

Abstract
Introduction:
Supraphysiologic stress induces a heat shock response, which may exert protection against ischemic necrosis. Herein we analyzed in vivo whether the induction of heat shock protein (HSP) 32 improves survival of chronically ischemic myocutaneous tissue, and whether this is based on amelioration of microvascular perfusion or induction of ischemic tolerance.
Methods:
The dorsal skin of mice was subjected to local heat preconditioning (n = 8) 24 hours before surgery. In additional heat-preconditioned animals (n = 8), HSP-32 was inhibited by tin-protoporphyrin-IX. Unconditioned animals served as controls (n = 8). A random-pattern myocutaneous flap was elevated in the back of the animals and fixed into a dorsal skinfold chamber. The microcirculation, edema formation, apoptotic cell death, and tissue necrosis were analyzed over a 10-day period using intravital fluorescence microscopy.
Results:
HSP-32 protein expression was observed only in heat-preconditioned but not in unconditioned flaps. Heat preconditioning induced arteriolar dilation, which was associated with a significant improvement of both arteriolar blood flow and capillary perfusion in the distal part of the flap. Further, heat shock reduced interstitial edema formation, attenuated apoptotic cell death, and almost completely abrogated the development of flap necrosis (4% ± 1% versus controls: 53% ± 5%; P[r] < 0.001). Most strikingly, inhibition of HSP-32 by tin-protoporphyrin-IX completely blunted the preconditioning-induced improvement of microcirculation and resulted in manifestation of 72% ± 4% necrosis.
Conclusion:
Local heat preconditioning of myocutaneous tissue markedly increases flap survival by maintaining adequate nutritive perfusion rather than inducing ischemic tolerance. The protection is caused by the increased arteriolar blood flow due to significant arteriolar dilation, which is mediated through the carbon monoxide-associated vasoactive properties of HSP-32.
Using a murine myocutaneous flap model and intravital microscopy, the study demonstrates that heat preconditioning significantly reduces ischemic necrosis by maintaining nutritional capillary perfusion. The protection is caused by the carbon monoxide-mediated arteriolar dilatory action of heat shock protein 32. Thus, heat preconditioning acts by preservation of adequate microcirculation rather than induction of ischemic tolerance.
A variety of surgical procedures bear the risk of development of ischemic tissue necrosis.1,2 These complications cause significant patient morbidity, including compromised functional results and prolonged hospital courses. The importance of this issue is emphasized by more aged patients, presenting with an increased risk for ischemic necrosis,3 as well as comorbidities such as peripheral vascular disease, which may aggravate the manifestation of tissue injury.4,5
Over the past 2 decades, several approaches have been studied to prevent ischemia-induced tissue injury.6–8 Lately, a novel concept has been introduced, which consists of preconditioning the tissue at risk prior to surgery to induce heat shock proteins (HSPs) by exposure to physical or pharmacologic stressors.9–12 While several HSPs of the 70-kDa family are known to act as molecular chaperones, escorting proteins targeted for other cellular compartments,13 or to prevent misfolding of newly synthesized proteins,14,15 HSP-32 has been identified as heme oxygenase (HO)-1,16 the rate-limiting enzyme in the catabolism of heme to biliverdin, free iron, and carbon monoxide. HSP-32 is the main endogenous source of carbon monoxide,17 which functions as a potent vasodilator.18
Recent experimental studies have elucidated that the induction of HSPs is capable of reducing ischemic necrosis in myocardial,19,20 neuronal,21 and renal tissue.22 Additional studies have indicated that HSP-32 may protect the microcirculation and thus tissue oxygenation in axial pattern osteomyocutaneous flaps.23 However, the protective potential of HSP-32 to prevent ischemic necrosis in randomly perfused tissue and its effects on the microcirculation are unknown yet. Although an adequate microcirculation is thought to be a prerequisite for tissue survival, it is still a matter of discussion whether protection from ischemic necrosis by heat shock preconditioning is mediated by the prevention of microvascular perfusion failure or by the induction of ischemic tolerance within the affected tissue. In this setting, ischemic tolerance is defined by a reduced oxygen demand of the tissue so that it survives despite a critically decreased nutritive perfusion and thus a limited oxygen supply.
