

Abstract
Objective:
To determine the role of p38 mitogen-activated protein kinase (MAPK) signaling in endotoxin-induced liver injury.
Background:
MAPKs have been reported to play a potential role in regulating inflammatory responses, but the role of p38 MAPK signaling in chemokine production, leukocyte recruitment, and hepatocellular apoptosis in the liver of endotoxemic mice is not known.
Methods:
Endotoxin-induced leukocyte-endothelium interactions were studied by use of intravital fluorescence microscopy in the mouse liver. Tumor necrosis factor-α (TNF-α) and CXC chemokines, liver enzymes, and apoptosis were determined 6 hours after endotoxin challenge. The specific p38 MAPK inhibitor SB 239063 was given immediately prior to endotoxin exposure. Phosphorylation and activity of p38 MAPK were determined by immunoprecipitation and Western blot.
Results:
Endotoxin increased phosphorylation and activity of p38 MAPK in the liver, which was markedly inhibited by SB 239063. Inhibition of p38 MAPK signaling dose-dependently decreased endotoxin-induced leukocyte rolling, adhesion, and sinusoidal sequestration of leukocytes. SB 239063 markedly reduced endotoxin-induced formation of TNF-α and CXC chemokines in the liver. Indeed, the endotoxin-provoked increase of liver enzymes and hepatocellular apoptosis were abolished and sinusoidal perfusion was restored in endotoxemic mice treated with SB 239063.
Conclusions:
This study demonstrates that p38 MAPK signaling plays an important role in regulating TNF-α and CXC chemokine production in endotoxemic liver injury and that inhibition of p38 MAPK activity abolishes endotoxin-induced leukocyte infiltration as well as hepatocellular apoptosis. These novel findings suggest that interference with the p38 MAPK pathway may constitute a therapeutic strategy against septic liver damage.
Cellular responses to stress signals are integrated by mitogen-activated protein kinases (MAPKs). We demonstrate that inhibition of p38 MAPK signaling protects against endotoxin-induced formation of chemokines, leukocyte recruitment, and hepatocellular apoptosis in the liver. Thus, targeting p38 MAPK may be an effective therapeutic strategy to treat septic liver injury.
Despite aggressive surgical interventions, antibiotic therapies, and immunoneutralization of tumor necrosis factor-α (TNF-α), sepsis and subsequent multiple organ failure remains the major cause of morbidity and mortality in intensive care units. Liver damage constitutes an insidious feature of septic patients, and numerous studies have demonstrated that leukocyte recruitment is a rate-limiting step in endotoxemic liver injury.1–3 Tissue accumulation of leukocytes is a multistep process initiated by tethering and rolling followed by subsequent firm leukocyte adhesion and transmigration.4,5 Endotoxin stimulation is known to increase formation of pro-inflammatory molecules, such as TNF-α, which in turn, up-regulate endothelial cell adhesion molecules, including P-selectin and intercellular adhesion molecule-1 (ICAM-1).6 P-selectin has been shown to support leukocyte rolling, which is a precondition for subsequent firm adhesion of leukocytes in postsinusoidal venules in the liver.7 Moreover, endotoxin-induced firm leukocyte adhesion in the hepatic microvasculature has been demonstrated to be mediated by lymphocyte function antigen-1 (LFA-1).8 LFA-1 belongs to the β2-integrin family of adhesion molecules expressed on leukocytes, which bind to members of the immunoglobulin supergene family expressed on endothelial cells, such as ICAM-1.5 A recent study demonstrated that endotoxin-induced extravascular infiltration of leukocytes into the liver is critically dependent on the formation and action of CXC chemokines, including macrophage inflammatory protein-2 (MIP-2) and cytokine-induced neutrophil chemoattractant (KC).9 Thus, the adhesive mechanisms behind hepatic recruitment of leukocytes are relatively well known, whereas the signaling pathways regulating these endotoxin-provoked pro-inflammatory activities in the liver remain elusive.
Signal transduction pathways through stress-activated mitogen-activated protein kinases (MAPKs) constitute major pathways by which extracellular stimuli are integrated and transmitted as an intracellular signal.10 MAPKs have been reported to play a key role in regulating cytokine production in response to a broad range of stimuli, including ischemia/reperfusion,11,12 ultraviolet light,13 heat shock,14 and microbial infection.15,16 The 3 major MAPK signaling pathways include p38 MAPK, extracellular signal-regulated protein kinases (ERK1/2), and c-Jun NH2-terminal protein kinases (JNKs).10 In particular, activation of the p38 MAPK pathway has been implicated in the generation of pro-inflammatory cytokines, such as TNF-α, interleukin-1 (IL-1), IL-6, and the CXC chemokine IL-8.17,18 Indeed, it has been shown that inhibition of p38 MAPK may protect against numerous and diverse disease processes, including models of arthritis,19 ischemia/reperfusion injury,11,12 and cardiac hypertrophy and dysfunction.20 Convincing data have shown that p38 MAPK can regulate LPS-induced secretion of TNF-α from macrophages in vitro.17 In addition, 2 previous studies have reported activation of p38 MAPK in the liver during endotoxemia.21,22 However, the role of the p38 MAPK pathway in endotoxin-induced leukocyte recruitment and hepatocellular injury is not known.
Based on the considerations above, the aim of the present study was to systematically define the regulatory mechanisms and functional importance of p38 MAPK signaling in endotoxin-induced CXC chemokine expression, leukocyte recruitment, and apoptosis in the liver.
MATERIALS AND METHODS
Animals
Adult male C57Bl/6 mice (22–27 g) were kept on a 12–12 hour light-dark cycle with free access to food and tap water. Animals were anesthetized by intraperitoneal (i.p.) administration of 7.5 mg ketamine hydrochloride (Hoffman-La Roche, Basel, Switzerland) and 2.5 mg xylazine (Janssen Pharmaceutica, Beerse, Belgium) per 100 mg body weight. The local ethics committee at Lund University approved all the experiments of this study.
Experimental Protocol
Mice were pretreated i.p. for 6 hours with or without (negative control animals) a combination of lipopolysaccharide (LPS, 10 μg/mouse, Sigma Chemical Co.) and d-galactosamine (Gal, 18 mg/mouse, Sigma Chemical Co., St. Louis, MO) dissolved in phosphate-buffered saline (PBS). To delineate the role of p38 MAPK in endotoxin-induced leukocyte recruitment and liver injury, we used a specific p38 MAPK inhibitor, SB 239063 [trans-1-(4-hydroxycyclohexyl)-4-(4-fluorophenyl)-5-(2-methoxypyridimidin-4-yl)imidazole] (Sigma Chemical Co.) in endotoxemic mice. SB 239063 is a second-generation p38 MAPK inhibitor that has been shown to be extremely selective for p38 MAPK (IC50 for inhibition of p38 MAPK: 0.44 μmol/L; for MEK1/2, ERK1/2, and JNK, >10 μmol/L).23 SB 239063 (4 and 40 mg/kg) or vehicle (acidified 0.5% tragacanth) was administered i.p. immediately prior to endotoxin challenge.
