]
- Carlini DB, Genut JE (2006) Synonymous SNPs provide evidence for selective constraint on human exonic splicing enhancers. J Mol Evol 62: 89–98 [DOI] [PubMed] [Google Scholar]
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, Kodzius R, Shimokawa K, Bajic VB, Brenner SE, Batalov S, Forrest AR, Zavolan M, Davis MJ, Wilming LG, Aidinis V, Allen JE, Ambesi-Impiombato A, Apweiler R, Aturaliya RN, Bailey TL, Bansal M, Baxter L, Beisel KW, Bersano T, Bono H, Chalk AM, Chiu KP, Choudhary V, Christoffels A, Clutterbuck DR, Crowe ML, Dalla E, Dalrymple BP, de Bono B, Della Gatta G, di Bernardo D, Down T, Engstrom P, Fagiolini M, Faulkner G, Fletcher CF, Fukushima T, Furuno M, Futaki S, Gariboldi M, Georgii-Hemming P, Gingeras TR, Gojobori T, Green RE, Gustincich S, Harbers M, Hayashi Y, Hensch TK, Hirokawa N, Hill D, Huminiecki L, Iacono M, Ikeo K, Iwama A, Ishikawa T, Jakt M, Kanapin A, Katoh M, Kawasawa Y, Kelso J, Kitamura H, Kitano H, Kollias G, Krishnan SP, Kruger A, Kummerfeld SK, Kurochkin IV, Lareau LF, Lazarevic D, Lipovich L, Liu J, Liuni S, McWilliam S, Madan Babu M, Madera M, Marchionni L, Matsuda H, Matsuzawa S, Miki H, Mignone F, Miyake S, Morris K, Mottagui-Tabar S, Mulder N, Nakano N, Nakauchi H, Ng P, Nilsson R, Nishiguchi S, Nishikawa S, Nori F, Ohara O, Okazaki Y, Orlando V, Pang KC, Pavan WJ, Pavesi G, Pesole G, Petrovsky N, Piazza S, Reed J, Reid JF, Ring BZ, Ringwald M, Rost B, Ruan Y, Salzberg SL, Sandelin A, Schneider C, Schonbach C, Sekiguchi K, Semple CA, Seno S, Sessa L, Sheng Y, Shibata Y, Shimada H, Shimada K, Silva D, Sinclair B, Sperling S, Stupka E, Sugiura K, Sultana R, Takenaka Y, Taki K, Tammoja K, Tan SL, Tang S, Taylor MS, Tegner J, Teichmann SA, Ueda HR, van Nimwegen E, Verardo R, Wei CL, Yagi K, Yamanishi H, Zabarovsky E, Zhu S, Zimmer A, Hide W, Bult C, Grimmond SM, Teasdale RD, Liu ET, Brusic V, Quackenbush J, Wahlestedt C, Mattick JS, Hume DA, Kai C, Sasaki D, Tomaru Y, Fukuda S, Kanamori-Katayama M, Suzuki M, Aoki J, Arakawa T, Iida J, Imamura K, Itoh M, Kato T, Kawaji H, Kawagashira N, Kawashima T, Kojima M, Kondo S, Konno H, Nakano K, Ninomiya N, Nishio T, Okada M, Plessy C, Shibata K, Shiraki T, Suzuki S, Tagami M, Waki K, Watahiki A, Okamura-Oho Y, Suzuki H, Kawai J, Hayashizaki Y, FANTOM Consortium, RIKEN Genome Exploration Research Group and Genome Science Group (Genome Network Project Core Group) (2005) The transcriptional landscape of the mammalian genome. Science 309: 1559–1563 [DOI] [PubMed] [Google Scholar]
- Cartegni L, Chew SL, Krainer AR (2002) Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 3: 285–298 [DOI] [PubMed] [Google Scholar]
- Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, Piccolboni A, Sementchenko V, Cheng J, Williams AJ, Wheeler R, Wong B, Drenkow J, Yamanaka M, Patel S, Brubaker S, Tammana H, Helt G, Struhl K, Gingeras TR (2004) Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 116: 499–509 [DOI] [PubMed] [Google Scholar]
- Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, Long J, Stern D, Tammana H, Helt G, Sementchenko V, Piccolboni A, Bekiranov S, Bailey DK, Ganesh M, Ghosh S, Bell I, Gerhard DS, Gingeras TR (2005) Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 308: 1149–1154 [DOI] [PubMed] [Google Scholar]
- Claverie JM (2005) Fewer genes, more noncoding RNA. Science 309: 1529–1530 [DOI] [PubMed] [Google Scholar]
- Cullen BR (2004) Transcription and processing of human microRNA precursors. Mol Cell 16: 861–865 [DOI] [PubMed] [Google Scholar]
- Eckstein F (2005) Small non-coding RNAs as magic bullets. Trends Biochem Sci 30: 445–452 [DOI] [PubMed] [Google Scholar]