The aim of the present study was therefore to examine in a chronic in vivo murine model of randomly perfused tissue24 whether 1) preconditioning by repetitive local heat application induces HSP-32 expression, 2) the activity of this enzyme improves tissue survival, and finally 3) HSP-32-induced protection from ischemic necrosis is mediated by improvement of the microcirculation or induction of ischemic tolerance.
MATERIALS AND METHODS
Animals
All experiments were performed according to the guiding principles for research involving animals and the German legislation on protection of animals. The experiments were approved by the local governmental animal care committee. A total of 24 mice (C57BL/6J; 12–24 weeks; 24–26 g body weight [BW]; Charles River Laboratories GmbH; Sulzfeld, Germany) were included in the study. The animals were housed in single cages at a room temperature of 22°C to 24°C and a relative humidity of 60% to 65% with a 12-hour day-night cycle. They were allowed free access to drinking water and standard laboratory chow (Altromin, Lage, Germany).
Anesthesia
For surgery and repetitive intravital fluorescence microscopy, the animals were anesthetized by intraperitoneal injection of 90 mg/kg BW ketamine hydrochloride (Ketavet, Parke Davis; Freiburg, Germany) and 25 mg/kg BW xylazine hydrochloride (Rompun, Bayer; Leverkusen, Germany).
Preparation of the Random Pattern Flap
Surgery was performed by the first 2 authors throughout the series. The flap was prepared at the dorsum of the animal and was incorporated into a dorsal skinfold chamber, which has been described previously in detail.25 In brief, after removal of the fur, the dorsal part of the titanium frame was temporarily placed on the double-layered skinfold. Then a laterally based random pattern skin flap with a width to length ratio of 15 mm × 11 mm, including the panniculus carnosus, was elevated perpendicularly to the spine, transecting bilaterally the nutritional blood supply. After extending the elevated tissue to the back side of the dorsal part of the titanium frame, the flap was sutured back to the surrounding skin laterally. To achieve direct view onto the back side of the elevated tissue, consisting of epidermis, subcutaneous tissue, and striated skin muscle (panniculus carnosus), an area of ∼90 mm2 of the overlaying skin layer was removed. Finally, the frame's counterpart was mounted, and the observation window was sealed with a coverglass for subsequent in vivo microscopy.24 Chambers (total weight ∼3 g) were well tolerated by the animals, which showed no changes in sleeping or feeding habits.
Intravital Fluorescence Microscopy
For in vivo microscopic analysis of the microcirculation, anesthetized mice were placed in left lateral decubital position on a plexiglas pad and received an intravenous injection of 0.05 mL 5% fluorescein-isothiocyanate-labeled dextran (molecular weight 150,000; Sigma Aldrich; Deisenhofen, Germany) via a tail vein. Subsequently, the mice were positioned under a Zeiss Axiotech microscope (Zeiss; Oberkocchen, Germany). The epi-illumination microscopic setup included a 100-W mercury lamp and filter sets for blue (450–490 nm excitation, >520 nm emission wavelength), green (530–560 nm/>580 nm) and ultraviolet (330–390 nm/>430 nm) light. Microscopic images were monitored by a charge-coupled device video camera (FK6990, Pieper; Schwerte, Germany) and recorded on video tape (Panasonic AG-7350-SVHS, Matsushita; Tokyo, Japan) for subsequent off-line evaluation.
Analysis of Microcirculation
The microscopic procedures for analysis of the microcirculation were performed at constant room temperature of 23°C. Different objectives (×4, NA [numerical aperture] = 0.16, ×10, NA = 0.30 and ×20 long distance, NA = 0.32; ×63 water immersion, NA = 0.90) were used for recordings. At each observation time point, the tissue within the window of the chamber was first scanned using the ×4 objective to determine the surface of nonperfused and necrotic tissue, respectively. Furthermore, second- or third-order arterioles and accompanying collecting venules with easily identifiable branching patterns were selected in the proximal, middle, and distal part of the flap. Using the ×10 and ×20 objective, video printouts of the above-described areas, including arteriolovenular bundles, were made to indicate the exact localization for repetitive measurements of red blood cell velocity, capillary diameter, individual volumetric blood flow and functional capillary density. Using the DNA-binding fluorochrome bisbenzimide H33342, apoptotic cells (n/mm2) were identified in vivo by the characteristic signs of nuclear condensation, fragmentation and margination. Recordings were made using a ×63 water immersion objective.
