

Abstract
Leprosy enables investigation of mechanisms by which the innate immune system contributes to host defense against infection, since in one form, the disease progresses, and in the other, the infection is limited. We report that Toll-like receptor (TLR) activation of human monocytes induces rapid differentiation into two distinct subsets: DC-SIGN+CD16+ macrophages and CD1b+DC-SIGN− dendritic cells. DC-SIGN+ phagocytic macrophages were expanded by TLR-mediated upregulation of IL-15/IL-15R. CD1b+ dendritic cells were expanded by TLR-mediated upregulation of GM-CSF/GM-CSFR, promoted T cell activation and secreted proinflammatory cytokines. While DC-SIGN+ macrophages were detected in lesions of all leprosy patients, CD1b+ dendritic cells were not detected in patients with the progressive lepromatous form, except during reversal reactions in which bacilli were cleared by Th1 responses. In T-lep lesions, DC-SIGN+ cells were positive for macrophage markers, but negative for dendritic cell markers. Thus, TLR-induced differentiation of monocytes into either macrophages or dendritic cells appears critically to influence effective host defenses in human infectious disease.
Introduction
The ability of the innate immune system to recognize microbial pathogens is mediated by highly conserved families of pattern recognition receptors that activate host defense pathways. One such family is comprised of the Toll-like receptors (TLRs) that are expressed by a variety of cells of the innate immune system with each family member endowed with the ability to recognize a distinct class of conserved microbial molecules. The expression and activation of TLRs contributes to host defense against infection in drosophila, mice, and humans1,2,3,4,5. There are two different mechanisms by which TLR activation can contribute to host defense. First, activation of TLRs can directly mediate innate responses by regulating phagocytosis and triggering antimicrobial activity6,7,8. Second, activation of TLRs can trigger the release of cytokines and the differentiation of immature to mature dendritic cells, enabling the innate immune system to instruct the adaptive immune response9,10,11.
Cells of the innate immune system are characterized in normal and diseased tissues according to various cell surface receptors. The group I CD1 molecules, CD1a and CD1b, are specifically expressed by tissue and cytokine-derived dendritic cells and facilitate the presentation of nonpeptide antigens to T cells12,13,14,15. The calcium-dependent (C-type) lectins, dendritic cell-specific ICAM grabbing nonintegrin (DC-SIGN) and the mannose receptor are expressed on both dendritic cells and macrophages, functioning in phagocytosis and antigen presentation16,17. DC-SIGN contains a carbohydrate-recognition domain enabling cytokine-derived dendritic cells to recognize and mediate phagocytosis/uptake of a broad range of pathogens including the human immunodeficiency virus and species of mycobacteria, leishmania, candida, and helicobacter17,18,19,20,21,22.
The disease leprosy provides an extraordinary opportunity to investigate mechanisms by which the innate immune system contributes to host defense against infection. The disease presents as a clinical spectrum that correlates with the level of the immune response to the pathogen23. At one pole of the disease, patients with the tuberculoid form (T-lep) are relatively resistant to the pathogen, the infection is localized and the lesions characterized by expression of Th1 cytokines characteristic of cell-mediated immunity24,25. In contrast, at the opposite pole, patients with lepromatous leprosy (L-lep) are relatively susceptible to the pathogen, the infection is systemically disseminated and the lesions characterized by Th2 cytokines characteristic of humoral responses. The spectrum of human leprosy is not static as those patients with the L-lep form of the disease can upgrade towards the T-lep pole during a reversal reaction, cell-mediated immune reactions characterized by the reduction of bacilli in lesions and the influx of CD4+ Th1 cells into disease lesions26. We therefore studied leprosy as a model to investigate general mechanisms by which the innate immune system regulates direct and indirect effector pathways, specifically by exploring the differentiation, distribution and function of CD1+ and DC-SIGN+ cells.
Results
Expression of DC-SIGN and CD1b in situ and in vitro
Given the distinct functions of DC-SIGN (antigen uptake) and CD1b (antigen presentation) on cells of the innate immune system, we examined the in situ distribution of DC-SIGN and CD1b in human lymphoid tissue. Based on studies of cytokine-derived dendritic cells, we expected to find cells co-expressing DC-SIGN and CD1b. Surprisingly, DC-SIGN and CD1b were expressed on distinct non-overlapping cell populations (Fig. 1a).
Figure 1.
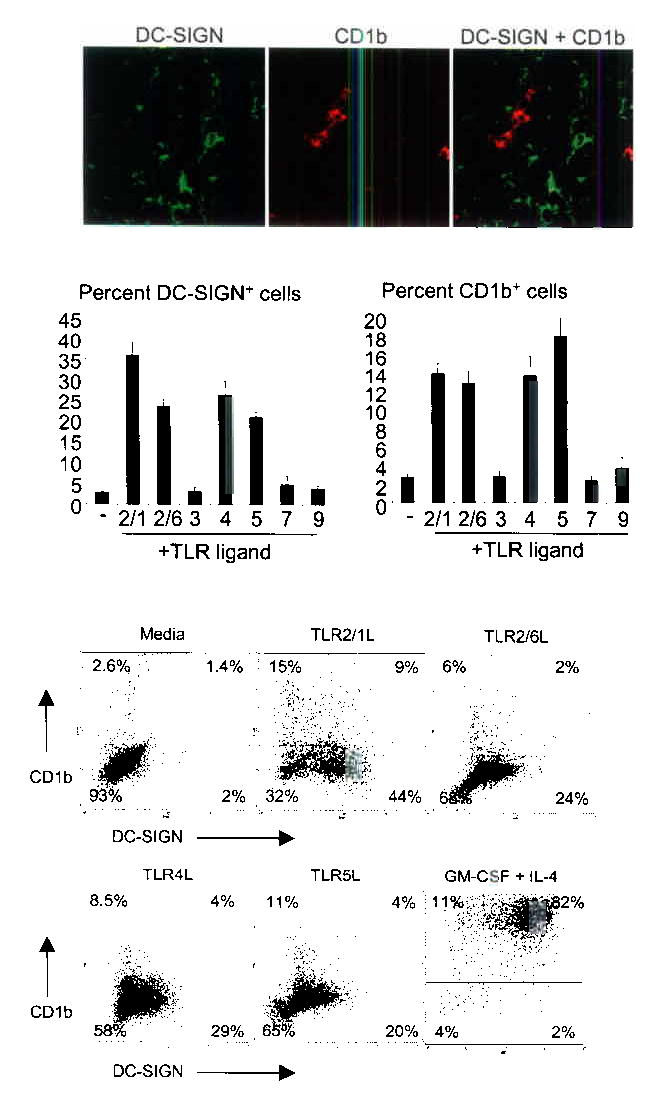
DC-SIGN and CD1b are expressed on distinct subsets of cells and are induced by TLR activation. (a) Human tonsil tissue sections were labeled with specific antibodies and visualized using confocal laser microscopy. Original magnification, 40X. (b) Human peripheral monocytes were activated with TLR ligands for 48 h and labeled with specific antibodies. Data are shown as the average ± SEM of between four and fourteen independent experiments. (c) Human peripheral monocytes were stimulated with TLR ligands for 48 h and double-labeled. Percent of cells in each quadrant are indicated. Data are representative of between three and 14 independent experiments.