- Eisenberg E, Nemzer S, Kinar Y, Sorek R, Rechavi G, Levanon EY (2005) Is abundant A-to-I RNA editing primate-specific? Trends Genet 21: 77–81 [DOI] [PubMed] [Google Scholar]
- Farh KK, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, Bartel DP (2005) The Widespread impact of mammalian microRNAs on mRNA repression and evolution. Science 310: 1817–1821 [DOI] [PubMed] [Google Scholar]
- Fedor MJ, Williamson JR (2005) The catalytic diversity of RNAs. Nat Rev Mol Cell Biol 6: 399–412 [DOI] [PubMed] [Google Scholar]
- Ferea TL, Brown PO (1999) Observing the living genome. Curr Opin Genet Dev 9: 715–722 [DOI] [PubMed] [Google Scholar]
- Filipowicz W, Pogacic V (2002) Biogenesis of small nucleolar ribonucleoproteins. Curr Opin Cell Biol 14: 319–327 [DOI] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391: 806–811 [DOI] [PubMed] [Google Scholar]
- Graveley BR (2001) Alternative splicing: increasing diversity in the proteomic world. Trends Genet 17: 100–107 [DOI] [PubMed] [Google Scholar]
- Graveley BR (2005) Mutually exclusive splicing of the insect Dscam pre-mRNA directed by competing intronic RNA secondary structures. Cell 123: 65–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon GJ, Rossi JJ (2004) Unlocking the potential of the human genome with RNA interference. Nature 431: 371–378 [DOI] [PubMed] [Google Scholar]
- Harrington ED, Boue S, Valcárcel J, Reich JG, Bork P (2004) Estimating rates of alternative splicing in mammals and invertebrates. Nat Genet 36: 916–917 [DOI] [PubMed] [Google Scholar]
- Hastings KE (2005) SL trans-splicing: easy come or easy go? Trends Genet 21: 240–247 [DOI] [PubMed] [Google Scholar]
- He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM (2005) A microRNA polycistron as a potential human oncogene. Nature 435: 828–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollbrook SR (2005) RNA structure: the long and the short of it. Curr Opin Struct Biol 15: 302–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood L, Galas D (2003) The digital code of DNA. Nature 421: 444–448 [DOI] [PubMed] [Google Scholar]
- Hüttenhofer A, Cavaille J, Bachellerie JP (2004) Experimental RNomics: a global approach to identifying small nuclear RNAs and their targets in different model organisms. Methods Mol Biol 265: 409–428 [DOI] [PubMed] [Google Scholar]
- Hüttenhofer A, Schattner P, Polacek N (2005) Non-coding RNAs: hope or hype? Trends Genet 21: 289–297 [DOI] [PubMed] [Google Scholar]
- Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin SC, Gram H, Han J (2005) Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell 120: 623–634 [DOI] [PubMed] [Google Scholar]
- Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD (2003) Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 302: 2141–2144 [DOI] [PubMed] [Google Scholar]
- Johnson JM, Edwards S, Shoemaker D, Schadt EE (2005) Dark matter in the genome: evidence of widespread transcription detected by microarray tiling experiments. Trends Genet 21: 93–102 [DOI] [PubMed] [Google Scholar]
- Johnston RJ Jr, Chang S, Etchberger JF, Ortiz CO, Hobert O (2005) MicroRNAs acting in a double-negative feedback loop to control a neuronal cell fate decision. Proc Natl Acad Sci USA 102: 12449–12454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapranov P, Cawley SE, Drenkow J, Bekiranov S, Strausberg RL, Fodor SP, Gingeras TR (2002) Large-scale transcriptional activity in chromosomes 21 and 22. Science 296: 916–919 [DOI] [PubMed] [Google Scholar]
