

Abstract
We previously reported that acetaminophen (APAP) caused apoptosis of C6 glioma cells. Therefore, we hypothesized that the level of p53, which usually stimulates apoptosis, might be increased after APAP exposure. However, APAP exposure for 24 h markedly decreased the p53 content and its down-stream target p21 in a concentration-dependent manner. Reduction of p53 was not accompanied by a decrease in p53 mRNA in C6 glioma cells, suggesting that p53 was mainly affected at the protein level. Unexpectedly, APAP stimulated phosphorylation of p53 at Ser15, Ser20 and Ser37, which usually elevates p53 content. However, phosphorylation of these residues did not prevent APAP-induced decrease in p53. The p53 reduction was independent from the level of phospho-Akt, which is known to promote p53 degradation. Immunoblot analysis of the immunoprecipitated p53 revealed that increased amounts of mdm2 and ubiquitin were bound to p53 during its degradation. Lactacystin and MG132, inhibitors of proteasomal proteolysis, prevented the decrease, supporting the proteasomal degradation of p53 upon APAP exposure. Pretreatment with chlormethiazole, an inhibitor of ethanol-inducible CYP2E1, significantly lowered the CYP2E1 enzyme activity and the rate of APAP-induced cell death while it prevented the reduction of p53 and p21 in C6 glioma cells. A non-toxic analog of APAP (4-hydroxyacetanilide), 3-hydroxyacetanilde, did not reduce p53 and p21 contents in C6 glioma cells and LLC-PK1 porcine kidney cells. Taken together, our results show that APAP, or its reactive metabolite(s), can directly reduce the p53 content through mdm2-mediated ubiquitin conjugation, despite phosphorylation of p53 at its N-terminus.
Abbreviations: APAP, acetaminophen; CYP2E1, ethanol-inducible cytochrome P450 2E1 isoform; CMZ, chlormethiazole; DMSO, dimethyl sulfoxide; 5-FU, 5-fluorouracil; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HRP, horse radish peroxidase; mdm2, murine double minute 2; NAPQI, N-acetyl-p-benzoquinoneimine; PBS, phosphate buffered saline
Introduction
Acetaminophen (APAP, paracetamol, 4-hydroxyacetanilide, N-acetyl-p-aminophenol) is a widely used analgesic drug and generally considered safe under therapeutic doses. However, it is known that simultaneous exposure to alcohol and APAP can cause severe liver damage (Seeff et al., 1986; Whitcomb and Block, 1994). Acute exposure of large doses of APAP can also cause liver damage (Thomas, 1993) while APAP toxicities caused by overdose and long-term addictive usage are not rare (Shayiq et al., 1999; Lee, 2004). Although N-acetyl-p-benzoquinoneimine (NAPQI), a toxic metabolite of APAP produced by various cytochrome P450 isozymes such as CYP2E1, CYP1A2, and CYP3A (Thomas, 1993; Sinclair et al., 2000), is believed to cause the toxicity, the mechanism for APAP-induced toxicity is still under investigation. Despite strong correlations between the levels of NAPQI-protein adducts and the severity and regional location of the tissue damage (Cohen et al., 1997), other factors such as lipid peroxidation, Ca2+ homeostasis, reactive oxygen and nitrogen species (Knight et al., 2002), may also play a role in APAP-mediated toxicity. Recent data suggest that mechanisms other than NAPQI may also be important in causing toxicity, since NAPQI-protein binding was still observed after hepatic damage was significantly alleviated (Michael et al., 1999), and that 3-hydroxyacetanilide, a non-cytotoxic analog of APAP, also exhibited patterns of protein binding similar to that of NAPQI (Myers et al., 1991; Fountoulakis et al., 2000).
The p53 gene encodes a 393-amino acid protein which possesses five conserved regions: an N-terminal transcriptional activation domain, a SH3-domain, a specific DNA binding domain, a C-terminal oligomerization domain, and a basic domain. p53 protein is a transcription factor which plays a critical role in regulating cell growth, DNA repair and apoptosis in response to stressful conditions (Lakin and Jackson, 1999; Vogelstein et al., 2000). However, p53 is a short-lived protein under complex regulation including reversible cycles of post-translational modification such as phosphorylation, acetylation, and ubiquitination. Under physiological conditions, p53 can be conjugated with ubiquitin, primarily by the murine double minute (mdm2) protein (Lakin and Jackson, 1999; Ogawara et al., 2002), a major ubiquitin ligase for p53, leading to its degradation in an ubiquitin-dependent manner. After DNA damage or under apoptotic conditions, p53 is phosphorylated at several sites including Ser15 and Ser37 near the binding site for mdm2. Phosphorylation of p53 at its N-terminus reduces the interaction between p53 and mdm2, resulting in p53 stabilization due to decreased ubiquitin binding. Depending on the severity of DNA damage from various stressful conditions, p53 is activated to induce growth arrest to allow cells to repair damaged DNA. When DNA damage is too severe and thus impossible to be repaired, p53 can promote apoptosis to eliminate damaged cells (Giaccia and Kastan, 1998; Vogelstein et al., 2000). Growth arrest induced by p53 was shown to be mediated by the down-stream targets of p53 such as p21 and Gadd45, both of which regulate cell cycling through modulation of cyclin dependent kinases and its regulatory proteins (Zhan et al., 1998). Alternatively, p53 could promote cell death by up-regulating pro-apoptotic Bax protein upon withdrawal of a neurotrophic factor (Aloyz et al., 1998). Furthermore, a p53-mdm2 feedback loop exists to control the levels of p53 and mdm2 (Lev Bar-Or et al., 2000).
We previously reported that APAP can cause apoptosis (Bae et al., 2001) and oxidative DNA damage in C6 glioma cells (Wan et al., 2004). Therefore, we hypothesized that the level of p53 would be up-regulated by APAP, similar to the levels observed after exposure to chemotherapeutic agents and other DNA damaging agents (Vogelstein et al., 2000) because APAP can induce apoptosis (Bae et al., 2001), and wild type p53 is present in C6 glioma cells (Asai et al., 1994). Contrary to this hypothesis, APAP treatment markedly decreased the level of p53. Therefore, in this study, we investigated the mechanism of p53 reduction, and determined the level of p21, a down-stream target of p53, upon exposure to APAP. We also studied whether phosphorylation of p53 at its N-terminus affects the APAP-induced reduction of p53 protein. Finally, we also studied the differential effects of APAP and its non-toxic analogue 3-hydroxyacetanilide on the p53 levels in C6 glioma and LLC-PK1 porcine kidney cells.