All microcirculatory parameters were analyzed off-line using a computer-assisted image analysis system (CapImage, Zeintl Software; Heidelberg, Germany).26 The area of necrosis, which was identified as tissue with complete lack of perfusion, was determined planimetrically and is given in percent nonperfused tissue of the total flap size. The functional capillary density was defined as length of red blood cell perfused capillaries per observation field and expressed in cm/cm2. Capillary diameters were measured perpendicularly to the vessel path and are given in μm. Capillary red blood cell velocity was analyzed by the line shift method and is given in μm/s. Volumetric blood flow, given in pL/s, was calculated in arterioles, capillaries and venules from red blood cell velocity and vessel cross-sectional area (π * r2) according to the equation of Gross and Aroesty, ie, Q = V * π * r2 assuming a cylindrical vessel geometry.27
Histology
Formalin-fixed, paraffin-embedded full thickness longitudinal segments of the flaps obtained at day 10 after surgery were cut and stained with hematoxylin and eosin. The sections served for the examination of morphologic changes within the epidermis, the subcutaneous tissue, and the panniculus carnosus. Using computer-assisted image analysis, interstitial edema formation within the panniculus carnosus was quantified by determining the area of the interstitial space, given in percent of the overall tissue area.
Immunohistochemistry
Frozen sections in Tissue-Tek (Sakura Finetek, Zoeterwoude, NL) obtained at day of surgery were used to assess cell-type-specific expression and spatial pattern of HSP-32. After incubation of 4-μm-thick sections in 3% H2O2-methanol to block activity of endogenous peroxidase, the slides were incubated with normal 3% goat serum in phosphate-buffered saline, followed by a primary, cross-reacting polyclonal rabbit anti-rat HSP-32 antibody (dilution 1:100; StressGen Biotechnologies, Victoria, British Columbia, Canada) at 21°C for 1 hour. A POD-conjugated goat anti-rabbit antibody was used as secondary antibody (dilution 1:500; Amersham Biosciences, Freiburg i.B. Germany) and 3.3′-diamnino-benzidine was used as chromogen. Slides were counterstained with hematoxylin and examined by light microscopy (BX 60F, Olympus Optical Corp., Tokyo, Japan).
Chemicals
Tin-protoporphyrin-IX (SnPP-IX; Frontier Scientific, Lancashire; UK), was dissolved by mixing SnPP-IX with sodium-bicarbonate 8.4% and phosphate-buffered saline to achieve a final concentration of 5 μmol/mL. The solution was stored at a maximum temperature of 8°C in a light-protected tube for no longer than 1 hour until usage. SnPP-IX acts as a competitive inhibitor of HO-1. It is a synthetic nonmetabolized heme analogue, which binds to the active site of the enzyme.28 The enzyme is thought to specifically block the function of HO-1.29 There is no evidence from the literature that SnPP-IX interferes with the induction or activity of other HSPs than HSP-32.
Preconditioning
After removal of the fur, local heat shock priming was induced 24 hours before flap dissection. Therefore, the anesthetized mice were lying in a lateral decubital position on a heating pad. The right flank was then heated up to reach a skin temperature of 43.0°C for 3 × 30 minutes, interrupted by cycles of 30 minutes each at room temperature (23°C), during which the mice were taken off the heating pad. Twenty-four hours before surgery, sham animals were anesthetized for an equivalent period of time without heat shock priming.
Experimental Protocol
A total of 24 animals were assigned to 3 groups of 8 mice each. In groups 2 and 3, the animals were subjected to repetitive local heat shock. Animals in group 3 were additionally treated with intraperitoneal injections of SnPP-IX at a dosage of 50 μmol per kg BW, whereas animals in group 2 received an equivalent amount of saline. In analogy to local heat preconditioning, SnPP-IX was given 24 hours before surgery. To block the activity of HSP-32 during the progress of necrosis, the competitive antagonist was additionally injected at the day of surgery and at day 1, 2, and 3 after elevation of the flap. Unpreconditioned animals in group 1 received similar amounts of saline at the corresponding time points and served as controls. Repetitive microscopic observations were performed 24 hours as well as 3, 5, 7, and 10 days after surgery. At the end of the experiments, the animals were killed by injection of an overdose of the anesthetic.