This finding raised the question whether activation of the innate immune system could lead to the expansion of these two distinct cell populations expressing DC-SIGN or CD1b. Given the role of TLRs in recognizing microbial patterns and activating the innate immune system, we focused on this receptor family. Human peripheral monocytes were activated with ligands known to trigger specific TLRs: Mycobacterium tuberculosis 19 kDa lipopeptide (TLR2/1)5,10, MALP-2 (TLR2/6), poly(I:C) (TLR3), LPS (TLR4), flagellin (TLR5), imiquimod (TLR7), and bacterial CpG (TLR9). At time 0, less than 2% of the peripheral monocytes expressed DC-SIGN or CD1b. However, after 48 h, ligands for TLR2/1, TLR2/6, TLR4, and TLR5 induced expression of both DC-SIGN (18–53%) and CD1b (5–23%) (Fig.1b). Response to these ligands correlated with the TLR expression pattern of human peripheral monocytes27.
When TLR-activated monocytes were double-labeled with DC-SIGN and CD1b, two major and distinct populations of cells were identified: DC-SIGN+CD1b− and DC-SIGN−CD1b+ (Fig. 1c). In addition, a third population of DC-SIGN+CD1b+ were variably identified in vitro (1–9%), but never observed in lymphoid tissue. In contrast, after cotreatment with rGM-CSF and rIL-4, 80–90% of cells were DC-SIGN+CD1b+ (Fig. 1c). These data indicate that DC-SIGN+ cells and CD1b+ cells represent two distinct subsets in vivo, and can be rapidly induced by TLR activation of peripheral monocytes in vitro.
Mechanism of TLR-induced differentiation
To identify potential autocrine/paracrine signaling pathways that could mediate the TLR-induced differentiation, DNA microarray experiments were performed on monocytes treated with either media or the mycobacterial 19 kDa lipopeptide TLR2/1 ligand for 0, 3, 6, 12, or 24 h. Using the Database of Ligand-Receptor Partners, which contains 455 known interacting protein ligand and receptor pairs, we searched for those pairs with correlated gene expression in the TLR2/1-treated monocytes using the computational algorithm described by Graeber et. al28. Based on this analysis, 78 ligand/receptor pairs that demonstrated correlated gene expression were discovered. Focus was placed on those cytokine genes that displayed correlated gene expression and were significantly induced following TLR2/1 activation (P<0.05). These criteria identified five cytokine ligand/receptor pairs (Fig. 2a). We also identified cytokine ligand/receptor pairs that were correlatively downregulated or had an anti-correlated expression pattern (Supplementary Figure 1)
Figure 2.
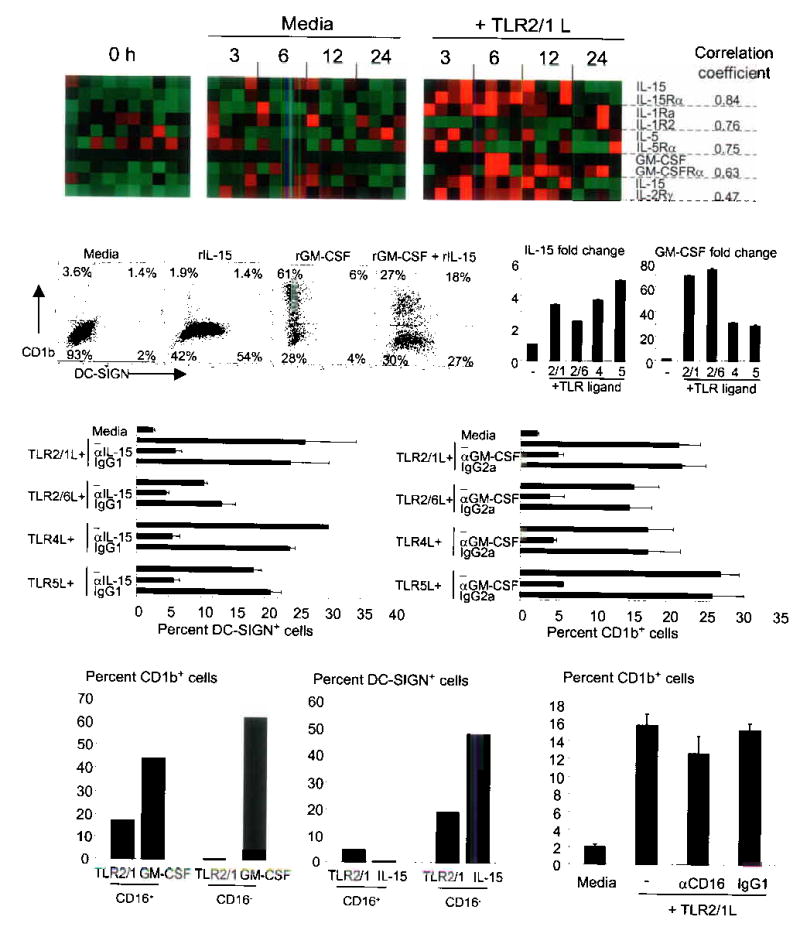
IL-15 and GM-CSF induce monocyte differentiation. (a) TLR2/1 activation triggers cytokine ligand/receptor pairs with correlated expression. Each column represents a single donor. (b) Monocytes were cultured with recombinant cytokines. Representative data shown (n=7). (c) mRNA levels of TLR-stimulated monocytes (average of duplicate wells ± SD and representative of two donors). (d) Monocytes were stimulated with TLR ligands. Data are represented as average percent positive cells ± SEM from between two and five experiments. (e) Monocyte sub-populations were activated with either the TLR2/1L or cytokines and represent data from two independent experiments. (f) Monocytes were activated with the 19 kDa TLR2/1L in various conditions. Data represent the average (n=3) ± SEM.
Monocytes stimulated with rIL-15 expressed DC-SIGN (25–60%) but not CD1b while monocytes stimulated with rGM-CSF expressed CD1b (60–80%) and very little or no DC-SIGN (Fig. 2b). Cells stimulated with both rGM-CSF and rIL-15 displayed an expression pattern similar to that found after TLR activation (Fig. 2b). rIL-1Ra and rIL-5 had no effect on the expression of DC-SIGN or CD1b (data not shown).
Identical to the pattern of DC-SIGN and CD1b induction in Fig 1b, we found that ligands for TLR 2/1, 2/6, 4, and 5 induced the expression of IL-15 and GM-CSF mRNA (Fig. 2c). Furthermore, addition of an IL-15 blocking antibody inhibited TLR-induced DC-SIGN by 60–90% (but not CD54, data not shown) and addition of a GM-CSF antibody blocked TLR-induced CD1b expression by 75–80% (Fig. 2d). Therefore, the mechanism by which TLR activation triggers differentiation of monocytes into DC-SIGN+ and CD1b+ populations is through the upregulation of cytokine/receptor pairs: IL-15/IL-15R are responsible for differentiation into DC-SIGN+ cells and GM-CSF/GM-CSFR are key for differentiation into CD1b+ cells.