Materials and Methods
Materials
C6 glioma and LLC-PK1 cells were obtained from American Type Culture Collection (Manassas, VA). Rabbit polyclonal antibody against p21, Akt, phospho-Akt, mdm2, goat polyclonal antibody against p53 or actin, and secondary anti-rabbit IgG or anti-goat IgG conjugated with horse radish peroxidase (HRP) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against phospho-p53 at various sites and APAP were from Cell Signaling Technology (Beverly, MA) and Biogenesis (Brentwood, NH), respectively. Dimethyl sulfoxide (DMSO, cell culture grade), APAP, 3-hydroxyacetanilide, polyclonal antibody against ubiquitin were from Sigma (St. Louis, MO). Cell culture media, antibiotics, Trizol, and protein G-bound agarose were purchased from Invitrogen (Carlsbad, CA). Lactacystin and MG132 were obtained from Biomol (Plymouth Meetings, PA). Chlormethiazole (CMZ) was kindly provided by Dr. Joong-Ick Yang at Dong-A Pharmaceutical Company, Seoul, Korea.
Cell Culture and Immunoblot Analyses of Various Proteins
C6 glioma or LLC-PK1 cells (1x 107 cells/150 mm-diameter culture dish) were grown in Dulbecco’s modified Eagle’s medium with 10% heat-inactivated fetal bovine serum and antibiotics before treatment with different concentrations (up to 5 mM) of APAP or 3-hydroxyacetanilide, as described (Bae et al., 2001). To prepare proteins for immunoblot analyses, harvested C6 and LLC-PK1 cells were homogenized in the lysis buffer as described (Bae et al., 2001). Cell debris and particulate fractions were removed by centrifugation at 5,000 x g for 10 min at 4 °C. Equal amounts of protein in the 5,000 x g supernatant fractions or whole homogenates were separated by 10% or 12% SDS-PAGE, transferred onto PVDF-Immobilon membranes, and subjected to immunoblot analysis using the respective antibody against: p53, phospho-p53, Akt, phospho-Akt, mdm2, p21, actin, or ubiquitin. Immunoreactive proteins were subsequently detected with appropriate secondary antibodies conjugated with HRP and enhanced chemiluminescence kits.
RT-PCR Analysis for p53 mRNA Expression
Total RNA was isolated by using the Trizol reagent kit. Purity and concentration of RNA were determined by measuring UV absorbance at 260 and 280 nm. RT-PCR was performed using SuperScript™ one-step RT-PCR kit (Invitrogen) following the manufacturer’s instruction. Total RNA (400 ng/assay) was used for each RT-PCR using a PE GeneAmp PCR system 9700: one cycle of reverse transcription at 37 °C for 30 min, 94 °C for 2 min, followed by 26 cycles of PCR at 94 °C (20 s), 55 °C (45 s), and 68 °C (60 s). DNA sequences of the oligonucleotide primer set for rat p53 mRNA (Soussi et al., 1988) were: sense, 5’- TCTGTCATCTTCCGTCCCTTC TC-3’ and anti-sense, 5’-AACACGAACCTCAA AGCTGTCCCG-3’. The primers used for amplification of glyceraldehyde 3-phosphate dehydrogenase (GAPDH, 194 bp) transcript were the same as described (Soh et al., 1996) and 23 cycles of PCR were used to amplify rat GAPDH transcript (as a loading control). Amplified DNA (10 μl PCR mixture) was resolved on 1% agarose gel for electrophoresis and visualized under UV illumination.
Immunoblot Analyses of Immunoprecipitated p53
To immunoprecipitate p53 protein, specific antibody to p53 was incubated for 2 h with the soluble proteins (500 μg/sample) from C6 cells treated with APAP for different times as indicated. To facilitate immunoprecipitation of p53, protein G-bound agarose (0.1 ml/sample) was added to each sample and incubated for another 4 h before centrifugation at 10,000 x g for 10 min. The immunoprecipitated p53 was washed twice with 1 x phosphate buffered saline (PBS) and subjected to 10% SDS-PAGE followed by immunoblot analysis using the specific antibody against p53, ubiquitin, or mdm2. In addition, the same membrane used for the first immunoblot for p53 was extensively washed with a buffer containing 62.5 mM Tris-HCl (pH 6.8), 100 mM 2-mercaptoethanol and 2.0% SDS. The second immunoblot analysis was then performed to determine the level of p53-bound ubiquitin.
Data processing and statistical analysis
The density of immunoreactive proteins or mRNA transcript was quantified using NIH image 1.61 software. The relative densities of p53, Akt, phospho-Akt, phospho-p53, ubiquitin, mdm2 and p21 to actin were calculated and compared for all samples with different treatments. Statistical analyses were performed using the Student’s t test and p <0.05 was considered statistically significant. All the data represent the results from at least three separate experiments, unless stated otherwise. Other materials and methods not described here were preformed as previously described (Bae et al., 2001; Bae and Song, 2003).
Results
APAP Concentration-Dependent Reduction of p53 and p21 Proteins
Because of the APAP-induced apoptosis (Bae et al., 2001), we expected up-regulation of p53 content. Contrary to this expectation, treatment with 5 mM APAP for 24 h markedly decreased the level of p53 in C6 glioma cells (Figure 1A, top) compared to the DMSO-treated control in an APAP concentration-dependent manner. Immunoblot results revealed that 1.25 mM APAP treated for 24 h markedly decreased the level of p53. In general, greater reduction in p53 content was observed after exposure to higher concentrations of APAP than lower concentrations (Fig. 1A, top panel). The levels of p21 (Fig. 1A, second panel) and Gadd45 (data not shown), downstream targets of p53, markedly decreased at 24 h after treatment with APAP. In contrast, the level of actin (bottom), used as a loading control, did not change after APAP exposure. Our results indicate that the reduced p21 level is likely to result from the decreased level of p53 protein following exposure to APAP.
Fig. 1.
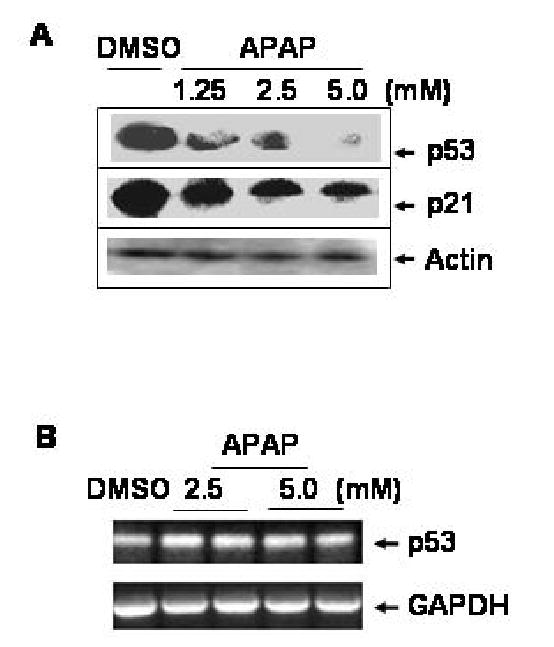
APAP concentration-dependent reduction in p53 and p21 proteins. (A) Whole cell lysates were prepared after C6 cells treated with DMSO alone or varying concentrations of APAP for 24 h as indicated. Equal amounts of the soluble extracts from different treatments (100 μg protein/well) were separated on SDS-PAGE followed by immunoblot analysis using the specific antibody, as indicated: p53, p21, and actin. Each lane represents the mixture of two different samples from the same treatment. This Figure represents a typical result from three independent experiments. (B) RT-PCR analyses for p53 and GAPDH mRNA transcripts were performed as described (Soh et al., 1996) using total cellular RNA (400 ng/assay) prepared from C6 cells treated with DMSO, 2.5, or 5.0 mM APAP for 24 h. The respective primers were the same as described for p53 (Soussi et al., 1988) or GAPDH transcript (Soh et al., 1996). Each amplified DNA band represents a mixture of three samples.