Intravital Microscopy
The right jugular vein was cannulated with a polyethylene catheter for intravenous (i.v.) administration of test substances. A transverse subcostal incision was performed, and the ligamentous attachments from the liver to the diaphragm and the abdominal wall were gently released. Animals were positioned on their left side and the left liver lobe was carefully exteriorized onto an adjustable stage for analysis of hepatic microcirculation by use of intravital fluorescence microscopy as described previously.9 Briefly, we used a modified Olympus microscope (BX50WI, Olympus Optical Co. GmbH, Hamburg, Germany), and the image was televised using a charge-coupled device video camera (FK 6990 Cohu, Pieper GmbH, Schwerte, Germany) and recorded on videotape for subsequent off-line evaluation. Blood perfusion within individual microvessels was studied after contrast enhancement by FITC-dextran (0.1 mL, 2 μmol/kg, Sigma Chemical Co.). In vivo labeling of leukocytes with rhodamine-6G (0.1 mL, 0.05 mg/mL, Sigma Chemical Co.) enabled quantitative analysis of leukocyte flow behavior in the hepatic microcirculation. Five postsinusoidal venules with connecting sinusoids were evaluated in each animal. Venular leukocyte rolling was measured by counting the number of cells rolling in the venule during 30 seconds and expressed as cells/min. Leukocyte adhesion was measured by counting the number of cells that adhered along the venular endothelium and remained stationary during the observation period of 30 seconds, and expressed as cells/mm venule length. Sinusoidal trapping was determined in 20 to 30 high power fields and given as cell/10 high power fields. The number of perfused sinusoids is given as a percentage of the total number of sinusoids observed (ie, sinusoidal perfusion). Hepatocyte apoptosis was determined by topical application of the fluorochrome Hoechst 33342 (0.02 mL, 0.2 μg/mL, Molecular Probes, Leiden, the Netherlands) onto the liver surface for staining of hepatocyte DNA. Hoechst 33342 is a fluorescent dye that enables analysis of nuclear morphology, eg, nuclear condensation and fragmentation in cultured hepatocytes and endothelial cells.24 Hepatocyte apoptosis is given as the percentage of the number of hepatocyte nuclei showing apoptotic features from the total number of hepatocyte nuclei observed. Blood was drawn and alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were analyzed using standard spectrophotometric procedures.
Caspase-3 Protease Activity
Caspase-3 protease activity in the liver tissue was measured using a caspase-3 colorimetric assay kit (R & D Systems) according to the manufacturer's instructions. Briefly, after homogenization of whole liver tissue in cell lysis buffer, homogenates were centrifuged and the supernatant was incubated with DEVD-pNA and reaction buffer for 90 minutes at 37°C. Levels of the chromophore pNA released by caspase-3 activity were quantified spectrophotometrically. The data are given as fold increases in caspase-3 activity of test livers relative to PBS-treated control livers.
ELISA
Levels of TNF-α, KC, and MIP-2 in the liver and serum were determined by use of double antibody Quantikine ELISA kits (R & D Systems) according to the manufacturer's instructions using recombinant murine TNF-α, KC, and MIP-2 as standards. The minimal detectable protein concentrations are less than 0.5 pg/mL.
Immunoprecipitation and p38 MAPK Activity Assay
To analyze the kinase activity of p38 MAPK and the inhibitory effect of p38 MAPK by SB 239063 in the liver, an in vitro kinase assay was used according to the manufacturer's instructions (Cell Signaling Technology, Beverly, MA). Liver sections were weighed and homogenized in lysing buffer. Activated p38 MAPK was immunoprecipitated by incubating lysates with a p38 MAPK antibody immobilized by crosslinking to agarose hydrazide beads (Cell Signaling Technology). Next, the beads were pelleted, and the precipitated p38 MAPK was incubated with activating transcription factor-2 (ATF-2) fusion protein and ATP for 30 minutes at 30°C. After the reaction was terminated by boiling in SDS sample buffer, samples were centrifuged and the pellets discarded. Samples were separated by electrophoresis and transferred onto nitrocellulose membrane. Membranes were probed with an antibody specific to phosphorylated (Thr71) ATF-2 and a secondary horseradish peroxidase-conjugated antibody, and the resultant signal was detected by adding diaminobenzidine.
Histology
Samples were taken from the left lobe of liver and fixed in 4% formaldehyde phosphate buffer overnight. Dehydrated, paraffin-embedded, 6-μm sections were stained with hematoxylin and eosin and analyzed under light microscopy.
Statistical Analyses
Data are presented as mean values ± SEM. Statistical evaluations were performed using Kruskal-Wallis one-way analysis of variance on ranks followed by multiple comparisons versus control group (Dunn's method). P < 0.05 was considered significant, and n represents the number of animals.
RESULTS
Phosphorylation and Activity of p38 MAPK
Liver tissue proteins were extracted, and p38 MAPK was examined by Western blot analysis. We observed that challenge with LPS caused clear-cut phosphorylation of p38 MAPK in the liver (not shown). To determine whether this increase in p38 MAPK phosphorylation in the liver actually represented a functional increase in p38 MAPK activity, phosphorylated p38 MAPK immunoprecipitated from liver lysates was examined for its capacity to phosphorylate the downstream p38 MAPK substrate, ATF-2. As shown in Figure 1, it was found that phosphorylation of ATF-2 was increased, suggesting that the hepatic p38 MAPK activity was elevated in LPS-treated mice. Notably, we found that ATF-2 phosphorylation (ie, p38 MAPK activity) was markedly reduced in endotoxemic animals pretreated with SB 239063 (Fig. 1). Administration of SB 239063 alone did not stimulate p38 MAPK activity in the liver (Fig. 1).
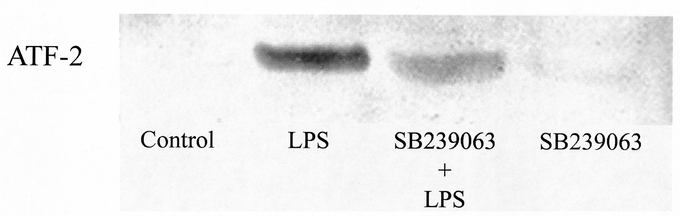
FIGURE 1. The activity of p38 MAPK in liver lysates was examined by analyzing phosphorylation (Thr71) of ATF-2 (see Materials and Methods for details). Mice were challenge with a combination of lipopolysaccharide (LPS, 10 μg) and d-galactosamine (Gal, 18 mg). Negative control animals received only PBS and vehicle (Control). Mice were pretreated i.p. with vehicle or an inhibitor of p38 MAPK (SB 239063, 40 mg/kg) immediately prior to LPS/Gal challenge.