Based on the expression of CD16, two distinct populations of human peripheral monocytes have been identified. TLR2/1-induced differentiation of monocytes into CD1b+ cells was restricted to the CD16+ versus CD16− subset while rGM-CSF-induced differentiation into CD1b+ cells was found in both monocyte populations (Fig. 2e and Supplementary Note). CD16 does not have a role in the differentiation process since addition of a CD16 neutralizing antibody had no effect on TLR2/1-induced CD1b expression (Fig. 2f). In contrast, the ability of TLR2/1 and rIL-15 to trigger differentiation of monocytes in DC-SIGN+ cells was enhanced in the CD16− vs. CD16+ subset.
Cells expressing DC-SIGN have a macrophage phenotype
The DC-SIGN positive cells had a macrophage-like phenotype, expressing higher levels of CD14 (TLR coreceptor), CD16 (FcγRIII), CD32 (FcγRII) and CD64 (FcγRI), even though they derived from CD16− precursors (Fig. 3a). In contrast, CD1b positive cells had an immature dendritic cell phenotype with higher expression of CD1a (antigen presentation molecule), CD206 (mannose receptor), CD86 (B7.2, costimulatory molecule), and CD40 (T cell co-stimulation) as well as low expression of CD14 and CD16 (Fig. 3a). The level of TLR2 was found to be higher on the DC-SIGN+ cells. Meanwhile, levels of MHCII (antigen presentation molecule), CD54 (ICAM-1, adhesion), CD80 (B7.1, costimulatory molecule), and CD11c (myeloid marker) were similar between the two populations while both populations were negative for the tissue macrophage marker CD163 and the mature dendritic cell markers CD83 and DEC-205. Identical expression patterns were observed upon activation with ligands for TLR2/6, TLR4, and TLR5, although we found that activation via TLR4 induced the expression of CD163 on both cell populations (Fig. 3b). Furthermore, activation of monocytes with a second TLR2/1 ligand, the tri-acylated lipopeptide Pam3CSK4, also resulted in distinct DC-SIGN+ and CD1b+ cell populations with a similar expression profile (Supplementary Figure 2). Similar cell surface expression profiles were observed when monocytes were cultured with rIL-15 or rGM-CSF, allowing us to use recombinant cytokines in subsequent experiments to differentiate monocytes into the distinct populations found following activation with TLR ligands (data not shown). These data indicate that TLR activation induces two distinct cell types; cells expressing DC-SIGN have a macrophage-like phenotype, while those expressing CD1b have an immature dendritic cell phenotype.
Figure 3.
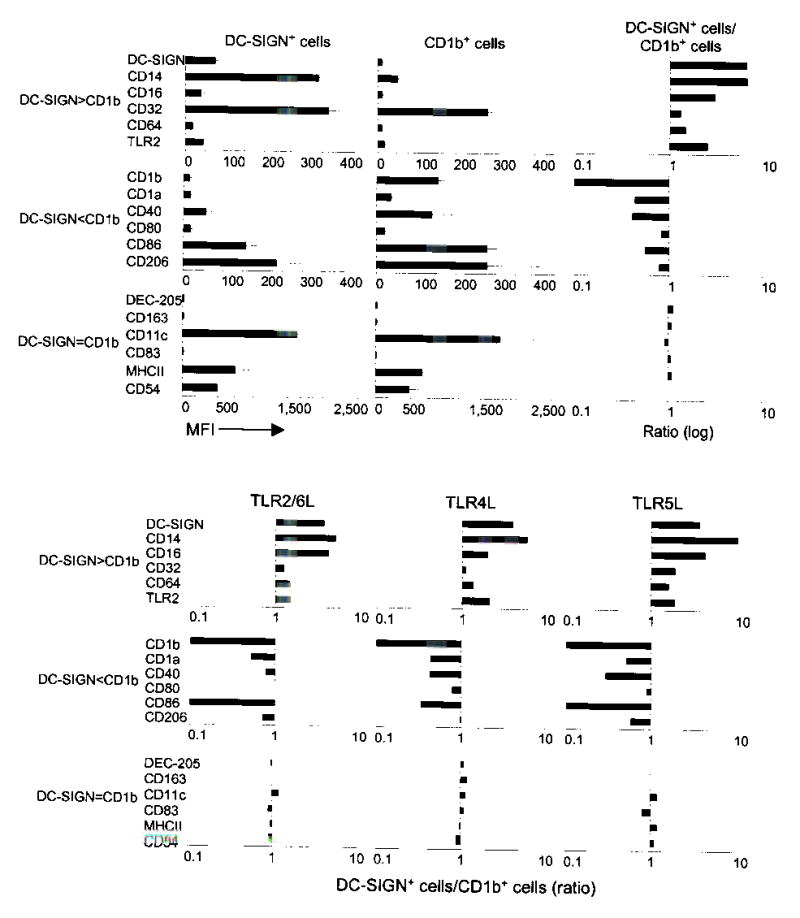
DC-SIGN+ cells have a macrophage phenotype while CD1b+ cells have a dendritic cell phenotype. (a) Monocytes stimulated with the 19 kDa TLR2/1 ligand were double labeled with DC-SIGN or CD1b together with the indicated markers. Data are shown as mean fluorescence intensity (MFI) of at least four independent experiments ± SEM (left and center). Data are also represented as a ratio of expression (right) on DC-SIGN+ cells versus CD1b+ cells. (b) Monocytes were stimulated with TLR ligands, labeled as in (a) and are represented as a ratio of expression based on two to three independent experiments.
DC-SIGN+ macrophages bind and phagocytose mycobacteria
DC-SIGN has been implicated in the direct binding of mycobacteria to DC-SIGN19,18. We therefore compared the ability of DC-SIGN+ macrophages and CD1b+ dendritic cells to bind Bacille Calmette-Guerin (BCG). Monocytes were differentiated with rGM-CSF or rIL-15 and then cultured with BCG expressing GFP. We found that across a multiplicity of infection (MOI) between 10 and 40, approximately 3-4% of DC-SIGN+ cells bound BCG while only 0.5–1.2% of CD1b+ cells bound BCG (Fig. 4a). This binding was blocked by 40–60% in the presence of a DC-SIGN neutralizing antibody (Fig. 4b). These data indicate that DC-SIGN facilitates the ability of macrophages to bind mycobacteria.
Figure 4.