Statistical Analysis
All values are expressed as mean ± standard error of the mean (SEM). For comparison between individual time points within each group, ANOVA for repeated measures followed by the appropriate posthoc test was performed, including correction of the alpha-error according to Bonferroni probabilities. Comparison between the groups included ANOVA and the appropriate post hoc test to compensate for multiple comparisons (SigmaStat, Jandel, San Rafael, CA). A value of P < 0.05 was taken to indicate statistical significance.
RESULTS
HSP-32 Protein Expression
The myocutaneous tissue of unconditioned control animals did not express HSP-32 (Fig. 1A). In contrast, preconditioned tissue showed a distinct staining for HSP-32 at 24 hours after heat exposure, involving predominantly fibroblasts and macrophages within the subcutaneous tissue. Of interest, muscle cells of the panniculus carnosus also showed slight expression of HSP-32 (Fig. 1B).
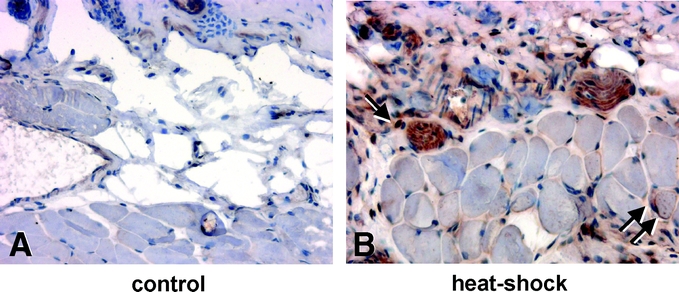
FIGURE 1. Longitudinal sections of flap tissue 24 hours after preconditioning stained for heat shock protein (HSP)-32 expression in control animals (A) and animals after local heat shock exposure (B). Note the nuclear and cytosolic staining of heat shock-treated flaps within the subcutaneous (arrow) and muscle tissue (double arrow) (original magnification ×80).
Flap Necrosis
Macroscopic analysis of the chambers' windows at day 10 after surgery demonstrated morphologic changes of the microvasculature within the middle and distal areas of the flaps (Fig. 2). Unconditioned controls showed a clear demarcation of the vital tissue from the distal necrosis, which was separated by a hyperemic red zone indicating microvascular remodeling (Fig. 2A). In contrast, flaps of heat shock-preconditioned animals presented with almost full preservation of the elevated tissue (Fig. 2B). However, heat shock-preconditioned flaps, which additionally received SnPP-IX, showed an increase of flap necrosis when compared with that of controls, and, most interestingly, revealed a lack of vascular remodeling within the zone of demarcation (Fig. 2C).

FIGURE 2. Chamber tissue at day 10 after flap elevation. Control animals show a distinct area of demarcation with a red zone, which indicates a hyperemic response and microvascular remodeling (arrows) adjacent to the distal necrosis (*) (A). Flaps after local heat shock preconditioning demonstrate almost complete tissue survival (B). In contrast, heat shock-preconditioned flaps, which were treated with SnPP-IX, also show a zone of demarcation and distal necrosis (*). This, however, developed more proximally, which indicates increased flap necrosis and lacks hyperemia and vascular remodeling (C) (original magnification ×16).
In controls, the initial microcirculatory perfusion failure at day 1 after flap elevation was associated with an area of nonperfused tissue of 47% ± 4%. Along with the persisting microcirculatory dysfunction, this resulted in a 52% ± 5% necrosis at day 10 after flap elevation. Local heat shock preconditioning significantly reduced the area of initial microcirculatory perfusion failure (2% ± 1%; P < 0.001 versus control) and resulted in an only 4% ± 1% flap necrosis at day 10 (P < 0.001). Functional inhibition of HSP-32 by administration of SnPP-IX completely blunted the heat shock-induced protection from initial microcirculatory perfusion failure (53% ± 5%; P < 0.05 versus control) and was associated with a final flap necrosis, which was even increased (72% ± 4%; P < 0.05) when compared with that observed in unpreconditioned controls (Fig. 3).

FIGURE 3. Time course of necrosis (given in percent of the total flap area) in control flaps (white circles), heat shock-preconditioned flaps (gray circles) and heat shock-preconditioned flaps treated with SnPP-IX (black circles). Note the significant protection of heat shock preconditioning against flap necrosis. Functional inhibition of HSP-32 by SnPP-IX does not only blunt the heat shock preconditioning-induced protection but further increases the manifestation of necrosis. Data are mean ± SEM. *P < 0.05, **P < 0.001 versus control. ##P < 0.001 versus heat shock.