Hepatocellular Injury and Apoptosis
Endotoxin challenge provoked a significant liver injury, indicated by the prominent increase in liver injury enzymes (Fig. 2). Indeed, we found that ALT and AST increased by more than 57-fold and 50-fold, respectively, in endotoxemic mice (Fig. 2; P < 0.05 versus negative control, n = 5–8). Administration of SB 239063 significantly protected against the LPS-induced increase in liver enzymes in a dose-dependent fashion (Fig. 2). For example, pretreatment with 40 mg/kg of SB 239063 reduced ALT and AST from 35.2 ± 2.2 μKat/L and 40 ± 5.3 μKat/L down to 4.2 ± 1 μKat/L and 7.5 ± 1 μKat/L, respectively, in endotoxemic mice (Fig. 2; P < 0.05 versus vehicle, n = 5–8). Thus, inhibition of p38 MAPK signaling reduced liver enzymes by more than 80%. Apoptosis is a key feature of endotoxin-induced liver damage. Herein, we evaluated apoptosis using 2 different methods. First, we determined the level of apoptosis by use of the DNA-binding fluorescent dye Hoechst 33342, which stains the nuclei of hepatocytes and permits quantification of the percentage of cells with nuclear condensation and fragmentation.24 In negative control mice, we found that the percentage of apoptotic hepatocytes was 2.7% ± 0.2%, which was significantly increased by endotoxin to 31.8% ± 3%, ie, by almost 12-fold (Fig. 3; P < 0.05 versus negative control, n = 5–8). Notably, it was observed that pretreatment with SB 239063 dose-dependently reduced endotoxin-induced apoptosis (Fig. 3). For example, 40 mg/kg of SB 239063 decreased the percentage of apoptotic hepatocytes down to 6.0% ± 1.1% in endotoxemic mice, corresponding to an 88% reduction (Fig. 3; P < 0.05 versus vehicle, n = 5–8). Next, we determined the activation of the protease caspase-3, which is an important and early step in hepatic parenchymal cell apoptosis. Indeed, we found that caspase-3 activity in the liver was elevated almost 3-fold in animals exposed to endotoxin (Fig. 4; P < 0.05 versus negative controls, n = 5–8). Interestingly, this LPS-induced increase in caspase-3-activity was reduced by 81% in animals pretreated with 40 mg/kg of SB 239063 (Fig. 4; P < 0.05 versus vehicle, n = 5–8). Moreover, morphologic examination showed normal microarchitecture in livers from PBS-treated animals (Fig. 5A), whereas administration of LPS resulted in severe destruction of the liver tissue structure characterized by massive panlobular hemorrhage and necrosis as well infiltration of neutrophils (Fig. 5B). In line with the aforementioned data on apoptosis and liver enzymes, it was found that pretreatment with 40 mg/kg of SB 239063 almost completely protected against endotoxin-induced destruction of tissue architecture, hepatocellular damage, and neutrophil infiltration in the liver (Fig. 5C).
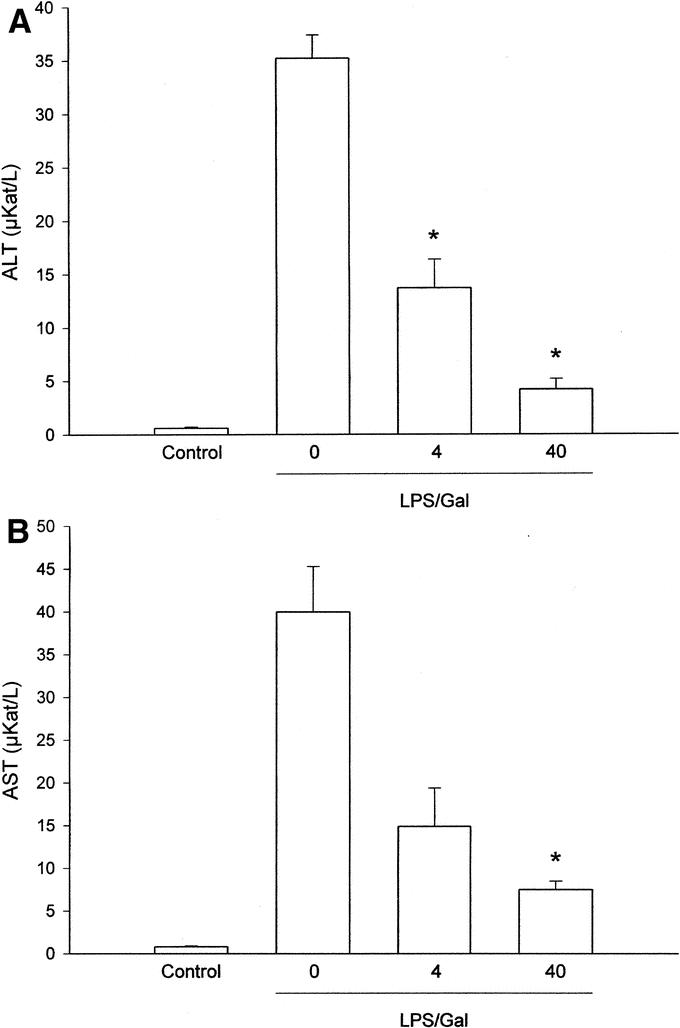
FIGURE 2. Alanine aminotransferase (ALT) (A) and aspartate aminotransferase (AST) (B) were determined 6 hours after treatment with a combination of lipopolysaccharide (LPS, 10 μg) and d-galactosamine (Gal, 18 mg) in mice. Negative control animals received only PBS and vehicle (Control). Mice were pretreated i.p. with vehicle (0) or an inhibitor of p38 MAPK (SB 239063, 4 and 40 mg/kg). Data represent mean ± SEM *P < 0.05 versus vehicle (0); and n = 5–8.
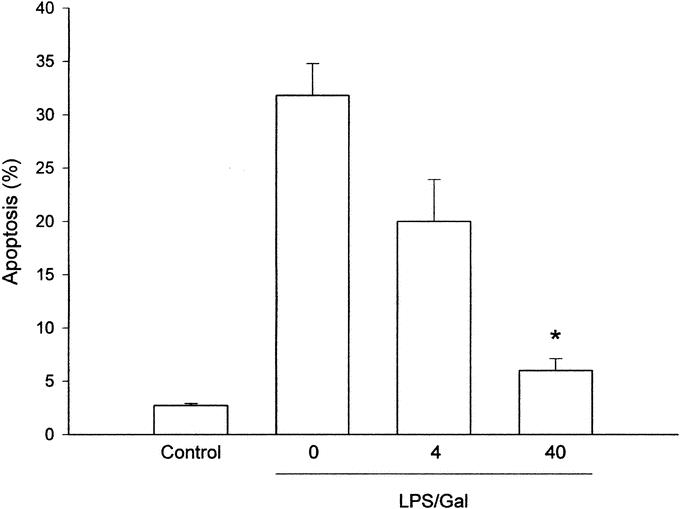
FIGURE 3. Apoptosis of hepatocytes 6 hours after treatment with a combination of lipopolysaccharide (LPS, 10 μg) and d-galactosamine (Gal, 18 mg) in mice. Negative control animals received only PBS and vehicle (Control). Mice were pretreated i.p. with vehicle (0) or an inhibitor of p38 MAPK (SB 239063, 4 and 40 mg/kg). Data represent mean ± SEM *P < 0.05 versus vehicle (0); and n = 5–8.
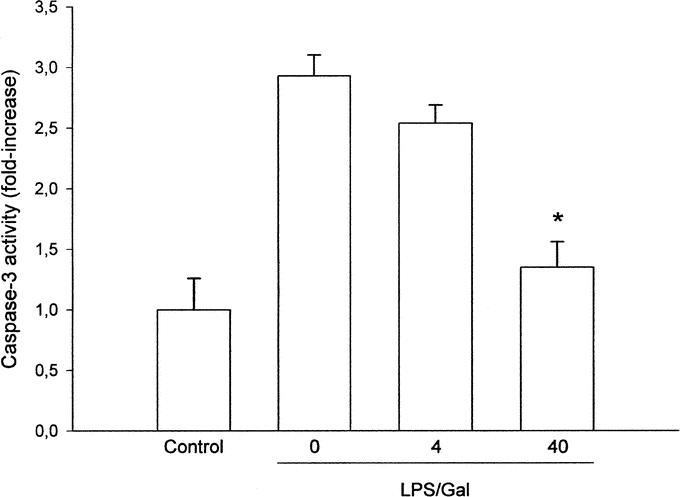
FIGURE 4. Activity of the protease caspase-3 in the liver 6 hours after treatment with a combination of lipopolysaccharide (LPS, 10 μg) and d-galactosamine (Gal, 18 mg). Negative control animals received only PBS and vehicle (Control). Mice were pretreated i.p. with vehicle (0) or an inhibitor of p38 MAPK (SB 239063, 4 and 40 mg/kg). Data represent mean ± SEM *P < 0.05 versus vehicle (0); and n = 5–8.
