

Abstract
Single-nucleotide polymorphisms (SNPs) constitute the great majority of variations in the human genome, and as heritable variable landmarks they are useful markers for disease mapping and resolving population structure. Redundant coverage in overlaps of large-insert genomic clones, sequenced as part of the Human Genome Project, comprises a quarter of the genome, and it is representative in terms of base compositional and functional sequence features. We mined these regions to produce 500,000 high-confidence SNP candidates as a uniform resource for describing nucleotide diversity and its regional variation within the genome. Distributions of marker density observed at different overlap length scales under a model of recombination and population size change show that the history of the population represented by the public genome sequence is one of collapse followed by a recent phase of mild size recovery. The inferred times of collapse and recovery are Upper Paleolithic, in agreement with archaeological evidence of the initial modern human colonization of Europe.
Information on the demographic history of a species is imprinted in the distribution of sequence variations in its genome. The completion of a draft sequence for the human genome provides a useful substrate for both the detection of sequence variants and a study of their distribution. To date, the number of publicly available single-nucleotide polymorphisms (SNPs) well exceeds two million (dbSNP build 105). The main data sources for computational SNP discovery have been expressed sequence tags (ESTs) (1, 2), genomic restriction fragments (3), sequences aligned to genome both from the ends of bacterial artificial chromosomes (BACs) and from random shotgun sequences of clone sequence, and overlapping regions of genomic clone sequences themselves (4, 5). Generally, SNPs from these data were detected in surveys of a few chromosomes, an ascertainment strategy that biases allele frequency patterns toward common variations (6), and thus these data are expected to fall into a range that is unlikely to contain the majority of clinically important mutations (7, 8). Under the “common disease, common allele” hypothesis, however, these common variants may be of special importance. In either case, to assess the potential utility of these data for inferences of gene function or population history, one must first understand its overall structure and distribution in the genome. Statistical power in such analyses requires a large amount of data, ascertained under uniform, well-characterized conditions. Clone overlaps and their derived variations are well suited for this task, as long (up to 100 kb) regions of redundant sequence coverage distributed in roughly even intervals (5), covering nearly a quarter of the genome. The fact that regions in a wide range of overlap length are available makes this set especially suited for studying the effects of recombination and demographic size fluctuation on the spatial (density) distribution of genomic sequence variations. To this end, we built a set of reagents (pairwise sequence alignments and their corresponding sets of variation) by analyzing the overlapping regions of large-insert clones sequenced as part of the human genome project. These data provided marker density observations grouped by overlap fragment length. Extending previous methods (9, 10), we implemented simulation and numerical techniques to estimate population genetic parameters that best describe these observed data. We report results indicating that both the effects of recombination and substantial changes in effective population size are required to fit models of neutral sequence evolution to observed marker densities.
Methods
Overlap Detection, SNP Discovery, and Tabulation of Observed Marker Density.
The initial data consisted of genomic clones of either finished or draft quality that were part of the September 5, 2000, genome data freeze. Regions of known human repeats and low complexity sequence were masked with repeatmasker (Arian Smit, http://repeatmasker.genome.washington.edu). Candidate sequence overlaps were determined by a fast initial similarity search with megablast (11), followed by pairwise alignment with the dynamic programming algorithm cross_match (Phil Green, www.phrap.org). Draft quality sequence is often composed of unordered fragments; hence an overlap between two such clones is broken up into a set of partial overlap fragments. Overlaps were retained for further analysis if: (i) both clones resided on the same chromosome, as could be determined by physical mapping; and (ii) total overlap length was >6 kb, counting only overlap fragments longer than 3 kb in the total. Overlap fragments were analyzed with the polybayes SNP-discovery program (12). An observed mismatch was called a candidate SNP if the corresponding polybayes probability value was at least 0.80, and there were no discrepancies in the five base pairs immediately flanking either side. To avoid false positive predictions caused by the erroneous alignment of divergent copies of segmental duplications (sequence paralogy) we have excluded overlap fragments with >1 SNP per 400 nucleotides. This censorship procedure, necessary to maintain a high quality for the candidate set, also removes overlap fragments in which the inherent polymorphism rate was genuinely high. The resulting bias was estimated in subsequent analysis. An additional bias was introduced when regions of low-quality sequences were analyzed. These regions cannot be effectively evaluated for SNPs, as sequence differences are more likely to represent sequencing errors than true polymorphisms. We rectified this bias by adjusting the overlap interval to include only the high-quality portions of the overlaps [i.e., where the base quality value (13) was >35 in both sequences]. This procedure discarded ≈5% of the total overlap length.