- Kapranov P, Drenkow J, Cheng J, Long J, Helt G, Dike S, Gingeras TR (2005) Examples of the complex architecture of the human transcriptome revealed by RACE and high-density tiling arrays. Genome Res 15: 987–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J, Suzuki H, Carninci P, Hayashizaki Y, Wells C, Frith M, Ravasi T, Pang KC, Hallinan J, Mattick J, Hume DA, Lipovich L, Batalov S, Engstrom PG, Mizuno Y, Faghihi MA, Sandelin A, Chalk AM, Mottagui-Tabar S, Liang Z, Lenhard B, Wahlestedt C, RIKEN Genome Exploration Research Group, Genome Science Group (Genome Network Project Core Group), FANTOM Consortium (2005) Antisense transcription in the mammalian transcriptome. Science 309: 1564–1566 [DOI] [PubMed] [Google Scholar]
- Kim H, Klein R, Majewski J, Ott J (2004) Estimating rates of alternative splicing in mammals and invertebrates. Nat Genet 36: 915–916 [DOI] [PubMed] [Google Scholar]
- Kishore S, Stamm S (2006) The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science 311: 230–232 [DOI] [PubMed] [Google Scholar]
- Konarska MM, Query CC (2005) Insights into the mechanisms of splicing: more lessons from the ribosome. Genes Dev 19: 2255–2260 [DOI] [PubMed] [Google Scholar]
- Kornblihtt AR (2005) Promoter usage and alternative splicing. Curr Opin Cell Biol 17: 262–268 [DOI] [PubMed] [Google Scholar]
- Kuwabara T, Hsieh J, Nakashima K, Taira K, Gage FH (2004) A small modulatory dsRNA specifies the fate of adult neural stem cells. Cell 116: 779–793 [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ, International Human Genome Sequencing Consortium (2001) Initial sequencing and analysis of the human genome. Nature 409: 860–921 [DOI] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843–854 [DOI] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM (2005) Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433: 769–773 [DOI] [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR (2005) MicroRNA expression profiles classify human cancers. Nature 435: 834–838 [DOI] [PubMed] [Google Scholar]
- Lucchesi JC, Kelly WG, Panning B (2005) Chromatin remodeling in dosage compensation. Annu Rev Genet 39: 615–651 [DOI] [PubMed] [Google Scholar]
- Mabon SA, Misteli T (2005) Differential recruitment of pre-mRNA splicing factors to alternatively spliced transcripts in vivo. PLoS Biol 3: e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T, Tasic B (2002) Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature 418: 236–243 [DOI] [PubMed] [Google Scholar]
- Matlin AJ, Clark F, Smith CWJ (2005) Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol 6: 386–398 [DOI] [PubMed] [Google Scholar]
- Mattick JS (2004a) The hidden genetic program of complex organisms. Sci Am 291: 60–67 [DOI] [PubMed] [Google Scholar]
- Mattick JS (2004b) RNA regulation: a new genetics? Nat Rev Genet 5: 316–323 [DOI] [PubMed] [Google Scholar]
- Mattick JS, Makunin IV (2005) Small regulatory RNAs in mammals. Hum Mol Genet 14 (Spec No. 1): R121–R132 [DOI] [PubMed] [Google Scholar]
- Meister G, Tuschl T (2004) Mechanisms of gene silencing by double-stranded RNA. Nature 431: 343–349 [DOI] [PubMed] [Google Scholar]
- Nagasaki H, Arita M, Nishizawa T, Suwa M, Gotoh O (2005) Species-specific variation of alternative splicing and transcriptional initiation in six eukaryotes. Gene 364: 53–62 [DOI] [PubMed] [Google Scholar]
- Nilsen TW (2003) The spliceosome: the most complex macromolecular machine in the cell? BioEssays 25: 1147–1149 [DOI] [PubMed] [Google Scholar]
- Noller HF (2005) RNA structure: reading the ribosome. Science 309: 1508–1514 [DOI] [PubMed] [Google Scholar]
- Novina CD, Sharp PA (2004) The RNAi revolution. Nature 430: 161–164 [DOI] [PubMed] [Google Scholar]