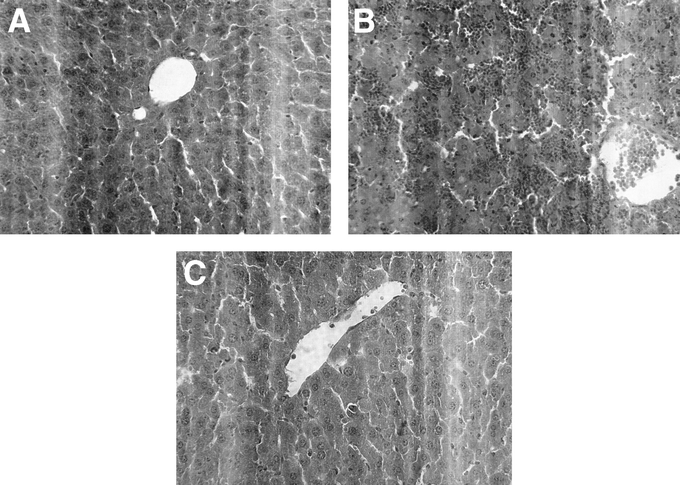
FIGURE 5. Sections of the liver were stained with hematoxylin and eosin and representative pictures are shown. Animals were treated with a combination of lipopolysaccharide (LPS, 10 μg) + d-galactosamine (Gal, 18 mg) for 6 hours. A) Negative control animals received only PBS and vehicle. Mice were pretreated i.p. with vehicle (B) or an inhibitor of p38 MAPK (SB 239063, 40 mg/kg) (C) immediately prior to LPS/Gal challenge (original magnification ×500).
Leukocyte Responses and Sinusoidal Perfusion
Leukocyte recruitment is a rate-limiting step in endotoxin-induced liver injury.2,3 The number of rolling and adherent leukocytes was 3 ± 0.3 cells/min and 2 ± 0.1 cells/mm, respectively, in negative control mice (Fig. 6; n = 5). Administration of LPS greatly enhanced leukocyte responses in postsinusoidal venules (Fig. 6). Thus, endotoxin challenge increased leukocyte rolling to 22.8 ± 3.1 cells/min and firm leukocyte adhesion to 18.4 ± 2.8 cells/mm (Fig. 6; P < 0.05 versus negative control, n = 5). Notably, pretreatment with SB 239063 (40 mg/kg) significantly decreased leukocyte-endothelium interactions in the liver. Indeed, inhibition of p38 MAPK activity decreased leukocyte rolling and adhesion down to 8.6 ± 1.5 cells/min and 8.6 ± 1.2 cells/mm, respectively (Fig. 6; P < 0.05 versus vehicle, n = 5), which corresponds to a more than 60% reduction in leukocyte responses. Endotoxin-induced hepatic injury is also characterized by reduced perfusion and increased sequestration of leukocytes in the sinusoids.3 Thus, it was observed that the percentage of perfused sinusoids decreased from 98.1% ± 0.2% down to 80.4% ± 2.5% in response to LPS challenge (P < 0.05 versus negative control, n = 5). Pretreatment with 40 mg/kg of SB 239063 significantly increased sinusoidal perfusion up to 91.6% ± 1.8% (P < 0.05 versus vehicle, n = 5). Moreover, LPS stimulation increased sinusoidal trapping of leukocytes by more than 5-fold, ie, from 5.0 ± 0.5 cells/10 high power fields up to 38.8 ± 3.5 cells/10 high power fields (P < 0.05 versus negative control, n = 5). We found that 40 mg/kg of SB 239063 significantly improved sinusoidal perfusion by 63% and decreased sinusoidal sequestration of leukocytes by 84% in endotoxemic mice (P < 0.05 versus vehicle, n = 5).
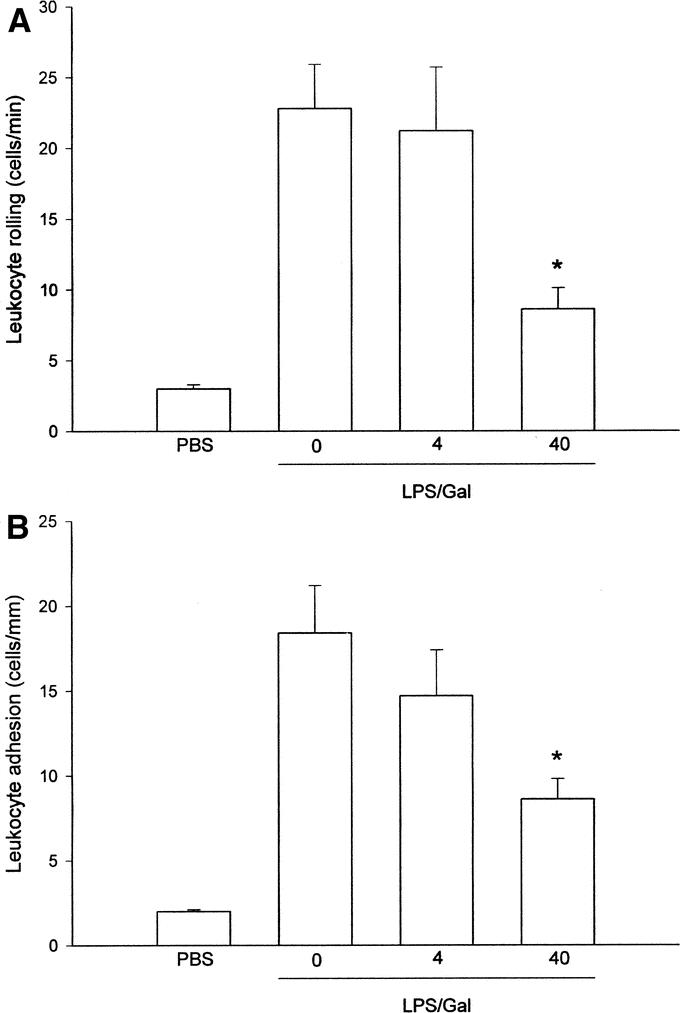
FIGURE 6. Leukocyte rolling (A) and firm (B) adhesion in hepatic postsinusoidal venules 6 hours after treatment with a combination of lipopolysaccharide (LPS, 10 μg) + d-galactosamine (Gal, 18 mg) in mice. Negative control animals received only PBS and vehicle (Control). Mice were pretreated i.p. with vehicle (0) or an inhibitor of p38 MAPK (SB 239063, 4 and 40 mg/kg). Data represent mean ± SEM *P < 0.05 versus vehicle (0); and n = 5.