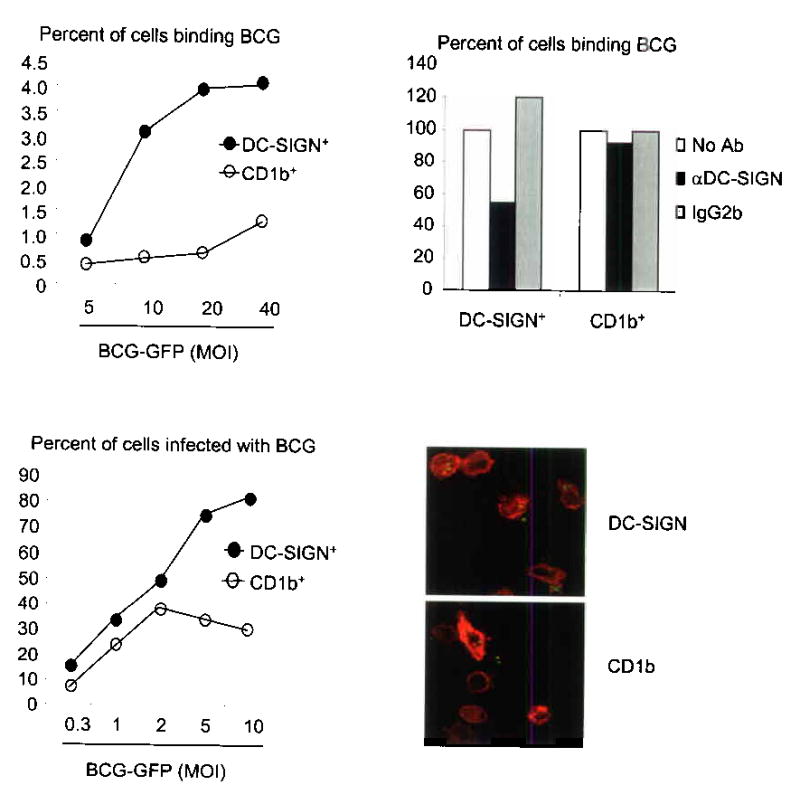
DC-SIGN+ macrophages bind and phagocytose mycobacteria. (a) Cytokine-differentiated monocytes were cultured with M. bovis BCG. Data represent percent of cells that are double positive for either DC-SIGN and BCG-GFP (filled circle) or CD1b and BCG-GFP (open circle) from two independent experiments. (b) Binding was measured in the presence of a DC-SIGN blocking antibody or controls. Data are represented as percent of binding relative to media control. (c) DC-SIGN+ and CD1b+ cells were incubated with BCG-GFP and uptake was measured by double labeling. Data is representative from two independent experiments. (d) Confocal images of cells cultured and labeled as in (c).
Since binding of the bacteria to the cell is necessary but not sufficient for phagocytosis, the ability of DC-SIGN+ and CD1b+ cells to phagocytose BCG was compared by incubating the different cell populations with mycobacteria for 18 h. Analogous to the binding experiments, DC-SIGN+ cells were more phagocytic than CD1b+ cells, particularly at a MOI of five or 10, at which up to 80% of the DC-SIGN+ cells had taken up BCG compared to 30% of the CD1b+ cells (Fig. 4c). Finally, by confocal laser microscopy, BCG was demonstrated to be intracellular, and not bound simply to the cell membrane (Fig. 4d). Together, these data suggest that DC-SIGN+ macrophages are more efficient at the binding and phagocytosis of mycobacteria than immature dendritic cells.
CD1b+ dendritic cells produce cytokine and activate T-cells
After TLR activation, CD1b+ cells were more potent producers of cytokine as compared to DC-SIGN+ cells, secreting 14-fold more IL-12p40 and six-fold more TNF-α (Fig. 5a). Neither cell subset produced measurable levels of IL-10 (data not shown).
Figure 5.
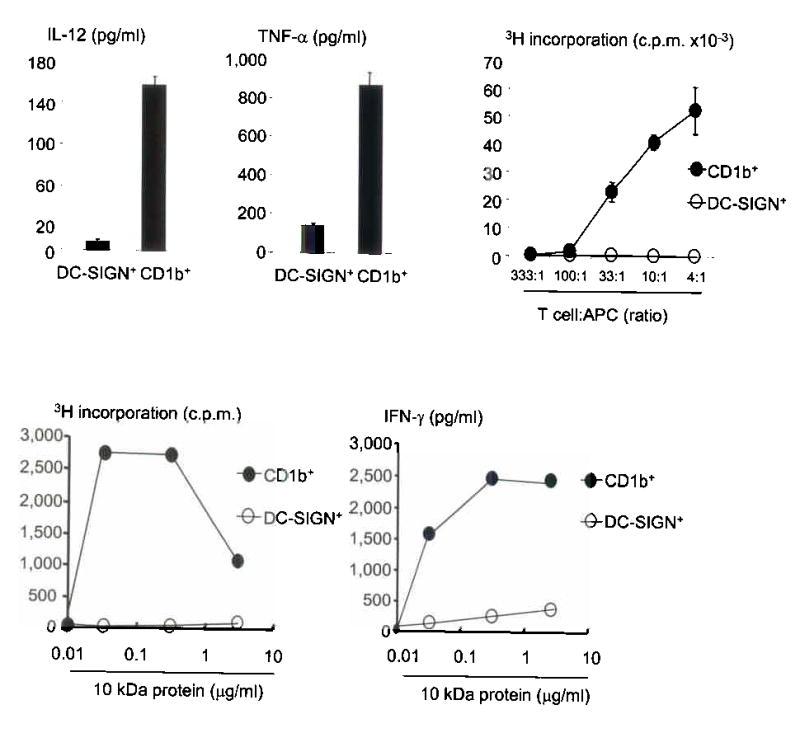
CD1b+ dendritic cells are producers of cytokine and potent T-cell activators. (a) Cytokine-differentiated monocytes were enriched for cells expressing DC-SIGN or CD1 and stimulated with the TLR2/1 ligand. Data represent the average of triplicate wells of two independent experiments ± SEM. (b) We obtained cells as in (a), plated them with un-matched T cells and measured proliferation (CD1b+ cells, filled circle; DC-SIGN+ cells, open circle). Data are representative of two independent experiments. (c) Antigen presenting cells added together with various concentrations of peptide and a class II-restricted T cell line, D103.5. Data are representative of two independent experiments.
In a mixed lymphocyte reaction CD1b+ cells were more potent than DC-SIGN+ cells, inducing between 10–700 fold greater T cell proliferation across a range of T cell:APC ratios (Fig. 5b). The ability of CD1b+ dendritic cells and DC-SIGN+ macrophages to present antigen to an MHCII-restricted T cell clone, D103.5, which recognizes a peptide from the M. leprae 10 kDa, GroES, was also tested. Despite having similar levels of MHCII on the cell surface, CD1b+ cells triggered approximately 20- to 50-fold more T cell proliferation and 10-fold more production of IFN-γ compared to DC-SIGN+ cells (Fig. 5c). These results suggest that CD1b+ dendritic cells are substantially more potent activators of adaptive T cell responses than DC-SIGN+ macrophages.