Integration with the Public Genome Assembly.
To ensure an unbiased evaluation of the density distributions with respect to the reference genome sequence, we included only those portions of our overlaps that were also present in the genome assembly based on the September 5, 2000, data (14). We evaluated repeat content in the genome, as well as in the clone overlap regions by using repeatmasker. We used custom software to compute G+C nucleotide and CpG dinucleotide content.
SNP Validation and Estimation of Allele Frequency.
The experimental methods and conditions used to assess the candidate SNPs were fully described previously (15).
Modeling Marker Density Distributions.
Mismatch distributions describe the likelihood of observing k (k = 0, 1, 2, …) polymorphic sites (mismatches) when n sample sequences of a given length, L, are compared (n = 2 in this study). Traditionally, the opposing effects of meiotic recombination and co-ancestry have been studied under two simple, yet extreme, scenarios (Fig. 1a). A simple (first-order) model that ignores any structure imposed by demographic history and assumes complete independence between the genealogies of neighboring sites because of recombination (infinite recombination model) predicts a Poisson mismatch distribution driven solely by the mutation rate (16). Conversely, a first-order model that accounts for genealogical structure only through static demographic history and ignores recombination (zero-recombination model) predicts a geometric distribution of mutational differences (17).
Figure 1.

Marker density distributions predicted under competing population-genetic models (for 10-kb pairwise aligned length, censored at 25 SNPs per alignment). (a) First-order, stationary history. (b) Second-order, expansion history. (c) Second-order, collapse history. (d) Third-order, “bottleneck”-shaped history. r indicates the per nucleotide, per generation recombination rate.
A detailed demographic history described by the time evolution of effective population size, Ne, profoundly affects the distribution of polymorphic sites shared between individuals. In particular, a large increase of the effective population size yields an overabundance of new lineages that increase the likelihood that random sequence pairs will harbor one or more mutational differences (9) (Fig. 1b). Alternatively, a sharp decrease in effective population size raises the likelihood of relatedness between random pairwise DNA samples, resulting in the opposite effect: an overrepresentation of sequence identity (zero difference) as seen in Fig. 1c. Both models represent a second-order changing population size dynamics characterized by different effective population sizes in each of two epochs: an ancestral size N2, followed by a size change to N1, happening T1 generations ago. It is possible to go to higher-order models by increasing the number of epochs in a population history. An example third-order (three-epoch) model is the “bottleneck” dynamics (i.e., a collapse followed by a phase of recent population recovery) depicted in Fig. 1d.
While the density distribution can be computed explicitly for zero-recombination models even with high-order population dynamics (9), no explicit formulas are available for models with realistic levels of recombination. In these cases, we are reliant on numerical simulations that use the coalescent process with recombination (18). Implementing this technique with custom software, we were able to study the counterparts of the previous (zero-recombination) models with realistic levels of recombination (Fig. 1). Models used a uniform genome-averaged mutation rate, μ = 2.0 × 10−8 per site per generation [obtained as a compromise between prominent estimates (19, 20)] and a uniform genome-averaged recombination rate of r = 1.0 × 10−9 per nucleotide per generation [obtained from recombination frequencies measured across the genome (21)] when appropriate. We note that an alternative estimate of μ = 1.0 × 10−8, although less conventional, is perhaps more plausible, as it accounts for a larger ancestral anthropoid population size and older separation times (20, 22). The latter rate estimate will yield a human effective size estimate of 20,000 rather than 10,000 (below), and double the time estimates for demographic events. Initial simulations were run with 100,000 replicates per parameter set, and refinements for the best-performing parameter sets were reevaluated with one million replicates.