- Nudler E, Mironov AS (2004) The riboswitch control of bacterial metabolism. Trends Biochem Sci 29: 11–17 [DOI] [PubMed] [Google Scholar]
- O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT (2005) c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435: 839–843 [DOI] [PubMed] [Google Scholar]
- Ostriker JP, Steinhardt P (2003) New light on dark matter. Science 300: 1909–1913 [DOI] [PubMed] [Google Scholar]
- Pan Q, Shai O, Misquitta C, Zhang W, Saltzman AL, Mohammad N, Babak T, Siu H, Hughes TR, Morris QD, Frey BJ, Blencowe BJ (2004) Revealing global regulatory features of mammalian alternative splicing using a quantitative microarray platform. Mol Cell 16: 929–941 [DOI] [PubMed] [Google Scholar]
- Pang KC, Frith MC, Mattick JS (2006) Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet 22: 1–5 [DOI] [PubMed] [Google Scholar]
- Parmley JL, Chamary JV, Hurst LD (2006) Evidence for purifying selection against synonymous mutations in mammalian exonic splicing enhancers. Mol Biol Evol 23: 301–309 [DOI] [PubMed] [Google Scholar]
- Patil C, Walter P (2001) Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol 13: 349–355 [DOI] [PubMed] [Google Scholar]
- Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, Bertrand E, Filipowicz W (2005) Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science 309: 1573–1576 [DOI] [PubMed] [Google Scholar]
- Prasanth KV, Prasanth SG, Xuan Z, Hearn S, Freier SM, Bennett CF, Zhang MQ, Spector DL (2005) Regulating gene expression through RNA nuclear retention. Cell 123: 249–263 [DOI] [PubMed] [Google Scholar]
- Ravasi T, Suzuki H, Pang KC, Katayama S, Furuno M, Okunishi R, Fukuda S, Ru K, Frith MC, Gongora MM, Grimmond SM, Hume DA, Hayashizaki Y, Mattick JS (2006) Experimental validation of the regulated expression of large numbers of noncoding RNAs from the mouse genome. Genome Res 16: 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relògio A, Ben-Dov C, Baum M, Ruggiu M, Gemund C, Benes V, Darnell RB, Valcárcel J (2005) Alternative splicing microarrays reveal functional expression of neuron-specific regulators in Hodgkin lymphoma cells. J Biol Chem 280: 4779–4784 [DOI] [PubMed] [Google Scholar]
- Schmucker D, Clemens JC, Shu H, Worby CA, Xiao J, Muda M, Dixon JE, Zipursky SL (2000) Drosophila Dscam is an axon guidance receptor exhibiting extraordinary molecular diversity. Cell 101: 671–684 [DOI] [PubMed] [Google Scholar]
- Seitz H, Youngson N, Lin SP, Dalbert S, Paulsen M, Bachellerie JP, Ferguson-Smith AC, Cavaille J (2003) Imprinted microRNA genes transcribed antisense to a reciprocally imprinted retrotransposon-like gene. Nat Genet 34: 261–262 [DOI] [PubMed] [Google Scholar]
- Shin C, Manley JL (2004) Cell signalling and the control of pre-mRNA splicing. Nat Rev Mol Cell Biol 5: 727–738 [DOI] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, Weinstock GM, Wilson RK, Gibbs RA, Kent WJ, Miller W, Haussler D (2005) Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Valcárcel J (2005) Building specificity with nonspecific RNA-binding proteins. Nat Struct Mol Biol 12: 645–653 [DOI] [PubMed] [Google Scholar]
- Smith CW, Valcárcel J (2000) Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem Sci 25: 381–388 [DOI] [PubMed] [Google Scholar]
- Sontheimer EJ, Carthew RW (2005) Silence from within: endogenous siRNAs and miRNAs. Cell 122: 9–12 [DOI] [PubMed] [Google Scholar]
- Springel V, White SD, Jenkins A, Frenk CS, Yoshida N, Gao L, Navarro J, Thacker R, Croton D, Helly J, Peacock JA, Cole S, Thomas P, Couchman H, Evrard A, Colberg J, Pearce F (2005) Simulations of the formation, evolution and clustering of galaxies and quasars. Nature 435: 629–636 [DOI] [PubMed] [Google Scholar]