Expression of DC-SIGN and CD1b in human leprosy
To understand the significance of TLR-induced monocyte differentiation in a disease model we investigated the capacity of peripheral monocytes from leprosy patients to differentiate following activation with the mycobacterial TLR2/1 ligand. Similar to healthy donors, monocytes from T-lep patients were able to differentiate into both DC-SIGN+ macrophages (28–61%) and CD1b+ dendritic cells (8–10%) (Fig. 6a). Strikingly, while peripheral monocytes from L-lep patients differentiated into DC-SIGN+ macrophages (33–66%), they failed to differentiate into CD1b+ dendritic cells (1–3%) (Fig. 6a). However in those L-lep patients undergoing reversal reaction, both DC-SIGN+ macrophages (38–61%) and CD1b+ dendritic cells (6–25%) were detected at frequencies similar to T-lep patients. In all patients, cytokine-stimulated monocytes were able to differentiate into CD1b+ dendritic cells (Fig. 6b and Supplementary Figure 3). Clearly, peripheral monocytes from L-lep patients activated through TLR receptors are able to differentiate into DC-SIGN+ macrophages but not CD1b+ dendritic cells.
Figure 6.
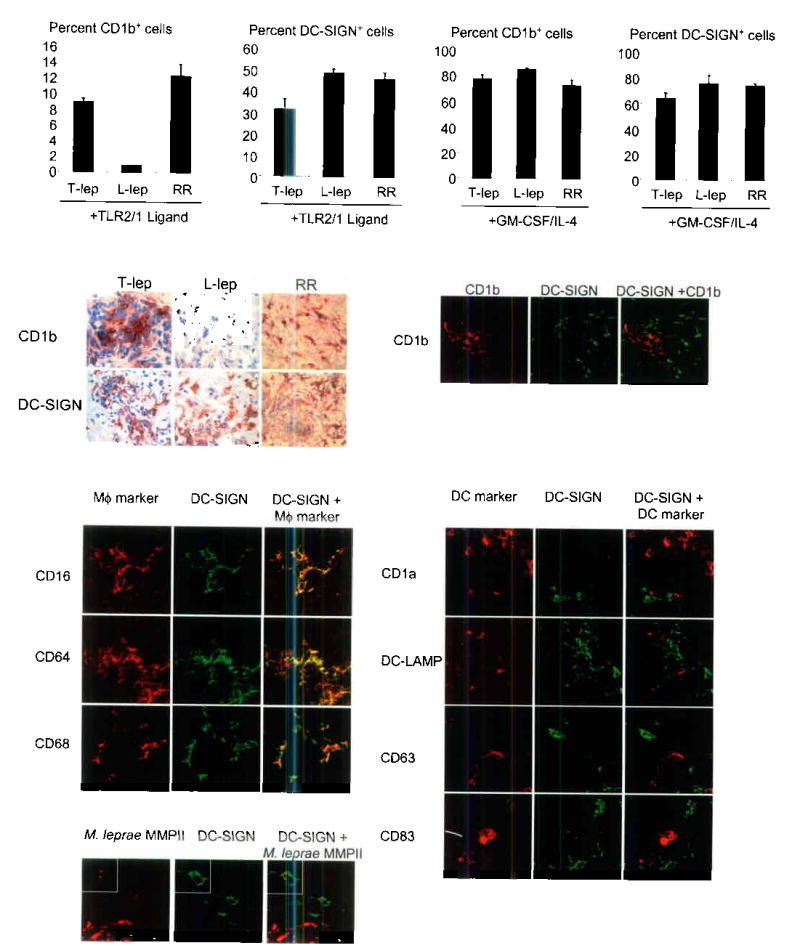
Macrophage and dendritic cell subsets in leprosy. We stimulated monocytes with either (a) the 19 kDa TLR2/1L or (b) rGM-CSF and rIL-4. Data is represented as cells expressing either DC-SIGN or CD1b relative to unactivated control ± SEM (L-lep, n=6, T-lep, n=4, RR, n=6). (c) We labeled skin biopsy sections from leprosy lesions by the immunoperoxidase method. (d) Immunofluorescence confocal images from T-lep lesions. (e) Two color confocal images with macrophage markers. (f) Confocal images with dendritic cell makers. (g) Two color confocal images from L-lep lesions for the M. leprae bacterioferritin major membrane protein II (MMPII) and DC-SIGN.
The in vivo relevance of these differentiation pathways was tested according to the expression of DC-SIGN and CD1b in leprosy skin lesions. Both DC-SIGN+ and CD1b+ cells were present in T-lep lesions (Fig. 6c). In contrast, DC-SIGN+ but not CD1b+ cells were found in L-lep lesions. However in those L-lep patients undergoing reversal reaction, both DC-SIGN+ and CD1b+ cells were detected at frequencies similar to T-lep lesions, consistent with the observed gain of cell-mediated immunity and local CD4+ Th1 responses.
Further examination of leprosy skin lesions using confocal laser microscopy revealed that within T-lep lesions, DC-SIGN and CD1b are expressed on distinct, non-overlapping cell populations (Fig. 6d). The DC-SIGN+ cells were found to express the monocyte/macrophage markers CD14, CD16, CD64 and CD68 (Fig. 6e), but did not express the dendritic cells markers CD1a, CD1b, CD63, CD83, or DC-LAMP (Fig. 6f). Our analysis of T-lep lesions revealed only a small number (<5%) of DC-SIGN+ cells that were positive for the dendritic cell markers examined. Identical results were obtained during our analysis of human tonsil (Supplementary Figure 4). These data indicate that the majority of DC-SIGN+ cells express cell surface markers typically found on monocytes/macrophages.
To ascertain whether DC-SIGN+ macrophages identified in L-lep lesions contained M. leprae, lesions were double-labeled using monoclonal antibodies against DC-SIGN and the M. lepare 22 kDa bacterioferritin major membrane protein II (MMPII). Confocal laser microscopy revealed that indeed DC-SIGN+ macrophages containing M. leprae were found in L-lep lesions (Fig. 6g). However, M. leprae was also found in CD16+DC-SIGN− cells, indicating that there are different subsets of macrophages in leprosy lesions. As expected, mycobacteria were not detected in T-lep lesions. These data indicate important differences between the forms of leprosy: T-lep lesions contain DC-SIGN+ macrophages and CD1b+ dendritic cells, whereas, L-lep lesions contained M. leprae-infected DC-SIGN+ macrophages but lacked CD1b+ dendritic cells.
Discussion
The innate immune system triggers both direct and indirect effector pathways to combat microbial pathogens. First, the innate immune response acts directly to localize the invading pathogen so it can be destroyed by antimicrobial mechanisms. Second, the innate immune response acts indirectly, through cytokine release and upregulation of costimulatory molecules, to induce adaptive T and B cell response. Here we demonstrate that activation of Toll-like receptors causes the rapid differentiation of distinct precursors of human peripheral monocytes into DC-SIGN+ and CD1b+ cells. The DC-SIGN+ cells have a macrophage-like phenotype, are phagocytic, and utilize DC-SIGN to facilitate the uptake of bacteria. In contrast, CD1b+ cells have an immature dendritic cell phenotype, release pro-inflammatory cytokines and function as efficient antigen presenting cells. These data provide evidence that the dual host defense roles of the innate immune system are mediated by the induction of two distinct phenotypic and functional populations, DC-SIGN+ macrophages and CD1b+ dendritic cells (see schematic diagram in Supplementary Figure 5). Furthermore, the DC-SIGN+ macrophages derived in vitro were identical in phenotype to the DC-SIGN+ cells found in situ in human disease lesions, CD16+, CD64+, but CD1a−, CD1b−, CD83−, providing in vivo relevance for these distinct subsets in innate immunity.