Model Parameterization.
A given model is specified by a recombination rate (r = 0 or 1.0 × 10−9) and a vector of population sizes (Ni) and epoch durations (Ti) determined by the model's order (for examples of such parameter sets see Table 1). Values of Ni (ranging from 1,000 to 100,000) were sampled in units of 100 for numerical calculations and for one-epoch model simulations, and in units of 1,000 for higher-epoch model simulations. Values of Ti (ranging from 100 to 10,000) were sampled in units of 100 for all cases. Predicted marker density distributions were generated for each length scale analyzed (L = 4, 6, 8, 10, 12, 14, and 16 kb) for each parameter set considered from the multidimensional parameter space defined above.
Table 1.
Performance of population genetic models of various complexities for fitting marker density data observed in interindividual BAC overlap fragments
| Model structure | Recombination rate (r) | Best-fitting model parameters | Model log likelihood | Improvement because of inclusion of recombination df = 1 | Improvement because of inclusion of extra epoch df = 2 |
|---|---|---|---|---|---|
| Free combination | ∞ | N = 9,200 | −13,576.89 | — | — |
| One-epoch | 0 | N1 = 12,000 | −626.11 | — | — |
| 10−8 | N1 = 10,300 | −566.25 | 2 ln λ = 119.72 (P < 1 × 10−7) | — | |
| Two-epoch | N2 = 13,200 | ||||
| 0 | T1 = 200 | −559.75 | — | 2 ln λ = 132.72 (P < 1 × 10−7) | |
| N1 = 2,000 | |||||
| N2 = 11,000 | |||||
| 10−8 | T1 = 700 | −466.82 | 2 ln λ = 185.86 (P < 1 × 10−7) | 2 ln λ = 198.86 (P < 1 × 10−7) | |
| N1 = 4,000 | |||||
| Three-epoch | N3 = 9,000 | ||||
| T2 = 7,000 | |||||
| 0 | N2 = 50,000 | −469.64 | — | 2 ln λ = 180.22 (P < 1 × 10−7) | |
| T1 = 7,000 | |||||
| N1 = 9,000 | |||||
| N3 = 11,000 | |||||
| T2 = 400 | |||||
| 10−8 | N2 = 5,000 | −463.07 | 2 ln λ = 13.14 (P < 2.89 × 10−4) | 2 ln λ = 7.5 (P < 0.023) | |
| T1 = 1,200 | |||||
| N1 = 6,000 |
For each model, we report the population parameter values within a given model structure that produced the best fit to the observations. We also report the corresponding log likelihood, ln P(data|model). The penultimate column reports the statistical significance of model improvement attributable to the introduction of a genome average recombination rate into our models (adding one extra model parameter). The final column reports the significance of the model improvement attributable to the introduction of an additional epoch (two extra model parameters). Significance of the improvement was evaluated with statistical hypothesis testing for nested model structures (see Methods).
Model Evaluation.
Parameter sets within a fixed model structure were compared by computing, for each competing model, a degree of fit between the observed (o) marker density distribution and the probability distribution predicted by each of the models (m), using the log likelihood of the data given the model in question. Because observations between different overlap fragment length classes, as well as observations for each of the number of differences, k, were independent, this likelihood is described by a multinomial distribution:
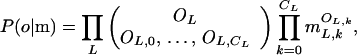 |
where OL is the number of overlap fragments in class L, OL,k is the number of fragments with k differences, CL is the maximum number of differences permitted by the censorship procedure for length L, and mL,k is the marker density probability predicted by the model, at length class L, for k differences. In evaluating an alternative goodness of fit for a given model, we used the χ2 metric (see Discussion):
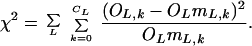 |
Using either of the above metrics requires the model-predicted probability distributions to be calculated very accurately, especially in mismatch categories with low predicted probabilities. In those cases where the distribution can be calculated only with simulations, accuracy is constrained by the practical upper limit on the number of simulation replicates. To avoid numerical instability, we restricted fit testing to the first KL categories within each length class such that categories k = 0, 1, … , KL contain 95% of all fragments for that class.