To further study the mechanism for APAP-induced p53 reduction, RT-PCR analysis was performed on rat p53 mRNA to compare with that of GAPDH. The level of p53 transcript increased in a linear manner between 26 and 30 PCR cycles, while GAPDH transcript elevated linearly between 22 and 28 PCR cycles (data not shown). Therefore, 26 PCR cycles were used to amplify p53 transcript and 23 cycles for GAPDH mRNA. The levels of p53 mRNA transcripts (546 bp, Fig. 1B, top panel), which were further confirmed by a second set of PCR primers, remained unchanged by treatment with 2.5 or 5.0 mM APAP for 24 h in C6 glioma cells. In addition, APAP did not change the levels of GAPDH transcripts (194 bp, Fig. 1B, bottom panel). These results indicate that APAP mainly affects p53 at the protein level without changing the steady state level of p53 mRNA.
Time- and Ubiquitin-Dependent p53 Degradation upon APAP Exposure
It is well established that p53 is rapidly degraded through ubiquitin-mediated proteolysis following interaction with mdm2, a major ubiquitin ligase for p53 (Lakin and Jackson, 1999). Because of the APAP-induced p53 reduction at the protein level, we then investigated whether APAP promotes p53 degradation through ubiquitin-dependent proteolysis. The levels of p53 in the soluble fraction were compared after C6 cells were exposed to APAP in the presence and absence of 1 μM lactacystin, a potent inhibitor of proteasome-dependent proteolysis (Fenteany and Schreiber, 1998). Lactacystin alone slightly affected the level of p53 (Fig. 2A, top panel, lane 4) as compared to that of the DMSO-treated control (lane 1). However, lactacystin appeared to prevent APAP-induced p53 degradation (lane 5), compared to the level of p53 in C6 glioma cells treated with APAP alone (lanes 2 and 3) while the level of actin, used as a loading control, did not change during each treatment (Fig. 2A, bottom panel). Similar results were also observed with MG132 (up to 1 μM final concentration), another inhibitor of proteasomal proteolysis (Maki et al., 1996) (data not shown).
Fig. 2.
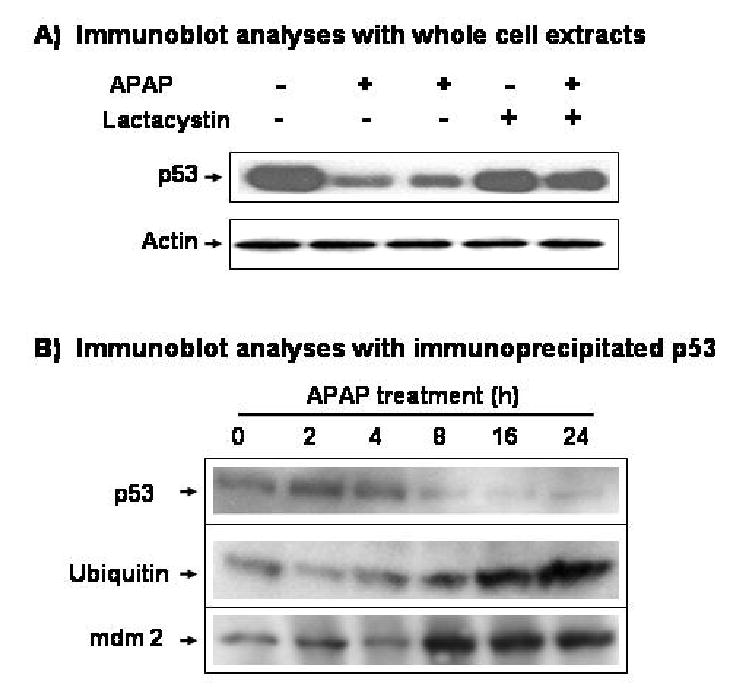
Time- and ubiquitin-dependent degradation of p53 after exposure to APAP. (A) Effect of lactacystin on APAP-induced p53 degradation. C6 cells were pretreated with 1 μM lactacystin for 4 h before cells were exposed to APAP for additional 24 h in the presence of 1 μM lactacystin. The level of p53 in the soluble fractions (100 μg protein/well) was then determined by immunoblot analysis with the specific antibody against p53 (top) or actin (bottom). (B) Immunoblot analyses of immunoprecipitated p53. Immunoprecipitation of p53 was performed as described under Materials and Methods. The immunoprecipitated p53 protein was washed twice with 1 x PBS and subjected to 10% SDS-PAGE followed by immunoblot analysis with the respective antibody against p53 (top), ubiquitin (middle), or mdm2 (bottom). These results represent a similar pattern of three independent experiments of p53 immunoprecipitation.
To further investigate the role of mdm2 and ubiquitin in APAP-induced p53 degradation, we also determined the respective level of p53, p53-bound ubiquitin, or p53-bound mdm2 after immunoprecipitation of p53 protein. Under our experimental conditions, a single or doublet of p53 protein band was recognized with the specific antibody against p53 in the immunoprecipitated protein with the same antibody (Fig. 2B, top panel). The amounts of p53 protein started to decline at 8 h and remained very low at 16 and 24 h after APAP treatment. However, increasing amounts of ubiquitin bound to immunoprecipitated p53 were detected with the specific anti-ubiquitin antibody at 8, 16, and 24 h after APAP exposure (Fig. 2B, middle panel), despite the decreased levels of p53 at these time points (top panel). Similar results of increased ubiquitin binding to p53 were also observed after extensive washing and re-blotting of the same membrane previously used for the first immunoblot analysis of p53 (data not shown). The levels of p53-bound mdm2 also increased at 8, 16, and 24 h after APAP exposure (Fig. 2B, bottom panel), despite markedly reduced amounts of p53 at these time points (top panel). These results, combined with the effects of lactacystin and MG132 on APAP-induced p53 degradation (Fig 2A), demonstrate that p53 protein is most likely degraded through mdm2-mediated ubiquitin conjugation upon exposure to APAP.