Expression of TNF-α and CXC Chemokines
Numerous studies have shown that TNF-α plays a dominant role in LPS-induced liver injury.25 Indeed, we found that endotoxin challenge provoked clear-cut induction of TNF-α both in the liver and circulation (Fig. 7). Baseline levels of TNF-α in the liver were 8.2 ± 4.6 pg/g tissue (Fig. 7A). LPS exposure increased liver TNF-α to 858 ± 174 pg/g (Fig. 7A; P < 0.05 versus negative control, n = 5–8). In mice treated with 40 mg/kg of SB 239063, the level LPS-induced TNF-α in the liver was reduced down to 366 ± 60 pg/g (Fig. 7A; P < 0.05 versus vehicle, n = 5–8). Moreover, LPS increased the serum levels of TNF-α from 9.9 ± 3.7 pg/mL to 228 ± 55 pg/mL (Fig. 7B; P < 0.05 versus negative control, n = 5–8). Pretreatment with SB 239063 (40 mg/kg) decreased serum levels of TNF-α down to 96 ± 23 pg/mL in endotoxemic animals (Fig. 7B; P < 0.05 versus vehicle, n = 5–8). Together, it was found that inhibition of p38 MAPK signaling reduced endotoxin-induced production of TNF-α in the liver and serum by more than 58%. CXC chemokines have been shown to play a critical role in the hepatic infiltration of leukocytes.9 The liver content of CXC chemokines in negative controls was low but detectable (Fig. 7C, D). Notably, the expression of MIP-2 and KC in livers of endotoxin-treated mice increased markedly, ie, from 0.08 ± 0.03 ng/g and 0.24 ± 0.04 ng/g at baseline up to 20.6 ± 2.6 ng/g and 36.3 ± 12.4 ng/g liver tissue, respectively (Fig. 7C, D, P < 0.05 versus negative controls, n = 5–8). Interestingly, pretreatment with 40 mg/kg of SB 239063 reduced MIP-2 and KC levels down to 5.0 ± 1.1 ng/g and 11.2 ± 4 ng/g liver tissue, respectively, in endotoxemic mice (Fig. 7C and D; P < 0.05 versus vehicle, n = 5–8). Thus, we observed that inhibition of p38 MAPK signaling decreased LPS-provoked expression of CXC chemokines by more than 69% in the liver.

FIGURE 7. Levels of tumor necrosis factor-α (TNF-α) in the liver (A), serum (B), and hepatic (C) levels of macrophage inflammatory protein-2 (MIP-2) and cytokine-induced neutrophil chemoattractant (KC) (D) 6 hours after treatment with a combination of lipopolysaccharide (LPS, 10 μg) and d-galactosamine (Gal, 18 mg). Negative control animals received only PBS and vehicle (Control). Mice were pretreated i.p. with vehicle (0) or an inhibitor of p38 MAPK (SB 239063, 4 and 40 mg/kg). Data represent mean ± SEM *P < 0.05 versus vehicle (0); and n = 5–8.
DISCUSSION
This study demonstrates an important role of p38 MAPK signaling in endotoxin-induced liver injury. Our data show that phosphorylation of p38 MAPK regulates hepatic expression of TNF-α and CXC chemokines in endotoxemic mice. Moreover, we found that inhibition of p38 MAPK activity protected against LPS-induced leukocyte rolling, adhesion, and sequestration in the liver microvasculature. Indeed, this inhibition of leukocyte responses exerted by p38 MAPK inhibition significantly decreased endotoxin-induced hepatocellular apoptosis and damage. Taken together, these novel findings suggest that the p38 MAPK signaling pathway plays a central role in the generation of pro-inflammatory mediators, leukocyte recruitment, and hepatotoxicity in the liver and that p38 MAPK may be an attractive target to protect against septic liver injury.
MAPKs play a key role in inflammation by integrating and processing extracellular stress signals and regulate fundamental cellular responses, such as pro-inflammatory cytokine production, cytoskeletal reorganization, and apoptosis.10 In the present study, we found that endotoxin challenge increased phosphorylation and activity of p38 MAPK in the liver. Moreover, it was observed that LPS exposure caused p38 MAPK-dependent phosphorylation of ATF-2 in the liver. Notably, administration of SB 239063, which is second-generation p38 MAPK inhibitor,23 not only blocked the kinase activity of p38 MAPK (ie, ATF-2 phosphorylation) but also reduced liver enzymes (ie, ALT and AST) by more than 80% in endotoxemic mice. These findings suggest that inhibition of the p38 MAPK signaling pathway significantly protects against endotoxin-induced hepatic damage and that septic liver injury may thus be added to the list of conditions, including reperfusion injury,11,12 joint inflammation,19 asthma,26 and glomerulonephritis27 that are ameliorated by interfering with p38 MAPK activity. Convincing data have demonstrated that TNF-α plays a key role in the pathogenesis of LPS-induced hepatocellular injury.25 Indeed, we found that inhibition of p38 MAPK signaling reduced the levels of TNF-α in the liver and circulation by nearly 60% in endotoxemic mice, supporting the notion that interference with p38 MAPK signaling exerts a protective effect on endotoxin-induced hepatotoxicity. One important role of pro-inflammatory cytokines in sepsis is to activate endothelial cells and support the recruitment of leukocytes.1–3 Tissue infiltration of leukocytes constitutes a key feature in host-defense reactions; but under certain situations, such as ischemia-reperfusion injury, graft rejection, and endotoxemia, activation and infiltration of leukocytes may cause organ damage.27 Indeed, numerous studies have documented that leukocyte recruitment constitutes a rate-limiting step in septic liver injury by demonstrating that LPS-provoked liver injury is markedly attenuated in adhesion molecule (P-selectin and LFA-1)-targeted and in neutrophil-depleted animals.7,8 By use of intravital fluorescence microscopy, we found herein that inhibition of p38 MAPK activity markedly decreased leukocyte rolling and firm adhesion in postsinusoidal venules as well as trapping of leukocytes in sinusoids in the liver of endotoxemic mice, suggesting that hepatic leukocyte responses are regulated by the p38 MAPK signaling pathway in response to LPS challenge. Indeed, we found that the inhibitory effect of SB 239063 on leukocyte recruitment (60%–84% reduction) correlated very well to the reduction in liver enzymes (80% reduction) and apoptosis (81%–88% reduction). Chemokines have emerged as a dominating group of molecules regulating tissue infiltration of leukocytes in several disease models.9,30–32 We have previously shown that CXC chemokines play a critical role in endotoxin-induced extravasation of leukocytes in the liver.9 In this study, we observed that inhibition of p38 MAPK activity markedly suppressed hepatic formation of MIP-2 and KC in endotoxemic mice. Indeed, we found that inhibition of p38 MAPK not only abolished CXC chemokine formation but also markedly decreased LPS-induced leukocyte responses in the liver. Knowing that inhibition of CXC chemokine function substantially protects against LPS-induced liver injury,9,33 our novel data suggest that attenuation of CXC chemokine formation may help explain this protective effect in the liver exerted by SB 239063. Thus, considered together, this is the first study showing that the p38 MAPK signaling pathway plays a critical role in LPS-induced expression of CXC chemokines and leukocyte recruitment in the liver. In this context, it is worth noting that these findings do not necessarily exclude a potential role of other MAPK subfamilies, such as ERKs and JNKs, in LPS-induced hepatotoxicity. Considering that the ERK signaling pathway does not appear to play a significant role in mediating LPS-provoked TNF-α production in macrophages,34,35 whereas both JNK and p38 MAPK are required for maximal TNF-α biosynthesis in macrophages,36–38 it may be worthwhile to also investigate the potential role of JNK signaling in this liver model in the future.
It is widely held that apoptosis constitutes a central feature in sepsis,39 and numerous studies have confirmed prominent apoptosis of hepatocytes in endotoxin-induced liver injury.7,9,40 In this study, we analyzed apoptosis morphologically by Hoechst 33324 staining and biochemically by determining caspase-3 activity in the liver. Both methods demonstrated that LPS increased the level of apoptosis in the liver. Herein, we observed that inhibition of the p38 MAPK pathway markedly reduced the number of apoptotic hepatocytes by 88%, which corresponded very well to the 81% decrease in hepatic levels of caspase-3 in endotoxemic mice. TNF-α may induce hepatocellular apoptosis directly or indirectly via stimulating hepatic infiltration of leukocytes. In addition, based on recent findings,40,41 it has been suggested that there may be an escalating mechanism in the liver, involving chemokine-induced neutrophil recruitment, which causes apoptosis on one hand, and apoptosis-induced secretion of chemokines, which causes neutrophil infiltration on the other.9 The fact that inhibition of p38 MAPK activity decreased both TNF-α formation and subsequent leukocyte infiltration may together help to explain the highly potent antiapoptotic effect exerted by SB 239063 in endotoxin-induced liver damage as observed in the present study.