Model Comparison.
The performances of different model structures were compared based on the maximum likelihood parameter estimates for each model structure. Standard tools of normal hypothesis testing could be used (23) when two nested models were compared, by calculating the likelihood ratio, λ, between the less restricted and more restricted model. The quantity 2 ln(λ) is expected to be asymptotically χ2 distributed with degrees of freedom equal to the difference in the number of parameters. Increasing the number of epochs by one adds two parameters to the model (the effective population size, and the duration of the new epoch). Considering recombination adds one extra parameter to the model structure. The less restricted model (the one with more free parameters) was accepted when the χ2 value yielded P ≤ 0.05 in a one-tailed test.
Monte Carlo Testing of Data Fit.
To analyze the behavior of our models in the face of increasing amounts of observed data we generated random subsets of the observed data set of given fractions. At each fraction, we generated 1,000 subsets. For each subset, and for a given model, we determined whether the fit between the predicted marker density distribution and the observation subset could be rejected as statistically insignificant by the described χ2 test. We calculated the proportion of subsets for which the model prediction could not be rejected, and tabulated these proportions for each of the data fractions analyzed.
Results
Data Collection and Assessment.
We analyzed 25,901 genomic clones consisting of 7,122 finished and 18,779 draft sequences for which phrap (13) base-quality scores were available. We identified 21,020 clone overlaps (Methods) comprising 124,356 overlap fragments (see Methods). The total pairwise length of these overlaps was 1,105 megabases. Using the polybayes SNP discovery tool (12), we detected and submitted to dbSNP (24) 507,152 candidate SNPs (Methods). When the data were restricted to overlaps also present in the genome assembly (Methods), the number of overlaps reduced to 18,074 overlaps (average length of 51.1 ± 35.5 kb) containing 399,067 candidate SNPs. Measures of G+C, CpG, and repeat content in the overlap set were generally equivalent to average genome values, indicating it to be representative of the complete genome assembly (see Supporting Text, which is published as supporting information on the PNAS web site, www.pnas.org). To evaluate the quality of candidate SNPs, we tested for segregation in human populations and evaluated the sequence data for intrinsic error (see supporting information on the PNAS web site for details). Verification experiments show that the computational SNP predictions from the BAC overlap sequences are high quality, and the majority of SNPs are informative in one or more world populations (15).
Estimation of Genomewide Nucleotide Diversity.
By comparing the number of SNPs to the total length of pairwise overlaps analyzed, we estimated the overall value of pairwise nucleotide diversity, θ̂, for our complete dataset as 5.047 × 10−4 per nucleotide. This value, however, is biased by the inclusion of overlaps derived from the same source chromosome from a single individual. For the remainder of this study, we thus used only clone overlaps derived from interindividual libraries where both clone sequences were of draft quality. There were 3,174 such overlaps (18,152 overlap fragments, total overlap length 144 megabases). The nucleotide diversity value observed in this set (θ̂ = 7.571 × 10−4) is in excellent agreement with the value observed for shotgun reads aligned to the human genome (25). Our value for θ̂ indicates an expectation of one SNP in every 1,321 bp of paired sequence (n = 2). This average value, however, must be treated with caution, as the actual number of SNPs in an overlap of a given length is highly variable at all length scales examined (Fig. 2a). Our value for θ̂ corresponds to an effective size estimate of Ne = 9,464, if the mutation rate is 2.0 × 10−8. Ne should be doubled if the mutation rate is 1.0 × 10−8, putting it in line with the figure of 17,500 estimated from Alu diversity in the human genome (6).