Additional immunoblot analysis showed that the levels of p53 and p21 in the whole cell homogenates did not change at 2 and 4 h after APAP exposure but started to decrease at 8 and 16 h (Supplemental Fig. 1). Subsequently, very low levels of these proteins were observed at 24 h. In contrast, the levels of phospho-Ser15-p53 and mdm2 appeared to increase at 8 h after APAP treatment and increased amounts of phosphorylated p53 and mdm2 were detected at 24 h (Supplemental Fig. 1).
Effects of Akt activity and p53 Phosphorylation on APAP-Induced p53 Reduction
The activated form of Akt (phospho-Akt) is known to phosphorylate mdm2 at Ser186, leading to mdm2-mediated ubiquitin conjugation and subsequent degradation of p53 (Ogawara et al., 2002). Because of the increased mdm2 binding to p53 protein, we determined whether the level of Akt or phospho-Akt was elevated after APAP treatment. Immunoblot results showed that APAP did not affect the levels of both Akt (Fig. 3A, second panel) and phospho-Akt (bottom panel), indicating that APAP-induced mdm2 binding to p53 and subsequent reduction of p53 protein were independent from the alterations of Akt and phospho-Akt. These results further suggest that the increased mdm2 binding and ubiquitin conjugation to p53 protein most likely result from direct interaction between p53 and APAP or its reactive metabolite(s).
Fig. 3.
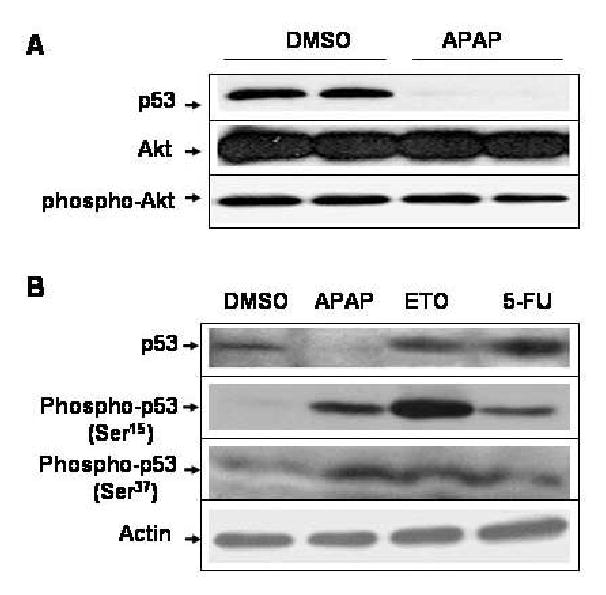
APAP-induced p53 reduction is independent from Akt or phospho-Akt and the phosphorylation status of p53 N-terminus. (A) Equal amounts of the soluble extracts (100 μg protein/well) from C6 cells treated with DMSO alone or 5 mM APAP for 24 h were separated on SDS-PAGE followed by immunoblot analysis using the specific antibody, as indicated: p53, Akt, and phospho-Akt, respectively. (B) C6 glioma cells were exposed to DMSO, 5 mM APAP, 10 μM etoposide (ETO), or 10 μM 5-fluorouracil (5-FU) for 24 h. Equal amounts of the soluble fraction from each treatment (100 μg protein/well) were separated on 10% SDS-PAGE followed by immunoblot analysis using the specific antibody against p53, phospho-p53 (Ser15 or Ser37), or actin.
It is also known that many DNA-damaging agents such as etoposide and 5-fluorouracil (5-FU) stabilize p53 through phosphorylation at its N-terminus, preventing p53 interaction with mdm2 and ubiquitin-dependent degradation (Tishler et al., 1993; Lakin and Jackson, 1999). Due to the APAP-induced p53 reduction (Figs. 1 and 2), it was hypothesized that APAP might interfere with the phosphorylation of p53. Therefore, we studied the degree of phosphorylation of p53 after C6 cells were exposed to APAP for 24 h, compared to that by etoposide or 5-FU used as a positive control. Under our experimental conditions, p53 levels were significantly reduced at 24 h after APAP treatment (Fig. 3B, top panel, lane 2) while actin level did not change (bottom panel). Contrary to our expectation, APAP stimulated phosphorylation of p53 at both Ser15 (second panel, lane 2) and Ser37 (third panel, lane 2), compared to the DMSO-treated control (lane 1). Treatment with etoposide and 5-FU stimulated phosphorylation of p53 at these Ser residues (Fig. 3B, second and third panels, lanes 3 and 4, respectively) with increased p53 content (top panel, lanes 3 and 4) compared to the DMSO-treated control (lane 1). Immunoblot analysis with a specific antibody which recognizes phospho-Ser20 revealed that APAP, etoposide and 5-FU, respectively, increased phosphorylation of p53 at Ser20 (data not shown), although the quality of this immunoblot was not as good as other immunoblots (second and third panels).
Role of CYP2E1 in APAP-Induced p53 Reduction
It is well established that APAP-induced toxicity results from P450-mediated metabolism (Thomas, 1993; Dai and Cederbaum, 1995; Holownia et al., 1997) and that pretreatment with P450 inhibitors can markedly reduce the rate of APAP-induced cell damage (Sinclair et al., 2000). Our previous results (Bae et al., 2001) supported this conclusion, which suggested an important role for CYP2E1 in APAP-induced cell damage. Therefore, we studied whether the degradation of p53 is also dependent on CYP2E1-mediated metabolism of APAP in C6 glioma cells. In this study, we determined the effect of a chemical inhibitor of CYP2E1 chlormethiazole (CMZ) (Hu et al., 1994) on the rate of cell death, the activity of CYP2E1, and the levels of p53 and p21. Cell death assay measured by MTT reduction (Bae et al., 2001) showed that less than 7% of DMSO-treated C6 glioma cells died and most cells looked normal under light microscopy (Fig. 4A, panel a). In contrast, approximately 35% of cells died at 24 h upon APAP treatment with many dead cells observed (Fig. 4A, panel b). However, pretreatment with 20 μM CMZ (Fig. 4A, panel c) significantly reduced the rate of APAP-induced cell death, where approximately 20% of cells died after APAP exposure in the presence of CMZ (p < 0.05, significantly different from the cells exposed to APAP alone). Under these experimental conditions, the baseline activity of CYP2E1 in C6 cells, as determined by N-nitrosodimethylamine demethylase (Bae et al., 2001), was approximately 4.8 pmol HCHO produced/mg protein of the whole cell extract/60 min. CMZ treatment reduced the activity of CYP2E1 by 50% (Fig. 4B). Consistent with the results shown in Figs. 1 – 3 and Supplemental Fig. 1, APAP markedly decreased p53 levels (Fig. 4C, top panel, lane 2) compared to that of the DMSO-treated control (lane 1). However, pretreatment with CMZ effectively prevented APAP-induced p53 degradation (lane 3) where CMZ alone did not change the level of p53 or p21 (Supplemental Fig. 2). A similar trend of APAP-mediated p21 reduction (lane 2) and recovery in the presence of CMZ (lane 3) was also observed (middle panel). In contrast, the levels of actin (bottom panel), used as a loading control, were similar in all samples.