Sepsis is a dynamic process characterized by an early pro-inflammatory phase associated with organ damage and later an anti-inflammatory phase characterized by immunosuppression and increased susceptibility to infectious complications.42 Our data show that pretreatment with the p38 MAPK inhibitor protects against liver injury early in the hyperinflammatory phase of septicemia. In this respect, it is interesting to note that previous studies have also suggested a role for p38 MAPK signaling in the induction of immune dysfunction at later stages of septicemia.43–45 For example, it has been shown that Th1-lymphocyte-mediated immune responses are suppressed 12 hours after induction of sepsis.43,45 Thus, Song et al45 reported that inhibition of p38 MAPK signaling attenuates IL-4 and IL-10 expression while increasing production of IL-2 and IFN-γ in lymphocytes and thus helps restoring Th1 responsiveness in the hypoinflammatory phase of septic mice. Considered together with our present findings, it may be suggested that inhibition the p38 MAPK pathway exerts protective effects in multiple phases of septicemia, although the actual mechanistic role of p38 MAPK signaling may be principally different in a specific phase of the immune response in sepsis. In this context, it is interesting to note that modulation of the major cytokines involved in sepsis, such as TNF-α and IL-10, may be more complex since these mediators exert diametrically opposite effects in the different phases of sepsis. For example, inhibition of TNF-α synthesis is protective in the early hyperinflammatory phase46,47 but may be deleterious in the later immunosuppressive phase when TNF-α is necessary to combat infections.48,49 Likewise, pretreatment with IL-10 is beneficial in the early hyperinflammatory phase of sepsis because of its anti-inflammatory capacity50,51 and immunoneutralization of IL-10 at the time of sepsis induction exacerbates mortality.52,53 In contrast, inhibition of IL-10 after induction of sepsis actually improves survival.52 Indeed, this immunosuppressive phase of sepsis has been reported to be induced by endogenous IL-10 secretion.42 Thus, TNF-α and IL-10 appear to be either protective or harmful depending on the time of intervention. Thus, it may be speculated that this complex balance of cytokine actions may be circumvented for therapeutic purposes by p38 MAPK inhibitors that appear to be protective in all phases of sepsis.
CONCLUSION
This study shows that phosphorylation and activity of p38 MAPK are increased in septic liver injury. Moreover, our data demonstrate that competitive inhibition of p38 MAPK activity by administration of SB 239063 inhibits endotoxin-induced CXC chemokine formation, leukocyte recruitment, and hepatocellular apoptosis in the liver. Thus, these novel findings suggest that p38 MAPK may be a useful target to treat pathologic inflammation in septic liver injury.
Discussions
Dr. Eggermont: I congratulate you on a comprehensive experimental study demonstrating the role of p38 kinase dependent pathways in endotoxemia-mediated liver damage and the number of cascades that it actually can down-regulate or up-regulate and how you can intervene with p38-kinase inhibitors.
My first question regards that you are the first one to show the role of leukocyte entrapment in this model for this particular kinase pathway. In TNF-mediated vascular damage models that we have in tumor models in my laboratory, we found that leukocyte entrapment plays an absolutely crucial role to achieve complete vascular damage and that IL-1β is a crucial cytokine. In your model, it is MIP2, macrophage inflammatory protein 2. My question is whether you have looked at a potential role of IL-1β. For in our tumor model, it is not just crucial for endothelial apoptosis and vascular destruction but also for the leukocyte invasion of the tumor parenchyma. So there might be a difference here between tumors and livers, and what you have demonstrated may be a liver-specific phenomenon.
My second question is: are the leukocytes crucial for the necrosis that you have shown? You quantify extensively the apoptosis and the direct TNF-mediated apoptosis, and you have evaluated that very nicely in 2 ways. But you are a little scant with your evaluation of the amount of necrosis that is actually observed, and I think that is crucial because that sets into motion the whole second wave of damaging effects in your liver endotoxemia model.
My third question is that I would like you to speculate on the potential use of these kinase inhibitors in the clinical setting because here you are mostly showing a protective effect when the drug is administered before the endotoxemia. Obviously, in the clinic, the question will be different. And will the drug be administered when endotoxemia is suspected or diagnosed? So what would be your time window, what is your opportunity to use these types of kinase inhibitors successfully in the clinical setting? When will it be too late?
Dr. Thorlacius: Thank you Dr. Eggermont for your questions.
The answer to question number 1 is no. It is known that this model is completely dependent on TNF, and interleukin-1 is probably also involved. When it comes to the role of leukocytes in causing the injury in this model, I want to emphasize that since inhibition of specific adhesion molecules, such as P-selectin and LFA-1, blocks leukocyte recruitment in this model and also protects against the liver damage, one may conclude that leukocyte recruitment plays a critical role in endotoxin-induced liver injury. Concerning the clinical setting and the time window for therapy, I would like to stress that sepsis has 2 phases: an early hyperinflammatory and a later hypoinflammatory phase, characterized by immunosuppression, in which most of the patients in clinical settings are observed. The interesting thing is that we have shown here that, in the early hyperinflammatory phase, p38 MAPK inhibition protects against the inflammatory organ damage. Others studies have shown using a similar model that the immunosuppressive phase after 12 hours characterized by lymphocyte depletion in secondary lymphatics due to apoptosis is also reversed by p38 MAPK inhibition. Moreover, in the late hypoinflammatory phase, you can see a shift from Th1 protective immunity against bacteria to a Th2 response and p38 MAPK inhibition actually restores the Th1 responsiveness by increasing interleukin 2 synthesis. So I think that for therapeutic purposes and considering the time window that MAPK inhibitors may actually work both in early and late phases of the septic liver injury. This is in contrast to the complex cytokine interactions observed in sepsis, ie, TNF-α is causing organ damage early but is necessary to combat infections in the late hypoinflammatory phase and vice versa IL-10 is protecting in the early phase but deleterious during the immunosuppressive late phase. Thus, p38 MAPK inhibition may for therapeutic purposes circumvent the complexity of cytokine actions, which are usually operating opposite directions in different phases of sepsis.
Dr. Senninger: I also enjoyed your paper very much. I have 2 short questions:
First is concerning your experiment: was this a consistent finding in all animals and how large was the group size? But secondly and more importantly, I would like to address the problem of marginal livers. You might be aware that Peter Friend in Oxford has a porcine transplant model running, whereby extended warm perfusion preservation you could modify the quality of the organ. Taking into account that a lot of our donors undergo intensive care for maybe 3 to 4 weeks, rendering the livers may be marginal and not suitable for splitting, I would be interested whether we could have a chance to introduce a treatment as you did here and improve liver function.
Dr. Thorlacius: The groups consist of 5 to 8 animals in each group, and it is, of course, speculative to answer your question whether it is feasible. It may be feasible because the drugs can be used clinically but are now mainly in early phases of rheumatoid arthritis with promising results. An advantage compared with other inhibitors like antibodies you have to deliver intravenously, is that this type of antagonists can be orally taken. But I cannot answer the question on marginal donors but only speculate that, since TNF-α is generally important in the liver pathophysiology, I would expect that there is reason good enough to test your suggestion.