Figure 2.

Comparison of model predictions to observed marker density data. (a) Marker density distributions observed in the interindividual overlap fragment data (ocher) and corresponding distributions predicted by our overall best-fitting, three-epoch bottleneck model (gray), at each analyzed length. (b) Predictions under the best-performing parameter set for each model structure studied, compared with observed (ocher) data (pairwise overlap length, 10 kb; censorship at 25 SNPs per alignment). r indicates models with recombination.
Of Two Extreme Models, Zero Recombination vs. Full Recombination, Zero Recombination Provides the Closest Fit to Our Data.
Based on the multikilobase scale marker density distributions in the overlap data, the “zero-recombination” model provides a clearly superior fit (Table 1) compared with the “infinite recombination” model, demonstrating that the inheritance of markers in close proximity is strongly correlated, and is consistent with extensive linkage disequilibrium observed in humans (26). This finding is an improvement over our previous study which, on the basis of marker density distribution measured in short read fragments aligned to genome sequence, did not carry sufficient power to distinguish between these competing models (25).
Examination of Second-Order Demographic Dynamics (Two-Epoch Model) Shows Population Collapse as the Dominant Effect.
Second-order models provided a superior fit compared with all stationary histories tested. The best-fitting parameters describe a severe, 2- to 7-fold, collapse of population size several hundred generations ago (Table 1). This result is consistent whether we used the zero-recombination models or a genome average recombination value. Additionally, within the class of all second-order models tested, models with a realistic recombination value fit significantly better than those that disregarded recombination effects.
Third-Order Models Show a Bottleneck History.
No third-order model that disregarded recombination could produce a fit superior to that of the best-fitting second-order (collapse) model with recombination (Table 1). However, the third-order models with recombination did produce an improved fit (see Fig. 2a) with the best-fitting parameters representing a “bottleneck”-shaped population history. A visual representation of the best-performing parameter sets within each model class is shown in Fig. 2b. These parameter values, together with the quantitative description of the fit values are given in Table 1. The third-order model structure with bottleneck parameters (Table 1) is our best description of the population history imprinted in the BAC overlap variation data. While all sets were qualitatively similar, the best-fitting parameter combination was slightly different for each overlap fragment length (data not shown). The overall optimum thus represents a compromise among the best-fitting parameter sets. We compared the predicted censored nucleotide diversity values predicted by the optimal model to the observed values at each length scale analyzed (Fig. 3). The fit is better at shorter sequence length than at longer lengths, as the majority of data available at shorter lengths were weighted heavier during the determination of a global optimum.
Figure 3.

Observed and predicted pairwise nucleotide diversity values at each overlap fragment length. Predicted values were based on the best-fitting bottleneck (three-epoch) model. Details of the censoring process (censored) and correction for censorship (uncensored) are described in the text.
Unbiased Estimates of Genomewide Nucleotide Diversity.
The direct measurement of pairwise nucleotide diversity is confounded with the effects of the censorship procedure (Methods). Using our best-fitting model, we projected the shape of the maker density curve beyond the censorship limit, and estimated the unbiased value of pairwise nucleotide diversity intrinsic to the overlap dataset as θ̂ = 8.25 × 10−4 per site per generation or one substitution-like polymorphism per 1,212 nucleotides. Assuming a genome average mutation rate of μ = 2.0 × 10−8 or 1.0 × 10−8 per site per generation, the corresponding long-term effective population size is Ne = 10,313 or 20,626, respectively.
Discussion
The dataset considered here, by virtue of its global nature, is expected to be robust against selection-induced distortions at individual loci and serves as a proper reagent to test theory describing the distribution of the number of mismatches in pairwise comparisons observed in a large number of different genomic regions.