- Stamm S, Ben-Ari S, Rafalska I, Tang Y, Zhang Z, Toiber D, Thanaraj TA, Soreq H (2005) Function of alternative splicing. Gene 344: 1–20 [DOI] [PubMed] [Google Scholar]
- Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM (2005) Animal microRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell 123: 1133–1146 [DOI] [PubMed] [Google Scholar]
- Storz G, Altuvia S, Wassarman KH (2005) An abundance of RNA regulators. Annu Rev Biochem 74: 199–217 [DOI] [PubMed] [Google Scholar]
- Stuart KD, Schnaufer A, Ernst NL, Panigrahi AK (2005) Complex management: RNA editing in trypanosomes. Trends Biochem Sci 30: 97–105 [DOI] [PubMed] [Google Scholar]
- Suzuki M, Hayashizaki Y (2004) Mouse-centric comparative transcriptomics of protein coding and non-coding RNAs. BioEssays 26: 833–843 [DOI] [PubMed] [Google Scholar]
- Takahara T, Tasic B, Maniatis T, Akanuma H, Yanagisawa S (2005) Delay in synthesis of the 3′ splice site promotes trans-splicing of the preceding 5′ splice site. Mol Cell 18: 245–251 [DOI] [PubMed] [Google Scholar]
- Tucker BJ, Breaker RR (2005) Riboswitches as versatile gene control elements. Curr Opin Struct Biol 15: 342–348 [DOI] [PubMed] [Google Scholar]
- Ule J, Ule A, Spencer J, Williams A, Hu JS, Cline M, Wang H, Clark T, Fraser C, Ruggiu M, Zeeberg BR, Kane D, Weinstein JN, Blume J, Darnell RB (2005) Nova regulates brain-specific splicing to shape the synapse. Nat Genet 37: 844–852 [DOI] [PubMed] [Google Scholar]
- Vaucheret H (2005) RNA polymerase IV and transcriptional silencing. Nat Genet 37: 659–660 [DOI] [PubMed] [Google Scholar]
- Verdel A, Moazed D (2005) RNAi-directed assembly of heterochromatin in fission yeast. FEBS Lett 579: 5872–5878 [DOI] [PubMed] [Google Scholar]
- Villa T, Pleiss JA, Guthrie C (2002) Spliceosomal snRNAs: Mg(2+)-dependent chemistry at the catalytic core? Cell 109: 149–152 [DOI] [PubMed] [Google Scholar]
- Vitali P, Basyuk E, Le Meur E, Bertrand E, Muscatelli F, Cavaille J, Huttenhofer A (2005) ADAR2-mediated editing of RNA substrates in the nucleolus is inhibited by C/D small nucleolar RNAs. J Cell Biol 169: 745–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson FL, Puttmann-Holgado R, Thomas F, Lamar DL, Hughes M, Kondo M, Rebel VI, Schmucker D (2005) Extensive diversity of Ig-superfamily proteins in the immune system of insects. Science 309: 1874–1878 [DOI] [PubMed] [Google Scholar]
- Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, Horvitz HR, Kauppinen S, Plasterk RH (2005) MicroRNA expression in zebrafish embryonic development. Science 309: 310–311 [DOI] [PubMed] [Google Scholar]
- Wienholds E, Plasterk RH (2005) MicroRNA function in animal development. FEBS Lett 579: 5911–5922 [DOI] [PubMed] [Google Scholar]
- Willingham AT, Orth AP, Batalov S, Peters EC, Wen BG, Aza-Blanc P, Hogenesch JB, Schultz PG (2005) A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science 309: 1570–1573 [DOI] [PubMed] [Google Scholar]
- Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M (2005) Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 434: 338–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeakley JM, Fan JB, Doucet D, Luo L, Wickham E, Ye Z, Chee MS, Fu XD (2002) Profiling alternative splicing on fiber-optic arrays. Nat Biotechnol 20: 353–358 [DOI] [PubMed] [Google Scholar]
- Zamore PD, Haley B (2005) Ribo-gnome: the big world of small RNAs. Science 309: 1519–1524 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary Table I