Fig. 4.
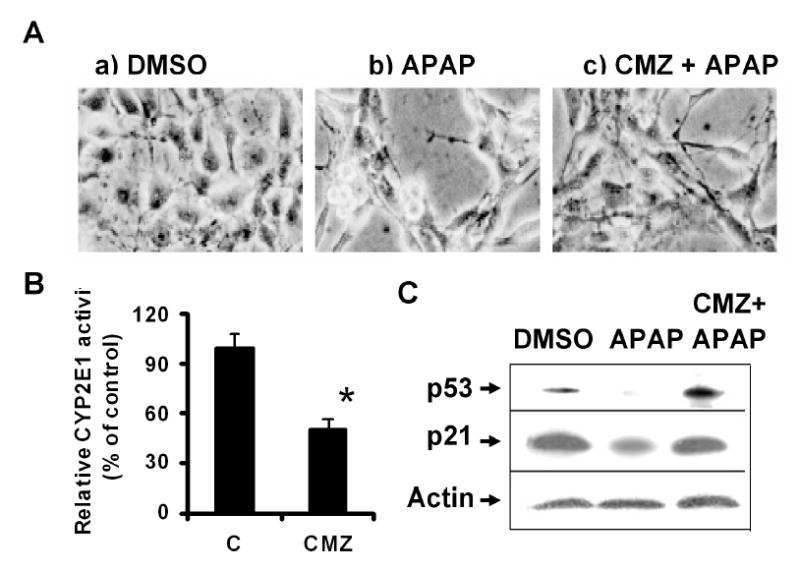
Protective effect of CMZ against APAP-induced change in cell morphology, cell death rate and the reduction of p53 and p21. (A) C6 glioma cells were pretreated with 20 μM CMZ for 16 h before APAP was treated for additional 24 h in the absence and presence of CMZ. Cell morphology, treated differently as indicated, was then determined under light microscopy (magnification, x 200). (B) CYP2E1 activity was determined by N-nitrosodimethylamine demethylase (Bae et al., 2001) with and without CMZ treatment. Each bar represents the average ± SD from three independent experiments. *, P < 0.01, significantly different from the DMSO-treated control. (C) Immunoblot analyses of p53, p21, and actin. The soluble fraction (100 μg/well) from C6 cells treated with APAP in the absence and presence of CMZ were subjected to 12% SDS-PAGE followed by immunoblot analysis using the respective antibody against p53 (top), p21 (middle), or actin (bottom).
Differential Effects of APAP and 3-Hydroxyacetanilide on Cell Death Rate and p53 Degradation
Consistent with earlier reports (Myers et al.,1991; Bae et al., 2001), APAP (4-hydroxyacetanilide) caused significantly more cell damage (about 35%) than DMSO alone or 3-hydroxyacetanilide, a non-toxic analog of APAP (<10%) (Fig. 5A). Under these conditions, we compared the effect of 3-hydroxyacetanilide or APAP (4-hydroxyacetanilde) on p53 reduction. Treatment with 5 mM 3-hydroxyacetanilide (Fig. 5B, top panel, lane 2) for 24 h did not reduce the p53 level, compared to the DMSO-treated vehicle control (top panel, lane 1). In contrast, exposure to 5 mM APAP markedly decreased the level of p53 (top panel, lane 3) in C6 glioma cells where the actin level did not change after each treatment (bottom panel).
Fig. 5.
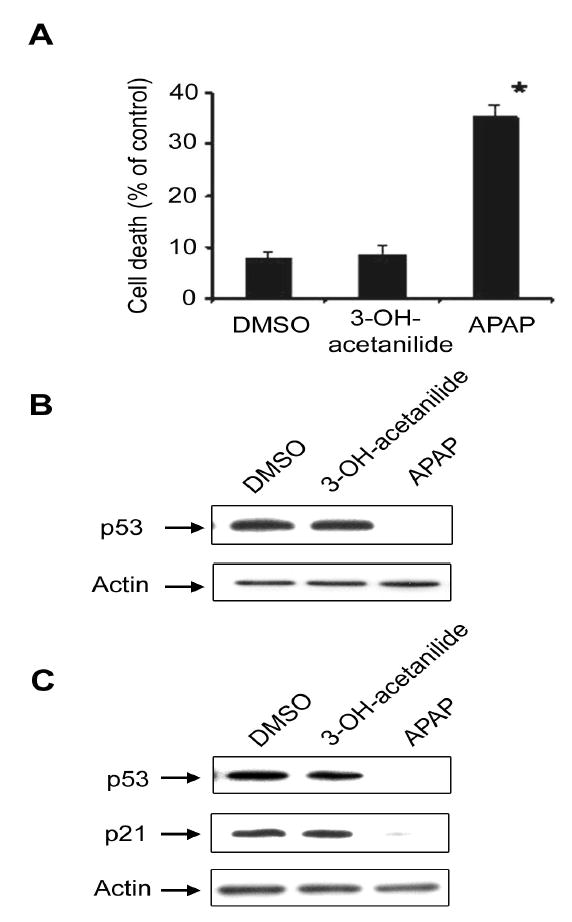
Differential effects of 3-hydroxyacetanilide and 4-hydroxyacetanilide on cell death rate and p53 reduction. (A) C6 glioma cells were treated with DMSO, 5 mM 3- hydroxyacetanilide, or 5 mM 4-hydroxyacetanilide (APAP) for 24 h. Cell death rate was then determined by MTT reduction. *, P < 0.01, significantly different from the cells treated with DMSO alone (vehicle control) or 3-hydroxyacetanilide. (B) Equal amounts of the soluble fraction (100 μg protein/well) were separated on 10% SDS-polyacrylamide gels followed by immunoblot analysis using the specific antibody against p53 (top) or actin (bottom). (C) LLC-PK1 cells were treated with DMSO, 5 mM 3-hydroxyacetanilide, or 5 mM APAP for 24 h before cell harvest. Equal amounts of the soluble fraction (20 μg/lane) were separated on 12% SDS-polyacrylamide gels and subjected to immunoblot analysis using the specific antibody against p53 (top), p21 (middle), or actin (bottom). This result represents a typical result of two independent experiments.
We also studied whether APAP-mediated reduction of p53 and its downstream target p21 could be observed in another type of cultured cells such as porcine kidney LLC-PK1 cells, since these cells seem to contain catalytically active CYP2E1 (Liu et al., 2002; Al-Ghamdi et al., 2004). Immunoblot analysis showed that 2.5 mM (data not shown) or 5.0 mM APAP treated for 24 h caused marked reduction of p53 and p21 contents in LLC-PK1 cells (Fig. 5C, lane 3, top and middle panels) while the levels of actin used as a loading control were similar in all lanes (bottom panel). In contrast, 5.0 mM 3-hydroxyacetanilide, a non-toxic analog of APAP, did not change the levels of p53 and p21 (lanes 2, top and middle panels).