Evidence from both archeological and genetic sources suggests that modern human populations are the product of an episode of explosive population growth beginning in the Pleistocene (9). Mismatch distributions from the hypervariable regions of the human mitochondrion exhibit a wavelike shape that has been interpreted as the sign of this expansion. However, limitations on the number of loci available for population genetic analysis have restricted a more detailed demographic inference (9). Our third-order analysis indicates that the dominant effect in our data is a collapse ca. 40,000 years ago (1,600 generations), consistent with the timing of the initial appearance of anatomically modern humans in Europe. To which population do our results refer? The ethnic composition of the DNA donors of the public human genome is not described, but genotyping of diallelelic, insertion-deletion type polymorphisms mined from the same BAC overlaps (27) suggests that the majority of these sequences represent donors of European origin. Similar patterns resulting in reduction of diversity and extension of linkage disequilibrium in European samples (26, 28–31), and reports of long invariable regions in the human genome (32) have been published. If our results indeed describe European chromosomes, then our estimated time of collapse is in good agreement with expansion time estimates from mitochondrial mismatch distributions (9).
How do we reconcile the signature of a population collapse seen in our data with the obvious recent explosive increase in population size? The recovery visible in our data is a very modest increase of effective population size during the Upper Paleolithic (Table 1). This finding suggests that recent population growth is not yet deeply imprinted in the nuclear marker density distribution, presumably because of the low average nuclear mutation rate.
Although our best, three-epoch, model produces a visually convincing fit to the observed data (Fig. 2a), application of a general χ2 test reveals that the fit can be rejected at the 5% (or even at the 1%) level, and the same is true for each of the other model structures (data not shown). Does this mean that we have to discard these models as inadequate descriptions of the observed data? The models are cartoon-like, and the marker density observed in the BAC overlap data was shaped by many unconsidered effects. If our models are not perfect, it is natural to ask how well they perform in an “absolute” sense, instead of relative terms, compared with each other. In all cases, model rejection is based on statistical significance, which in turn is always defined in the context of the test data at hand. Therefore, it is possible that a model that could not be rejected on the basis of a given dataset later proves inaccurate (rejected) when additional testing data become available. This consideration provides an alternative way to evaluate model accuracy, by examining how much data are necessary for the rejection of each of the competing models. The better the model, presumably, the larger the dataset that is required for its rejection as inaccurate. Accordingly, we have performed a computational experiment to examine how a χ2 measure of data fit between our observations and best-fitting models decays as more and more of the original data is considered. Results are shown in Fig. 4. Our best-fitting model (three-epoch history with uniform recombination) can be rejected only 50% of the time when subsets containing at least 15% (≈2,300 overlap fragments) of the original observed data are considered. We anticipate that evaluations of this sort will become increasingly useful in the analysis of genome-scale data (over-powered experiments) where numerical models will fail traditional significance tests when genome-sized datasets are considered.
Figure 4.

Model assessment based on the amount of data required for rejection by the general χ2 test. For each model, at each data fraction, we have plotted the percentage of successful trials (random data subsets for which the model cannot be rejected by the χ2 test at the 5% level). r indicates models with recombination.
What can we do to improve our models? We know that mutation rates are not uniform in the nuclear genome (19). There is also evidence for recombination hotspots (33). The existence of hotspots implies that, at least to some degree, recombination favors certain regions of the genome, a departure from the uniform distribution that we have assumed in our models. We also know that population history is far more complex than we can capture in our cartoon-like models invoking instantaneous stepwise changes of effective size. The same history may or may not be true for all chromosomal regions within the genome. There is also a large corpus of literature discussing nonneutral effects such as selective sweeps (34). It is desirable to refine our models by considering these effects. To confirm the generality of our results it will be necessary to evaluate similar data from non-European samples, analyze other characteristic distributions of SNPs such the allele frequency spectrum, and contrast our observations to data collected in molecular systems with alternative mutational mechanisms such as diallelic insertions/deletions, short tandem repeat polymorphisms (STRPs), and mitochondrial polymorphisms.