Since we previously demonstrated that M. leprae predominantly activates TLR2/1 we investigated activation of the heterodimer and whether the regulated expression of TLR2/1 in lesions correlated with the clinical form of the disease5. Strikingly, TLR2/1-activation of peripheral monocytes from L-lep patients triggered differentiation into DC-SIGN+ macrophages but not CD1b+ dendritic cells, whereas activation of monocytes from T-lep patients triggered differentiation into both effector populations (see schematic diagram in Supplementary Figure 5). Correspondingly, DC-SIGN+ macrophages but not CD1b+ dendritic cells were detected in skin lesions from L-lep patients, whereas both cell types were detected in T-lep lesions. Because M. leprae was found to be abundant in DC-SIGN+ macrophages in L-lep lesions we infer that the innate immune system can mediate its direct effect, phagocytosing the bacteria into macrophages, but is unable to mediate its indirect effect, inducing dendritic cells to stimulate the adaptive T cell response required to kill intracellular pathogens29. However, when L-lep patients upgraded their clinical presentation during “reversal reactions” characterized by a clearance of bacilli, CD1b+ cells were generated following TLR activation and found to be present in the cutaneous lesions. Since these lesions show an influx of CD4+ Th1 cells26 and dendritic cells are required to activate antigen-specific CD4+ Th1 cells, the ability of TLR activation to regulate differentiation of monocytes into CD1b+ dendritic cells is likely a key immunologic event for host defense.
Our in situ studies of leprosy lesions and human tonsil together with our in vitro studies of TLR-activated peripheral monocytes suggest that DC-SIGN+ cells have a macrophage phenotype (CD16+, CD64+) but not a dendritic cell phenotype (CD1a−, CD1b−, CD83−). This is consistent with other studies indicating that DC-SIGN is not expressed on human dendritic cells, but is found on monocytes differentiated in the presence of cyclic nucleotides and macrophages in the lymph nodes and at sites of inflammation32,33,34,35. Our data cannot preclude the possibility that the DC-SIGN+ cells may represent a subset of dendritic cells with a macrophage-like phenotype and function. Nevertheless, the role of DC-SIGN in the phagocytosis of microbes by macrophage-like cells is relevant for the studies of other infectious diseases such as HIV infection where the function of DC-SIGN mediated uptake is part of the host response36.
The mechanism by which TLR2/1 activation triggered monocyte differentiation was elucidated using gene expression data and a computational algorithm that identifies potential autocrine/paracrine loops of known receptor and ligand pairs. Together with functional studies, the data revealed two distinct regulatory mechanisms responsible for the rapid expansion of DC-SIGN+ cells and CD1b+ cells. The upregulation of GM-CSF and the GM-CSFR was responsible for the induction of CD1b. Additionally, the upregulation IL-15 and two of its receptor components, IL-15Rα and IL-2Rγ, leads to the induction of DC-SIGN, providing new information about the regulation of this C-type lectin37,32,38,39. Simultaneous addition of both rGM-CSF and rIL-15 led to the differentiation of monocytes into distinct DC-SIGN+ and CD1b+ cells, similar to the pattern we observed following TLR activation40. Our investigation of the expression and function of cytokines triggered by TLR2/1 activation demonstrates the powerful utility of using ligand/receptor gene expression-based analysis to identify novel autocrine/paracrine pathways of immune regulation.
In conclusion we show that the dual immunological functions of the innate immune system-- capturing and inhibiting pathogens, and presenting antigens to adaptive T and B cell responses-- are mediated by distinct phenotypic and functional subsets of cells. DC-SIGN+ macrophages carry out the phagocytic and presumably microbicidal or static activity and CD1b+ immature dendritic cells serve as are potent cytokine-producing antigen presenting cells. The study of human leprosy provides evidence that the regulation of these DC-SIGN+ macrophages and CD1b+ dendritic cells correlate and likely contributes to the resistant or susceptible outcome of human intracellular infection
Methods
TLR ligands.
LPS (Sigma) and MALP-2 was used as described41,42. Flagellin was a gift of Kelly Smith, University of Washington. TLR ligands were purchased: 19 kDa lipopeptide (EMC Microcollections), imiquimod (Sequioa Research), CpG (Invitrogen), Poly I:C (Amersham), and Pam3CSK4 (EMC Microcollections).
Microarrays.
We isolated RNA using TriZol reagent (Invitrogen) and prepared the probe according to the Affymetrix protocol. The UCLA Microarray Core Facility performed the hybridization to Affymetrix U133A Genechip. A total of 10 donors were used with time 0 h samples prepared for each. For timepoints, we used four of the 10 donors to prepare media and TLR2/1-stimulated samples.
Ligand-receptor pair algorithm.
The algorithm for identifying autocrine/paracrine signaling from gene expression data has been described28. Briefly, for each known cognate ligand-receptor pair from the DLRP measured on the gene microarry (434 of the 455 pairs), we determined the Pearson correlation between the ligand and receptor expression profiles.
Differentially expressed genes.
We ranked each gene by the probability that the means of its expression values are statistically distinct between media and TLR2/1 ligand treated populations using the Student’s t-test. We generally focused on genes meeting our criteria: P < 0.05 and fold change > 2.0. Diagrams were generated using the Cluster and TreeView software.
Antibodies and cytokines.
Antibodies for cell surface molecules: CD14 (Zymed), CD1a, CD1b (OKT6, Bcd3.1, ATCC), CD1a, CD68, (Dako), C11c, CD15, CD16, CD32, CD54, CD63, CD64, CD80, CD86, CD163, DC-SIGN, CD206 (BD Pharmingen), DEC-205, TLR2 (eBioscience), DC-LAMP (Immunotech), CD83, CD40, MHCII, and IgG controls (Sigma). Cytokines used: IL-4 (Peprotech), IL-5 (R&D Systems), IL-1Ra (R&D Systems), IL-15 (R&D Systems), and GM-CSF (Immunex).
Monocyte differentiation and enrichment.
We obtained whole blood from healthy donors (UCLA I.R.B. #92-10-591-31) with informed consent. We stimulated adherent monocytes5 with media, M. tb 19 kDa (10 μg/ml), Pam3CSK4 (0.1 μg/ml), Poly(I:C) (1 μg/ml), LPS (10 ng/ml), MALP-2 (10 ng/ml), flagellin (1–10 ng/ml), Imiquimod (5 μg/ml), CpG DNA (1 μg/ml), rGM-CSF (1–10 U/ml), rIL-15 (200 ng/ml), rIL-1Ra (200 ng/ml), or rIL-5 (200 ng/ml) for 48 h. To enrich for DC-SIGN+ or CD1b+ cells, we cultured monocytes with rIL-15 or rGM-CSF and labeled with a DC-SIGN or CD1 antibody. A MACS microbead-secondary antibody (Miltenyi Biotec) was added for 30 min at 4 °C. Flow-through was designated the negative population. Remaining cells were designated the positive population.