Prof. Clavien: Thank you very much for another exciting study from your group. I have 3 questions. The first question relates to the timing of administration of your inhibitor of p38 MAPK. Animals were pretreated with the drug before the LPS/Gal challenge, a strategy that is not applicable in the clinical setting, as we can intervene only after the endotoxemia. Did you look at the effects of the p38 MAPK inhibitors if given after the challenge with LPS/Gal? My second question relates to leukocyte–endothelium interaction investigated with intravital microscopy, which provides data only in the periphery of the liver. Do you have any evidence regarding leukocyte adhesion deeper in the liver? My third question is similar to the one I asked last year regarding your previous ESA paper. You looked at apoptosis only with the Hoechst staining, which additionally shows only a modest increase in positive cells. In your slide, we see mostly necrotic changes. Did you look at other markers of apoptosis, which may more convincingly support a mechanism involving TNF-mediated apoptosis?
Dr. Thorlacius: Thank you, Dr. Clavien, for your questions. The inhibitor of p38 MAPK was only given as pretreatment, but I do agree that is would have been of interest to also test late treatment in order to simulate the clinical setting. Concerning your question whether these intravital observations at the liver surface also represent deeper segments of liver, I think that in general there is correspondence and this is supported by the histology in our study. I agree that apoptosis should be evaluated by more than one method, and we have actually done that herein, ie, we used both caspase-3 and Hoechst 33324 staining, and the results indicate that there is around 40% apoptosis in the damaged liver, which in my opinion is not a low level of apoptosis.
Footnotes
Supported by grants from the Swedish Medical Research Council (2002-955, 2002-8012, 2003-4661), Crafoordska stiftelsen, Blanceflors stiftelse, Einar och Inga Nilssons stiftelse, Harald och Greta Jaenssons stiftelse, Greta och Johan Kocks stiftelser, Fröken Agnes Nilssons stiftelse, Franke och Margareta Bergqvists stiftelse för främjande av cancerforskning, Magnus Bergvalls stiftelse, Mossfelts stiftelse, Nanna Svartz stiftelse, Ruth och Richard Julins stiftelse, Svenska Läkaresällskapet (2001-907), Teggers stiftelse, Allmäna sjukhusets i Malmö stiftelse för bekämpande av cancer, MAS fonder, Malmö University Hospital, and Lund University.
Reprints: Henrik Thorlacius, PhD, Department of Surgery, Malmö University Hospital, Lund University, S-205, 02 Malmö, Sweden. E-mail: henrik.thorlacius@kir.mas.lu.se.
REFERENCES
- 1.Jaeschke H, Farhood A, Smith CW. Neutrophil-induced liver cell injury in endotoxin shock is a CD11b/CD18-dependent mechanism. Am J Physiol. 1991;261:G1051–G1056. [DOI] [PubMed] [Google Scholar]
- 2.Holman JM Jr, Saba TM. Hepatocyte injury during post-operative sepsis: activated neutrophils as potential mediators. J Leukoc Biol. 1988;43:193–203. [DOI] [PubMed] [Google Scholar]
- 3.Klintman D, Schramm R, Menger MD, et al. Leukocyte recruitment in hepatic injury: selectin-mediated leukocyte rolling is a prerequisite for CD18-dependent firm adhesion. J Hepatol. 2002;36:53–59. [DOI] [PubMed] [Google Scholar]
- 4.Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67:1033–1036. [DOI] [PubMed] [Google Scholar]
- 5.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. [DOI] [PubMed] [Google Scholar]
- 6.Hahne M, Jager U, Isenmann S, et al. Five tumor necrosis factor-inducible cell adhesion mechanisms on the surface of mouse endothelioma cells mediate the binding of leukocytes. J Cell Biol. 1993;121:655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klintman D, Li X, Thorlacius H. Important role of P-selectin in leukocyte recruitment, hepatocellular injury and apoptosis in endotoxemic mice. Clin Diagn Lab Immunol. 2004;11:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X, Klintman D, Weitz-Schmidt G, et al. Lymphocyte function antigen-1 mediates leukocyte adhesion and subsequent liver damage in endotoxemic mice. Br J Pharmacol. 2004;141:709–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X, Klintman D, Wang Y, et al. Critical role of CXC chemokines in the extravasation process of leukocytes in endotoxemia. J Leukoc Biol. 2004;75:443–452. [DOI] [PubMed] [Google Scholar]
- 10.Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signaling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov. 2003;2:717–726. [DOI] [PubMed] [Google Scholar]
- 11.Ma XL, Kumar S, Gao F, et al. Inhibition of p38 mitogen-activated protein kinase decreases cardiomyocyte apoptosis and improves cardiac function after myocardial ischemia and reperfusion. Circulation. 1999;99:1685–1691. [DOI] [PubMed] [Google Scholar]
- 12.Gao F, Yue TL, Shi DW, et al. p38 MAPK inhibition reduces myocardial reperfusion injury via inhibition of endothelial adhesion molecule expression and blockade of PMN accumulation. Cardiovasc Res. 2002;53:414–422. [DOI] [PubMed] [Google Scholar]
- 13.Bachelor M, Bowden GT. UVA-mediated activation of signaling pathways involved in skin tumor promotion and progression. Semin Cancer Biol. 2004;14:131–138. [DOI] [PubMed] [Google Scholar]
- 14.Demet T, Lei X, Tingcun Z, et al. Mitogen-activated protein kinases mediate heat shock-induced delayed protection in mouse heart. Am J Physiol. 2001;281:H523–H532. [DOI] [PubMed] [Google Scholar]
- 15.Ropert C, Closel M, Chaves AC, et al. Inhibition of a p38/stress-activated protein kinase-2-dependent phosphatase restores function of IL-1 receptor-associate kinase-1 and reverses Toll-like receptor 2- and 4-dependent tolerance of macrophages. J Immunol. 2003;171:1456–1465. [DOI] [PubMed] [Google Scholar]
- 16.Khan MA, Kang J, Steiner TS. Enteroaggregative Escherichia coli flagellin-induced interleukin-8 secretion requires Toll-like receptor 5-dependent p38 MAP kinase activation. Immunology. 2004;112:651–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JC, Laydon JT, McDonnell PC, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. [DOI] [PubMed] [Google Scholar]
- 18.Underwood DC, Osborn RR, Kotzer CJ, et al. SB 239063, a potent p38 MAP kinase inhibitor, reduces inflammatory cytokine production, airways eosinophil infiltration, and persistence. J Pharmacol Exp Ther. 2000;293:281–288. [PubMed] [Google Scholar]
- 19.Badger AM, Griswold DE, Kapadia R, et al. Disease-modifying activity of SB 242235, a selective inhibitor of p38 mitogen-activated protein kinase, in rat adjuvant-induced arthritis. Arthritis Rheum. 2000;43:175–183. [DOI] [PubMed] [Google Scholar]
- 20.Behr TM, Nerurkar SS, Nelson AH, et al. Hypertensive end-organ damage and premature mortality are p38 mitogen-activated protein kinase-dependent in a rat model of cardiac hypertrophy and dysfunction. Circulation. 2001;104:1292–1298. [DOI] [PubMed] [Google Scholar]