Our results observed in both C6 gloima and LLC-PK1 porcine kidney cells strongly suggest that APAP selectively decreased the level of p53 protein, likely through interaction between p53 and NAPQI or another reactive metabolite(s) produced from CYP2E1-mediated metabolism of APAP. In fact, increased levels of APAP moiety bound to p53 protein were recognized with the specific antibody to APAP when immunoprecipitated p53 protein with the p53-specific antibody was used in immunoblot analysis (Supplemental Fig. 3). This protein (54 kDa in lane 2) seemed to be absent in DMSO-treated control sample (lane 1), indicating that APAP and/or its related molecules appeared to bind to p53 protein after APAP exposure.
Discussion
We previously reported that C6 glioma cells, despite low levels of CYP2E1 expression, underwent apoptosis upon exposure to APAP (Bae et al., 2001) at the same concentration used in other studies (Dai and Cederbaum, 1995; Holownia et al., 1997). Therefore, we hypothesized that the level of p53 would increase after APAP treatment, because p53 normally promotes growth arrest or apoptotic cell death (Vogelstein et al., 2000). However, the level of p53 actually decreased before and during apoptosis of C6 glioma cells (Figs. 1 – 5 and Supplemental Fig. 1) after APAP treatment. Therefore, p53 cannot be directly involved in the APAP-induced apoptosis of C6 glioma cells. Based on this view, our current results support the importance of c-Jun N-terminal protein kinase-related cell death pathway in APAP-induced apoptosis (Bae et al., 2001) in a p53-independent manner, similar to many other cases of apoptosis without involvement of p53 (Lieberman et al., 1995; Conner et al., 1999). Consistent with the in vitro data, our preliminary results with alcohol-pretreated Balb/c mice also show that acute exposure of APAP reduces the amounts of p53 and p21 during APAP-mediated necrosis and apoptosis (data not shown). The current data also provide evidence for an important role of CYP2E1 in the metabolic activation of APAP and subsequent loss of p53 and p21, since CMZ, an inhibitor of CYP2E1, significantly prevented the APAP-induced degradation of p53 and p21 (Fig. 4C). The partial protection by CMZ against APAP-mediated cell death rate may result from the incomplete inhibition of CYP2E1 by the concentration of CMZ used in this experiment (Fig. 4B). Alternatively, this could result from the fact that APAP can be also metabolized by other P450 isozymes, besides CYP2E1, present in C6 glioma cells (Geng and Strobel, 1995).
It was shown that many cellular proteins can be covalently modified with NAPQI, an electrophilic metabolite of APAP (Myers et al., 1991; Fountoulakis et al., 2000). Some of these modified proteins became inactivated. For example, the covalent binding of APAP to N-10-formyl-tetrahydrofolate dehydrogenase led to a decrease in its catalytic activity (Pumford et al., 1997). APAP could also bind and reduce the activities of certain mitochondrial enzymes such as glutamate dehydrogenase (Halmes et al., 1996) and aldehyde dehydrogenase (Landin et al., 1996), respectively, although the content of each protein was not evaluated in those studies. In addition, certain P450 enzymes such as CYP2E1 and CYP1A2, involved in the APAP metabolism, were degraded after administration of toxic doses of APAP (Snawder et al., 1994; Sinclair et al., 2000), possibly through interaction between these P450 proteins and NAPQI or other reactive metabolites produced from P450-mediated metabolism of APAP. Furthermore, we recently observed that APAP causes degradation of nuclear Ogg1, a DNA repair enzyme, without changing its mRNA level (Wan et al., 2004). By similar mechanisms, p53 may interact directly with NAPQI or other reactive metabolites of APAP (Supplemental Fig. 3), prior to its degradation despite phosphorylation at its N-terminus.
Ray et al. (2001) previously reported that APAP slightly elevated the level of p53 in mouse liver. The level of p53 reported in that study, however, did not correspond with improvement in APAP-induced liver damage and normalization of other parameters such as hepatic lipid peroxidation, serum alanine aminotransferase activity, DNA fragmentation, and the level of Bcl-XL after treatment with chlorpromazine, 4-aminobenzamide, or nicotinamide. Those results suggest that the apoptotic role of p53 elevated by APAP may not be as important as decreased Bcl-XL content in APAP-induced liver damage. The results observed by Ray et al. (2001) are clearly different from our current results of APAP-induced p53 degradation in C6 and LLC-PK1 cells as well as preliminary results in APAP-treated mouse livers. Although the reason for the apparent differences is unknown, this difference remains to be clarified.
Results from this study demonstrate that p53 protein is likely to be degraded through increased mdm2 binding and ubiquitin-conjugation after exposure to APAP, despite multiple phosphorylations of p53 at its N-terminus. The mechanism by which APAP promotes mdm2 binding to phosphorylated p53 is unknown. It is conceivable that binding of NAPQI to p53 may cause conformational change in p53 with opening up another binding site(s) for mdm2 (Fig. 2B) in the NAPQI-p53 complex (Supplemental Fig. 3) regardless of the phosphorylation status at p53 N-terminus as demonstrated (Fig. 3B). Subsequently, increased mdm2 binding to NAPQI-p53 complex led to more ubiquitin conjugation and proteasomal degradation of p53 despite elevated phosphorylation of p53 protein. In contrast, 3-hydroxyacetanilide may not produce a reactive intermediate like NAPQI which can bind to p53 protein. As a consequence, little degradation of p53 and its downstream target protein p21 took place after exposure to 3-hydroxyacetanilide in C6 glioma cells (Fig. 5B) and LLC-PK1 porcine kidney cells (Fig. 6A). The detailed molecular mechanism for APAP-mediated p53 degradation observed in two different cell lines is unknown but is likely to result from binding of NAPQI to p53 followed by ubiquitin-dependent p53 degradation after treatment with a relatively high dose (2.5 or 5 mM) of APAP.
It is well established that p53 level is regulated by complex mechanisms. Depending on the nature of the DNA damaging agents, severity of damage, or cellular status, p53 level is elevated or reduced. For instance, p53 levels can be elevated through phosphorylation at its N-terminus after treatment with DNA damaging or apoptosis inducing agents (Tishler et al., 1993; Vogelstein et al., 2000). On the other hand, p53 degradation is mainly facilitated through mdm2, the major p53 ubiquitin ligase, leading to ubiquitin-dependent proteasomal degradation of p53 (Lakin and Jackson, 1999; Ogawara et al., 2002). Our results shown in this study particularly differ from the well-established relationship between p53 phosphorylation and mdm2 binding and p53 degradation. Therefore, our results represent a novel finding about p53 degradation by APAP. To our knowledge, our data represents the first report that APAP, possibly through the production of a highly reactive metabolite such as NAPQI, can reduce the level of p53 through ubiquitin-dependent proteolytic degradation. However, biological significance of the reduced levels of p53 and p21 after APAP exposure remains to be determined, because these proteins are known to be involved in many biological functions such as apoptosis, DNA repair and cell cycle arrest.