The amount of heterogeneity observed in the BAC overlaps should be a warning that average genome measures of nucleotide diversity should be used with caution. On the other hand, our computational experiments demonstrate that even relatively simple models of random drift are adequate to predict the range of variability in our data, suggesting that drift is an important (if not the most important) component of the resultant of forces that shape the regional distribution of human variability. It is striking, for example, that purely neutral forces can account for the fact that ≈10% of our 16-kb overlaps did not contain a single sequence variant (Fig. 2a). Observations such as this will require us to rethink our expectations when evaluating variation structure and its possible significance in specific genomic loci. The mature, fully annotated human reference sequence, together with an increase in well-characterized SNP markers, should afford us a high-resolution view to provide context for interpreting regional variation data, improving existing models of population history, and resolving the selective forces of genome evolution.
Supplementary Material
Acknowledgments
We thank James Weber for providing detailed recombination frequency data, Matt Minton and Rachel Donaldson for technical assistance, Stephen Altschul, Alexey Kondrashov, Raymond Miller, Ravi Sachidanandan, and John Spouge for useful discussion, and Andrew Clark for many useful comments on the manuscript. The work was supported in part by National Human Genome Research Institute Grant HG01720 (to P.-Y.K.).
Abbreviations
- SNPs
single-nucleotide polymorphisms
- BAC
bacterial artificial chromosome
Footnotes
Data deposition: SNPs discovered in this study are available from the dbSNP web site (http://ncbi.nlm.nih.gov/SNP), under the “KWOK” submitter handle (accession nos. ss1566252–ss2075206).
References
- 1.Clifford R, Edmonson M, Hu Y, Nguyen C, Scherpbier T, Buetow K H. Genome Res. 2000;10:1259–1265. doi: 10.1101/gr.10.8.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Irizarry K, Kustanovich V, Li C, Brown N, Nelson S, Wong W, Lee C J. Nat Genet. 2000;26:233–236. doi: 10.1038/79981. [DOI] [PubMed] [Google Scholar]
- 3.Altshuler D, Pollara V J, Cowles C R, Van Etten W J, Baldwin J, Linton L, Lander E S. Nature. 2000;407:513–516. doi: 10.1038/35035083. [DOI] [PubMed] [Google Scholar]
- 4.Mullikin J C, Hunt S E, Cole C G, Mortimore B J, Rice C M, Burton J, Matthews L H, Pavitt R, Plumb R W, Sims S K, et al. Nature. 2000;407:516–520. doi: 10.1038/35035089. [DOI] [PubMed] [Google Scholar]
- 5.Taillon-Miller P, Gu Z, Li Q, Hillier L, Kwok P Y. Genome Res. 1998;8:748–754. doi: 10.1101/gr.8.7.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sherry S T, Harpending H C, Batzer M A, Stoneking M. Genetics. 1997;147:1977–1982. doi: 10.1093/genetics/147.4.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Shaw N, Lane C R, Lim E P, Kalyanaraman N, et al. Nat Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- 8.Sunyaev S R, Lathe W C, III, Ramensky V E, Bork P. Trends Genet. 2000;16:335–337. doi: 10.1016/s0168-9525(00)02058-8. [DOI] [PubMed] [Google Scholar]
- 9.Harpending H, Rogers A. Annu Rev Genomics Hum Genet. 2000;1:361–385. doi: 10.1146/annurev.genom.1.1.361. [DOI] [PubMed] [Google Scholar]
- 10.Hudson R R. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Z, Schwartz S, Wagner L, Miller W. J Comput Biol. 2000;7:203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- 12.Marth G T, Korf I, Yandell M D, Yeh R T, Gu Z, Zakeri H, Stitziel N O, Hillier L, Kwok P Y, Gish W R. Nat Genet. 1999;23:452–456. doi: 10.1038/70570. [DOI] [PubMed] [Google Scholar]
- 13.Gordon D, Abajian C, Green P. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- 14.Lander E S, Linton L M, Birren B, Nusbaum C, Zody M C, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 15.Marth G, Yeh R, Minton M, Donaldson R, Li Q, Duan S, Davenport R, Miller R D, Kwok P Y. Nat Genet. 2001;27:371–372. doi: 10.1038/86864. [DOI] [PubMed] [Google Scholar]
- 16.Kimura M. Nature. 1968;217:624–626. doi: 10.1038/217624a0. [DOI] [PubMed] [Google Scholar]
- 17.Watterson G A. Theor Popul Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- 18.Hudson R R. In: Oxford Surveys in Evolutionary Biology. Futuyama D J, Antonovics J, editors. Vol. 7. Oxford: Oxford Univ. Press; 1990. pp. 1–44. [Google Scholar]
- 19. Kondrashov, A. S. (2003) Hum. Mutat., in press.