To study precursor populations, monocytes were enriched from blood using Rosette Sep monocyte enrichment cocktail (StemCell Technologies). CD16+ monocytes were isolated with a CD16 antibody (BD Pharmingen) and α-mouse IgG1-MACS beads (Miltenyi Biotech).
Patients and clinical specimens.
Patients with leprosy were classified according to the criteria of Ridley and Jopling43 and obtained as previously described5 (I.R.B., University of Southern California School of Medicine).
Cell surface labeling.
Surface expression was determined using specific antibodies. A PE-conjugated secondary antibody was used for CD1b. Cells were acquired and analyzed as described5. For blocking, monocytes were stimulated with TLR ligands in the presence of media, an IL-15 (R&D Systems) or GM-CSF antibody (BD Pharmingen) or an isotype control.
Cytokine secretion.
Monocytes were cultured with either recombinant IL-15 or GM-CSF for 48 h and enriched as above. Cells were activated with the TLR2/1 ligand for 24 h. IL-12 and TNF-α levels were measured by ELISA (Pharmingen).
T cell assays.
For the mixed lymphocyte reaction, DC-SIGN+ and CD1b+ cells were obtained as above, irradiated, and plated (3.3× 102 to 2.5× 105 cells/well). T cells from an unmatched donor were obtained with Rosette Sep T cell enrichment cocktail (StemCell Technologies). Media or 2× 105 T cells were added. IFN-γ levels were measured by ELISA (BD Pharmingen). Proliferation was measured as described14.
For MHCII studies, monocytes were differentiated, enriched and irradiated as above. Cells were cultured with MHCII (D103.5) restricted T-cells (1× 105) and the 10 kDa Ag derived from M. leprae as previously described44. IFN -γ and proliferation was measured.
Binding and phagocytosis.
DC-SIGN+ and CD1b+ cells were cytokine-derived as above. For binding, BCG-GFP was added at 4 °C for 4 h in antibiotic-free media. Cells were harvested, washed, and labeled with specific antibody. For blocking, cells were pre-treated with media, an IgG2b isotype, or a DC-SIGN-specific antibody (BD Pharmingen) (1 h) at 4 °C. For phagocytosis, we cultured cells with BCG (16 h) and performed double labeling.
Real time qPCR.
Monocytes were stimulated with either media or 19 kDa for 3 h. We isolated RNA and synthesized cDNA as described5. GM-CSF primers: forward 5’-GCCTCACCAAGCTCAAGGG-3’, reverse 5’-GGAGGGCAGTGCTGTTTGTAG-3’. IL-15 primers: forward 5’-TGTAGGAGGCATCGTGGATG-3’, reverse 5’-CCTTAAGTATTGAAGAAGAGCTGGCT-3’. Reactions use Sybr Green PCR Master Mix (BioRad). The relative quantities of GM-CSF and IL-15 per sample and normalization were calculated as described45.
Immunoperoxidase and immunofluorescence.
Immunoperoxidase-labeling was performed as described5. Double immunofluorescence was performed by serially incubating sections with antibodies against DC-SIGN followed by an isotype-specific fluorochrome (Caltag). Sections were washed, incubated with antibodies for CD1a, CD1b, CD16, CD63, CD64, CD68, CD83, DC-LAMP, or M. leprae 22 kDa for 1 h followed by a TRITC-conjugated secondary (Southern Biotechnology) and examined as described46.
URLs.
DLRP, http://dip.doe-mbi.ucla.edu/drlp; Cluster and TreeView software (http://rana.lbl.gov/).
Supplementary Material
Acknowledgments
We would like to thank P. Brennan (Colorado State University and NIAID Contract NO1 AI-75320) for the generous gift of the M. leprae bacterioferritin major membrane protein II (MMPII) monoclonal antibody. We would also like to thank K. Grossheider for her technical help. This work was supported in part by grants from the National Institutes of Health (AI07126, AI22553, AI47866 ).
References
- 1.Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–983. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 2.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to staphylococcus aureus infection. J Immunol. 2000;165:5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 3.O’Brien AD, et al. Genetic control of susceptibility to Salmonella typhimurium in mice: role of the LPS gene. J Immunol. 1980;124:20–24. [PubMed] [Google Scholar]
- 4.Alexopoulou L, et al. Hyporesponsiveness to vaccination with Borrelia burgdorferi OspA in humans and in. Nat Med. 2002;8:878–884. doi: 10.1038/nm732. [DOI] [PubMed] [Google Scholar]
- 5.Krutzik SR, et al. Activation and regulation of Toll-like receptors 2 and 1 in human leprosy. Nat Med. 2003;9:525–532. doi: 10.1038/nm864. [DOI] [PubMed] [Google Scholar]
- 6.Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 7.Doyle SE, et al. Toll-like Receptors Induce a Phagocytic Gene Program through p38. J Exp Med. 2004;199:81–90. doi: 10.1084/jem.20031237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thoma-Uszynski S, et al. Induction of direct antimicrobial activity through mammalian toll-like receptors. Science. 2001;291:1544–1547. doi: 10.1126/science.291.5508.1544. [DOI] [PubMed] [Google Scholar]
- 9.Medzhitov R, Preston-Hurlburt P, Janeway CAJ. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 10.Brightbill HD, et al. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science. 1999;285:732–736. doi: 10.1126/science.285.5428.732. [DOI] [PubMed] [Google Scholar]
- 11.Hertz CJ, et al. Microbial lipopeptides stimulate dendritic cell maturation via TLR2. J Immunol. 2000;166:2444–2450. doi: 10.4049/jimmunol.166.4.2444. [DOI] [PubMed] [Google Scholar]
- 12.Porcelli SA, Modlin RL. The CD1 system: antigen-presenting molecules for T cell recognition of lipids and glycolipids. Annu Rev Immunol. 1999;17:297–329. doi: 10.1146/annurev.immunol.17.1.297. [DOI] [PubMed] [Google Scholar]
- 13.Beckman EM, et al. Recognition of a lipid antigen by CD1-restricted αβ+ T cells. Nature. 1994;372:691–694. doi: 10.1038/372691a0. [DOI] [PubMed] [Google Scholar]
- 14.Sieling PA, et al. CD1-restricted T cell recognition of microbial lipoglycans. Science. 1995;269:227–230. doi: 10.1126/science.7542404. [DOI] [PubMed] [Google Scholar]
- 15.Moody DB, et al. Structural requirements for glycolipid antigen recognition by CD1b-restricted T cells. Science. 1997;278:283–286. doi: 10.1126/science.278.5336.283. [DOI] [PubMed] [Google Scholar]
- 16.Engering A, et al. The dendritic cell-specific adhesion receptor DC-SIGN internalizes antigen for presentation to T cells. J Immunol. 2002;168:2118–2126. doi: 10.4049/jimmunol.168.5.2118. [DOI] [PubMed] [Google Scholar]