In conclusion, results of this study demonstrate that a large dose of APAP can promote rapid degradation of p53 through mdm2-mediated ubiquitin-conjugation, but independent from Akt and phospho-Akt proteins, which are known to promote p53 degradation. In addition, APAP-induced p53 reduction is unrelated to the phosphorylation status of its N-terminus. APAP also decreased the level of p21 consistent with the reduced level of p53. Comparative experiments with APAP (4-hydroxyacetanilide) and 3-hydroxyacetanilide in the presence of a CYP2E1 inhibitor suggest that binding of NAPQI, produced during CYP2E1-mediated metabolism of APAP, to p53 is likely to lead to increased mdm2 binding and subsequently ubiquitin-dependent p53 degradation. These results represent another example of p53 modulation by a chemical compound such as APAP.
Supplementary Material
Acknowledgments
We thank Dr. Joong-Ik Yang for providing CMZ used in our experiments. We also thank Drs. Norman Salem, Jr. and James P. Hardwick for the support and critical reading of the manuscript, respectively.
Footnotes
The first two authors (YSL, JW) equally contributed to this work. Current address (YSL): Department of Endocrinology, School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania; Current address (JW): Department of Neurology, The Chengdu 416 Hospital (The Formal Suzhou Medical College 2nd Hospital), Chengdu, Sichuan, China; Current address (MAB): Laboratory of Molecular Pharmacology and Physiology, Korea Research Institute of Chemical Technologies, Daejon, Korea. This work was supported by the Intramural Research Fund at the National Institute on Alcohol Abuse and Alcoholism.
References
- Al-Ghamdi SS, Chatterjee PK, Raftery MJ, Thiemermann C, Yaqoob MM. Role of cytochrome P450 2E1 activation in proximal tubular cell injury induced by hydrogen peroxide. Ren Fail. 2004;26:103–110. doi: 10.1081/jdi-120038483. [DOI] [PubMed] [Google Scholar]
- Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, Kaplan DR, Miller FD. P53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J Cell Biol. 1998;143:1691–1703. doi: 10.1083/jcb.143.6.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai A, Miyagi Y, Sugiyama A, Gamanuma M, Hong SH, Takamoto S, Nomura K, Matsutani M, Takakura K, Kuchino Y. Negative effects of wild-type p53 and s-Myc on cellular growth and tumorigenicity of glioma cells. Implication of the tumor suppressor genes for gene therapy. J Neurooncol. 1994;19:259–268. doi: 10.1007/BF01053280. [DOI] [PubMed] [Google Scholar]
- Bae MA, Pie JE, Song BJ. Acetaminophen induces apoptosis of C6 glioma cells by activating the c-Jun N-terminal protein kinase-related cell death pathway. Mol Pharmacol. 2001;60:847–856. [PubMed] [Google Scholar]
- Bae MA, Song BJ. Critical role of c-Jun N-terminal protein kinase activation in troglitazone-induced apoptosis of human HepG2 hepatoma cells. Mol Pharmacol. 2003;63:401–408. doi: 10.1124/mol.63.2.401. [DOI] [PubMed] [Google Scholar]
- Cohen SD, Pumford NR, Khairallah EA, Boekelheide K, Pohl LR, Amouzadeh HR, Hinson JA. Selective protein covalent binding and target organ toxicity. Toxicol Appl Pharmacol. 1997;143:1–12. doi: 10.1006/taap.1996.8074. [DOI] [PubMed] [Google Scholar]
- Conner EA, Teramoto T, Wirth PJ, Kiss A, Garfield S, Thorgeirsson SS. HGF- mediated apoptosis via p53/bax-independent pathway activating JNK1. Carcinogenesis. 1999;20:583–590. doi: 10.1093/carcin/20.4.583. [DOI] [PubMed] [Google Scholar]
- Dai Y, Cederbaum AI. Cytotoxicity of acetaminophen in human cytochrome P450 2E1-transfected HepG2 cells. J Pharmacol Exp Ther. 1995;273:1497–1505. [PubMed] [Google Scholar]
- Fenteany G, Schreiber SL. Lactacystin, proteasome function, and cell fate. J Biol Chem. 1998;273:8545–8548. doi: 10.1074/jbc.273.15.8545. [DOI] [PubMed] [Google Scholar]
- Fountoulakis M, Berndt P, Boelsterli UA, Crameri F, Winter M, Albertini S, Suter L. Two-dimensional database of mouse liver proteins: Changes in hepatic protein levels following treatment with acetaminophen or its non-toxic regioisomer 3-acetamidophenol. Electrophoresis. 2000;21:2148–2161. doi: 10.1002/1522-2683(20000601)21:11<2148::AID-ELPS2148>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Geng J, Strobel HW. Identification of cytochromes P450 1A2, 2A1, 2C7, 2E1 in rat glioma C6 cell line by RT-PCR and specific restriction enzyme digestion. Biochem Biophys Res Commun. 1995;197:1179–1184. doi: 10.1006/bbrc.1993.2601. [DOI] [PubMed] [Google Scholar]
- Giaccia AJ, Kastan MB. The complexity of p53 modulation: emerging patterns from divergent signals. Gene Dev. 1998;12:2973–2983. doi: 10.1101/gad.12.19.2973. [DOI] [PubMed] [Google Scholar]
- Halmes NC, Hinson JA, Martin BM, Pumford NR. Glutamate dehydrogenase covalently binds to a reactive metabolite of acetaminophen. Chem Res Toxicol. 1996;9:541–546. doi: 10.1021/tx950158a. [DOI] [PubMed] [Google Scholar]
- Holownia A, Mapoles J, Menez JF, Braszko JJ. Acetaminophen metabolism and cytotoxicity in PC12 cells transfected with cytochrome P4502E1. J Mol Med. 1997;75:522–527. doi: 10.1007/s001090050137. [DOI] [PubMed] [Google Scholar]
- Hu Y, Mishin V, Johansson I, von Bahr C, Cross A, Ronis MJ, Badger TM, Ingelman-Sundberg M. Chlormethiazole as an efficient inhibitor of cytochrome P450 2E1 expression in rat liver. J Pharmacol Exp Ther. 1994;269:1286–1291. [PubMed] [Google Scholar]
- Knight TR, Ho Y-H, Farhood A, Jaeschke H. Peroxynitrite is a critical mediator of acetaminophen hepatotoxicity in murine livers: protection by glutathione. J Pharmacol Exp Ther. 2002;303:468–475. doi: 10.1124/jpet.102.038968. [DOI] [PubMed] [Google Scholar]