- 21.Oguro T, Takahashi Y, Ashino T, et al. Involvement of tumor necrosis factor alpha, rather than interleukin-1alpha/beta or nitric oxides in the heme oxygenase-1 gene expression by lipopolysaccharide in the mouse liver. FEBS Lett. 2002;516:63–66. [DOI] [PubMed] [Google Scholar]
- 22.Dahle MK, Overland G, Myhre AE, et al. The phosphatidylinositol 3-kinase/protein kinase B signaling pathway is activated by lipoteichoic acid and plays a role in Kupffer cell production of interleukin-6 (IL-6) and IL-10. Infect Immun. 2004;72:5704–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barone FC, Irving EA, Ray AM, et al. SB 239063, a second-generation p38 mitogen-activated protein kinase inhibitor, reduces brain injury and neurological deficits in cerebral focal ischemia. J Pharmacol Exp Ther. 2001;296:312–321. [PubMed] [Google Scholar]
- 24.Rauen U, Polzar B, Stephan H, et al. Cold-induced apoptosis in cultured hepatocytes and liver endothelial cells: mediation by reactive oxygen species. FASEB J. 1999;13:155–168. [DOI] [PubMed] [Google Scholar]
- 25.Josephs MD, Bahjat FR, Fukuzuka K, et al. Lipopolysaccharide and D-galactosamine-induced hepatic injury is mediated by TNF-alpha and not by Fas ligand. Am J Physiol. 2000;278:R1196–R1201. [DOI] [PubMed] [Google Scholar]
- 26.Choudhury BK, Wild JS, Alam R, et al. In vivo role of p38 mitogen-activated protein kinase in mediating the anti-inflammatory effects of CpG oligodeoxynucleotide in murine asthma. J Immunol. 2002;169:5955–5961. [DOI] [PubMed] [Google Scholar]
- 27.Stambe C, Atkins RC, Tesch GH, et al. Blockade of p38alpha MAPK ameliorates acute inflammatory renal injury in rat anti-GBM glomerulonephritis. J Am Soc Nephrol. 2003;14:338–351. [DOI] [PubMed] [Google Scholar]
- 28.Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- 29.Hewett JA, Jean PA, Kunkel SL, et al. Relationship between tumor necrosis factor-α and neutrophils in endotoxin-induced liver injury. Am J Physiol. 1993;265:G1011–G1015. [DOI] [PubMed] [Google Scholar]
- 30.Feng L, Xia Y, Yoshimura T, et al. Modulation of neutrophil influx in glomerulonephritis in the rat with anti-macrophage inflammatory protein-2 (MIP-2) antibody. J Clin Invest. 1995;95:1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmal H, Shanley TP, Jones ML, et al. Role for macrophage inflammatory protein-2 in lipopolysaccharide-induced lung injury in rats. J Immunol. 1996;156:1963–1972. [PubMed] [Google Scholar]
- 32.Diab A, Abdalla H, Li HL, et al. Neutralization of macrophage inflammatory protein 2 (MIP-2) and MIP-1α attenuates neutrophil recruitment in the central nervous system during experimental bacterial meningitis. Infect Immun. 1999;67:2590–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang P, Xie M, Zagorski J, et al. Attenuation of hepatic sequestration by anti-CINC antibody in endotoxic rats. Shock. 1995;4:262–268. [DOI] [PubMed] [Google Scholar]
- 34.Means TK, Pavlovich RP, Roca D, et al. Activation of TNF-alpha transcription utilizes distinct MAP kinase pathways in different macrophage populations. J Leukoc Biol. 2000;67:885–893. [DOI] [PubMed] [Google Scholar]
- 35.Weber SM, Chen JM, Levitz SM, et al. Inhibition of mitogen-activated protein kinase signaling by chloroquine. J Immunol. 2002;168:5303–5309. [DOI] [PubMed] [Google Scholar]
- 36.Swantek JL, Cobb MH, Geppert TD. Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor alpha (TNF-alpha) translation: glucocorticoids inhibit TNF-alpha translation by blocking JNK/SAPK. Mol Cell Biol. 1997;17:6274–6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brook M, Sully G, Clark AR, et al. Regulation of tumour necrosis factor alpha mRNA stability by the mitogen-activated protein kinase p38 signaling cascade. FEBS Lett. 2000;483:57–61. [DOI] [PubMed] [Google Scholar]
- 38.Nagahira A, Nagahira K, Murafuji H, et al. Identification of a novel inhibitor of LPS-induced TNF-alpha production with antiproliferative activity in monocyte/macrophages. Biochem Biophys Res Commun. 2001;281:1030–1036. [DOI] [PubMed] [Google Scholar]
- 39.Mahidhara R, Billiar TR. Apoptosis in sepsis. Crit Care Med. 2000;28:N105–N113. [DOI] [PubMed] [Google Scholar]
- 40.Lawson JA, Fisher MA, Simmons CA, et al. Parenchymal cell apoptosis as a signal for sinusoidal sequestration and transendothelial migration of neutrophils in murine models of endotoxin and Fas-antibody-induced liver injury. Hepatology. 1998;28:761–767. [DOI] [PubMed] [Google Scholar]
- 41.Faouzi S, Burckhardt BE, Hanson JC, et al. Anti-Fas induces hepatic chemokines and promotes inflammation by an NF-κB-independent, caspase-3-dependent pathway. J Biol Chem. 2001;276:49077–49082. [DOI] [PubMed] [Google Scholar]
- 42.Oberholzer A, Oberholzer C, Moldawer LL. Interleukin-10: A complex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit Care Med. 2002;30(suppl):58–63. [PubMed] [Google Scholar]
- 43.Ayala A, Deol ZK, Lehman DL, et al. Polymicrobial sepsis but not low dose endotoxin infusion causes decreased splenocyte IL-2/IFN-γ release while increasing IL-4/IL-10 production. J Surg Res. 1994;56:579–585. [DOI] [PubMed] [Google Scholar]
- 44.Cohen PS, Schmidtmayerova H, Dennis J, et al. The critical role of p38 MAP kinase in T cell HIV-1 replication. Mol Med. 1997;3:339–346. [PMC free article] [PubMed] [Google Scholar]
- 45.Song GY, Chung CS, Chaudry IH, et al. MAPK p38 antagonism as a novel method of inhibiting lymphoid immune suppression in polymicrobial sepsis. Am J Physiol. 2001;281:C662–C669. [DOI] [PubMed] [Google Scholar]
- 46.Beutler B, Millsark IW, Cerami AC. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effects of endotoxin. Science. 1985;229:869–871. [DOI] [PubMed] [Google Scholar]
- 47.Tracey KJ, Fong Y, Hesse DG, et al. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987;330:662–664. [DOI] [PubMed] [Google Scholar]
- 48.Nakane A, Minagawa T, Kato K. Endogenous tumor necrosis factor (cachectin) is essential to host resistance against Listeria monocytogenes infection. Infect Immun. 1988;56:2563–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Echtenacher B, Urbaschek R, Weigl K, et al. Treatment of experimental sepsis-induced immunoparalysis with TNF. Immunobiology. 2003;208:381–389. [DOI] [PubMed] [Google Scholar]
- 50.Berg DJ, Kuhn K, Rajewsky K, et al. Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartzman reaction but not endotoxin tolerance. J Clin Invest. 1995;96:2339–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Howard M, Muchamuel T, Andrade S, et al. Interleukin 10 protects mice from lethal endotoxemia. J Exp Med. 1993;177:1205–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song GY, Chung CS, Chaudry IH, et al. What is the role of interleukin 10 in polymicrobial sepsis: anti-inflammatory agent or immunosuppressant? Surgery. 1999;126:378–383. [PubMed] [Google Scholar]
- 53.Van der Poll TA, Marchant WA, Buurman L, et al. Endogenous IL-10 protects mice from death during septic peritonitis. J Immunol. 1995;155:5397–5401.7594556 [Google Scholar]