- 17.Geijtenbeek TB, et al. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell. 2000;100:575–585. doi: 10.1016/s0092-8674(00)80693-5. [DOI] [PubMed] [Google Scholar]
- 18.Geijtenbeek TB, et al. Mycobacteria target DC-SIGN to suppress dendritic cell function. J Exp Med. 2003;197:7–17. doi: 10.1084/jem.20021229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tailleux L, et al. DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. J Exp Med. 2003;197:121–127. doi: 10.1084/jem.20021468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Appelmelk BJ, et al. Cutting edge: carbohydrate profiling identifies new pathogens that interact with dendritic cell-specific ICAM-3-grabbing nonintegrin on dendritic cells. J Immunol. 2003;170:1635–1639. doi: 10.4049/jimmunol.170.4.1635. [DOI] [PubMed] [Google Scholar]
- 21.Cambi A, et al. The C-type lectin DC-SIGN (CD209) is an antigen-uptake receptor for Candida albicans on dendritic cells. Eur J Immunol. 2003;33:532–538. doi: 10.1002/immu.200310029. [DOI] [PubMed] [Google Scholar]
- 22.Colmenares M, Puig-Kroger A, Pello OM, Corbi AL, Rivas L. Dendritic cell (DC)-specific intercellular adhesion molecule 3 (ICAM-3)-grabbing nonintegrin (DC- SIGN, CD209), a C-type surface lectin in human DCs, is a receptor for Leishmania amastigotes. J Biol Chem. 2002;277:36766–36769. doi: 10.1074/jbc.M205270200. [DOI] [PubMed] [Google Scholar]
- 23.Wemambu SNC, Turk JL, Waters MFR, Rees RJW. Erythema nodosum leprosum: a clinical manifestation of the Arthus phenomenon. Lancet. 1969;2:933–935. doi: 10.1016/s0140-6736(69)90592-3. [DOI] [PubMed] [Google Scholar]
- 24.Salgame P, et al. Differing lymphokine profiles of functional subsets of human CD4 and CD8 T cell clones. Science. 1991;254:279–282. doi: 10.1126/science.254.5029.279. [DOI] [PubMed] [Google Scholar]
- 25.Yamamura M, et al. Defining protective responses to pathogens: cytokine profiles in leprosy lesions. Science. 1991;254:277–279. doi: 10.1126/science.254.5029.277. [DOI] [PubMed] [Google Scholar]
- 26.Yamamura M, et al. Cytokine patterns of immunologically mediated tissue damage. J Immunol. 1992;149:1470–1475. [PubMed] [Google Scholar]
- 27.Kadowaki N, et al. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med. 2001;194:863–869. doi: 10.1084/jem.194.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Graeber TG, Eisenberg D. Bioinformatic identification of potential autocrine signaling loops in cancers from gene expression profiles. Nat Genet. 2001;29:295–300. doi: 10.1038/ng755. [DOI] [PubMed] [Google Scholar]
- 29.Ochoa MT, et al. T-cell release of granulysin contributes to host defense in leprosy. Nat Med. 2001;7:174–179. doi: 10.1038/84620. [DOI] [PubMed] [Google Scholar]
- 30.Stenger S, Niazi KR, Modlin RL. Down-regulation of CD1 on antigen-presenting cells by infection with Mycobacterium tuberculosis. J Immunol. 1998;161:3582–3588. [PubMed] [Google Scholar]
- 31.Giuliani A, et al. Influence of Mycobacterium bovis bacillus Calmette Guerin on in vitro induction of CD1 molecules in human adherent mononuclear cells. Infect Immun. 2001;69:7461–7470. doi: 10.1128/IAI.69.12.7461-7470.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soilleux EJ, et al. Constitutive and induced expression of DC-SIGN on dendritic cell and macrophage subpopulations in situ and in vitro. J Leukoc Biol. 2002;71:445–457. [PubMed] [Google Scholar]
- 33.Chehimi J, et al. HIV-1 transmission and cytokine-induced expression of DC-SIGN in human monocyte-derived macrophages. J Leukoc Biol. 2003;74:757–763. doi: 10.1189/jlb.0503231. [DOI] [PubMed] [Google Scholar]
- 34.van Lent PL, et al. Expression of the dendritic cell-associated C-type lectin DC-SIGN by inflammatory matrix metalloproteinase-producing macrophages in rheumatoid arthritis synovium and interaction with intercellular adhesion molecule 3-positive T cells. Arthritis Rheum. 2003;48:360–369. doi: 10.1002/art.10786. [DOI] [PubMed] [Google Scholar]
- 35.Giordano D, Magaletti DM, Clark EA, Beavo JA. Cyclic nucleotides promote monocyte differentiation toward a DC-SIGN+ (CD209) intermediate cell and impair differentiation into dendritic cells. J Immunol. 2003;171:6421–6430. doi: 10.4049/jimmunol.171.12.6421. [DOI] [PubMed] [Google Scholar]
- 36.Geijtenbeek TB, et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100:587–597. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- 37.Relloso M, et al. DC-SIGN (CD209) expression is IL-4 dependent and is negatively regulated by IFN, TGF-beta, and anti-inflammatory agents. J Immunol. 2002;168:2634–2643. doi: 10.4049/jimmunol.168.6.2634. [DOI] [PubMed] [Google Scholar]
- 38.Judge AD, Zhang X, Fujii H, Surh CD, Sprent J. Interleukin 15 controls both proliferation and survival of a subset of memory-phenotype CD8(+) T cells. J Exp Med. 2002;196:935–946. doi: 10.1084/jem.20020772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schluns KS, Williams K, Ma A, Zheng XX, Lefrancois L. Cutting edge: requirement for IL-15 in the generation of primary and memory antigen-specific CD8 T cells. J Immunol. 2002;168:4827–4831. doi: 10.4049/jimmunol.168.10.4827. [DOI] [PubMed] [Google Scholar]
- 40.Pulendran B, et al. Dendritic cells generated in the presence of GM-CSF plus IL-15 prime potent CD8+ Tc1 responses in vivo. Eur J Immunol. 2004;34:66–73. doi: 10.1002/eji.200324567. [DOI] [PubMed] [Google Scholar]
- 41.Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J Immunol. 2000;165:618–622. doi: 10.4049/jimmunol.165.2.618. [DOI] [PubMed] [Google Scholar]
- 42.Takeuchi O, et al. Cutting edge: Preferentially the R-stereoisomer of the mycoplasmal lipopeptide macrophage-activating lipopeptide-2 activates immune cells through a toll-like receptor 2- and MyD88-dependent signaling pathway. J Immunol. 2000;164:554–557. doi: 10.4049/jimmunol.164.2.554. [DOI] [PubMed] [Google Scholar]
- 43.Ridley DS, Jopling WH. Classification of leprosy according to immunity. A five-group system. Int J Lepr. 1966;34:255–273. [PubMed] [Google Scholar]
- 44.Kim J, et al. Determinants of T cell reactivity to the Mycobacterium leprae GroES homologue. J Immunol. 1997;159:335–343. [PubMed] [Google Scholar]
- 45.Modlin RL, et al. Suppressor T lymphocytes from lepromatous leprosy skin lesions. J Immunol. 1986;137:2831–2834. [PubMed] [Google Scholar]
- 46.Stenger S, et al. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science. 1998;282:121–125. doi: 10.1126/science.282.5386.121. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.