- Lakin ND, Jackson SP. Regulation of p53 in response to DNA damage. Oncogene. 1999;18:7644–7655. doi: 10.1038/sj.onc.1203015. [DOI] [PubMed] [Google Scholar]
- Landin JS, Cohen SD, Khairallah EA. Identification of a 54-kDa mitochondrial acetaminophen-binding protein as aldehyde dehydrogenase. Toxicol Appl Pharm. 1996;141:299–307. doi: 10.1006/taap.1996.0287. [DOI] [PubMed] [Google Scholar]
- Lee WM. Acetaminophen and the U.S. Acute Liver Failure Study Group: lowering the risks of hepatic failure. Hepatology. 2004;40:6–9. doi: 10.1002/hep.20293. [DOI] [PubMed] [Google Scholar]
- Lev Bar-Or R, Maya R, Segel LA, Alon U, Levine AJ, Oren M. Generation of oscillations by the p53-Mdm2 feedback loop: a theoretical and experimental study. Proc Natl Acad Sci USA. 2000;97:11250–11255. doi: 10.1073/pnas.210171597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman DA, Hoffman B, Steinman RA. Molecular controls of growth arrest and apoptosis: p53-dependent and independent pathways. Oncogene. 1995;11:199–210. [PubMed] [Google Scholar]
- Liu H, Baliga M, Baliga R. Effect of cytochrome P450 2E1 inhibitors on cisplatin-induced cytotoxicity to renal proximal tubular epithelial cells. Anticancer Res. 2002;22:863–868. [PubMed] [Google Scholar]
- Maki CG, Huibregtse JM, Howley PM. In vivo ubiquitination and proteasome-mediated degradation of p53. Cancer Res. 1996;56:2649–2654. [PubMed] [Google Scholar]
- Michael SL, Pumford NR, Mayeux PR, Niesman MR, Hinson JA. Pretreatment of mice with macrophage inactivators decreases acetaminophen hepatotoxicity and the formation of reactive oxygen and nitrogen species. Hepatology. 1999;30:186–195. doi: 10.1002/hep.510300104. [DOI] [PubMed] [Google Scholar]
- Myers TG, Dietz EC, Anderson NL, Khairallah EA, Cohen SD, Nelson SD. A comparative study of mouse liver proteins arylated by reactive metabolites of acetaminophen and its nonhepatotoxic regioisomer, 3-hydroxyacetanilide. Chem Res Toxicol. 1991;8:403–413. doi: 10.1021/tx00045a012. [DOI] [PubMed] [Google Scholar]
- Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K, Masuyama N, Gotoh Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem. 2002;277:21843–21850. doi: 10.1074/jbc.M109745200. [DOI] [PubMed] [Google Scholar]
- Pumford NR, Halmes NC, Martin BM, Cook RJ, Wagner C, Hinson JA. Covalent binding of acetaminophen to N-1-formyltetrahydrofolate dehydrogenase in mice. J Pharmacol Exp Ther. 1997;280:501–505. [PubMed] [Google Scholar]
- Ray SD, Balasubramanian G, Bagchi D, Reddy CS. Ca2+-calmodulin antagonists chlorpromazine and poly(ADP-Ribose) polymerase modulators 4-aminobenzamide and nicotinamide influence hepatic expression of Bcl-XL and p53 and protect against acetaminophen-induced programmed and unprogrammed cell death in mice. Free Radic Biol Med. 2001;31:277–291. doi: 10.1016/s0891-5849(01)00562-7. [DOI] [PubMed] [Google Scholar]
- Seeff LB, Cuccherini BA, Zimmerman HJ, Adler H, Benjamin SB. Acetaminophen hepatotoxicity in alcoholics. A therapeutic misadventure. Ann Int Med. 1986;104:399–404. doi: 10.7326/0003-4819-104-3-399. [DOI] [PubMed] [Google Scholar]
- Shayiq RM, Roberts DW, Rothstein K, Snawder JE, Benson W, Ma X, Black M. Repeat exposure to incremental doses of acetaminophen provides protection against acetaminophen-induced lethality in mice: An explanation for high acetaminophen dosage in humans without hepatic injury. Hepatology. 1999;29:451–463. doi: 10.1002/hep.510290241. [DOI] [PubMed] [Google Scholar]
- Sinclair JF, Szakacs JG, Wood SG, Kostrubsky VE, Jeffery EH, Wrighton SA, Bement WJ, Wright D, Sinclair PR. Acetaminophen hepatotoxicity precipitated by short-term treatment of rats with ethanol and isopentanol: protection by triacetyloleandomycin. Biochem Pharmacol. 2000;59:445–454. doi: 10.1016/s0006-2952(99)00349-4. [DOI] [PubMed] [Google Scholar]
- Snawder JE, Roe AL, Benson RW, Robert DW. Loss of CYP2E1 and CYP1A2 activity as a function of acetaminophen dose: relation to toxicity. Biochem Biophys Res Commun. 1994;203:532–539. doi: 10.1006/bbrc.1994.2215. [DOI] [PubMed] [Google Scholar]
- Soh Y, Rhee HM, Sohn DH, Song BJ. Immunochemical detection of CYP2E1 in fresh rat lymphocytes and its pretranslational induction by fasting. Biochem Biophys Res Commun. 1996;227:541–546. doi: 10.1006/bbrc.1996.1542. [DOI] [PubMed] [Google Scholar]
- Soussi T, Caron de Fromentel C, Breugnot C, May E. Nucleotide sequence of a cDNA encoding the rat p53 nuclear oncoprotein. Nucl Acids Res. 1988;16:11384. doi: 10.1093/nar/16.23.11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SH. Paracetamol (acetaminophen) poisoning. Pharmacol Ther. 1993;60:91–120. doi: 10.1016/0163-7258(93)90023-7. [DOI] [PubMed] [Google Scholar]
- Tishler RB, Calderwood SK, Coleman CN, Price BD. Increases in sequence specific DNA binding by p53 following treatment with chemotherapeutic and DNA damaging agents. Cancer Res. 1993;53:2212–2216. [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Wan J, Bae MA, Song BJ. Acetaminophen-induced oxidative DNA damage through reduction of Ogg1 DNA repair enzyme in C6 glioma cells. Exp Mol Med. 2004;36:71–77. doi: 10.1038/emm.2004.10. [DOI] [PubMed] [Google Scholar]
- Whitcomb DC, Block GD. Association of APAP toxicity with fasting and ethanol use. JAMA. 1994;272:1845–1850. doi: 10.1001/jama.1994.03520230055038. [DOI] [PubMed] [Google Scholar]
- Zhan QM, Chen IT, Antimore MJ, Fornace AJ., Jr Tumor suppressor p53 can participate in transcriptional induction of GADD45 promoter in the absence of direct DNA binding. Mol Cell Biol. 1998;18:2768–2778. doi: 10.1128/mcb.18.5.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.