- 20.Nachman M W, Crowell S L. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu A, Zhao C, Fan Y, Jang W, Mungall A J, Deloukas P, Olsen A, Doggett N A, Ghebranious N, Broman K W, et al. Nature. 2001;409:951–953. doi: 10.1038/35057185. [DOI] [PubMed] [Google Scholar]
- 22.Brunet M, Guy F, Pilbeam D, Mackaye H T, Likius A, Ahounta D, Beauvilain A, Blondel C, Bocherens H, Boisserie J R, et al. Nature. 2002;418:145–151. doi: 10.1038/nature00879. [DOI] [PubMed] [Google Scholar]
- 23.Ott J. Analysis of Human Genetic Linkage. 2nd Ed. Baltimore: John Hopkins Univ. Press; 1991. [Google Scholar]
- 24.Sherry S T, Ward M, Sirotkin K. Genome Res. 1999;9:677–679. [PubMed] [Google Scholar]
- 25.Sachidanandam R, Weissman D, Schmidt S C, Kakol J M, Stein L D, Marth G, Sherry S, Mullikin J C, Mortimore B J, Willey D L, et al. Nature. 2001;409:928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- 26.Reich D E, Cargill M, Bolk S, Ireland J, Sabeti P C, Richter D J, Lavery T, Kouyoumjian R, Farhadian S F, Ward R, et al. Nature. 2001;411:199–204. doi: 10.1038/35075590. [DOI] [PubMed] [Google Scholar]
- 27.Weber J L, David D, Heil J, Fan Y, Zhao C, Marth G T. Am J Hum Genet. 2002;71:854–862. doi: 10.1086/342727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kimmel M, Chakraborty R, King J P, Bamshad M, Watkins W S, Jorde L B. Genetics. 1998;148:1921–1930. doi: 10.1093/genetics/148.4.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pereira L, Dupanloup I, Rosser Z H, Jobling M A, Barbujani G. Mol Biol Evol. 2001;18:1259–1271. doi: 10.1093/oxfordjournals.molbev.a003911. [DOI] [PubMed] [Google Scholar]
- 30.Goldstein D B, Weale M E. Curr Biol. 2001;11:R576–R579. doi: 10.1016/s0960-9822(01)00348-7. [DOI] [PubMed] [Google Scholar]
- 31.Gabriel S B, Schaffner S F, Nguyen H, Moore J M, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, et al. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 32.Miller R D, Taillon-Miller P, Kwok P Y. Genomics. 2001;71:78–88. doi: 10.1006/geno.2000.6417. [DOI] [PubMed] [Google Scholar]
- 33.Jeffreys A J, Kauppi L, Neumann R. Nat Genet. 2001;29:217–222. doi: 10.1038/ng1001-217. [DOI] [PubMed] [Google Scholar]
- 34.Nachman M W. Trends Genet. 2001;17:481–485. doi: 10.1016/s0168-9525(01)02409-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.