Arteriolar Perfusion
In controls, analysis of arteriolar diameters within the proximal area of the flaps showed a significant dilation over the 10-day observation period (Fig. 4). Within the middle area, dilation was less pronounced, and within the distal part of the flap it was completely abrogated. Flaps of heat shock-preconditioned animals showed also a significant arteriolar dilation within the proximal area of the flaps. In contrast to unpreconditioned controls, however, the arterioles of the distal part of the flaps were also found dilated throughout the entire study period (Fig. 4). Functional inhibition by SnPP-IX completely blunted the arteriolar dilation response, as indicated by a lack of dilation in the proximal area, arteriolar constriction within the middle area, and arteriolar unresponsiveness within the distal part of the flap (Fig. 4).
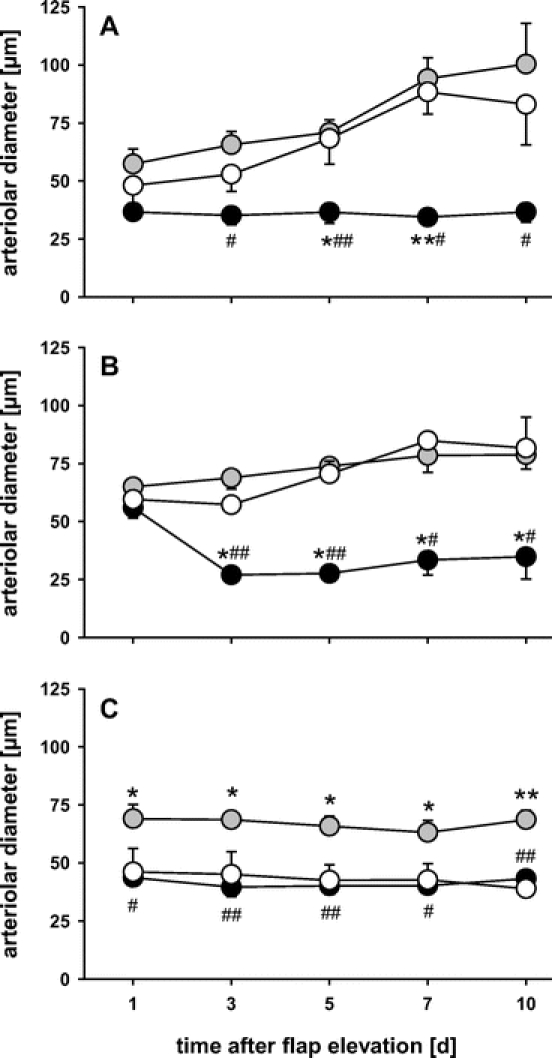
FIGURE 4. Arteriolar diameters over the 10-day observation period within the proximal (A), middle (B), and distal part (C) of control flaps (white circles), heat shock-preconditioned flaps (gray circles), and heat shock-preconditioned flaps treated with SnPP-IX (black circles). Note the dilation response in the proximal area of control and heat shock-preconditioned, but not SnPP-IX-treated flaps, and the prevention of arteriolar constriction in the distal part of heat shock-preconditioned, but not control and SnPP-IX-treated flaps. Data are mean ± SEM. *P < 0.05, **P < 0.001 versus control. #P < 0.05, ##P < 0.001 versus heat shock.
Analysis of arteriolar blood flow within the proximal and the middle area of unpreconditioned control flaps showed a progressive increase of perfusion until the end of the 10-day observation period. Within the distal part of the flap, however, arteriolar blood flow ceased initially after flap elevation and was not found reestablished during the subsequent 10-day period (Table 1). In contrast, heat shock preconditioned flaps showed a markedly increased arteriolar blood flow in all tissue areas analyzed, including the distal part of the flap (Table 1). SnPP-IX treatment of heat shock-preconditioned flaps did not only blunt the heat shock-associated increase in arteriolar blood flow but also resulted in a significantly decreased perfusion within the proximal and the middle area, and cessation of blood flow within the distal part of the flap (Table 1).
TABLE 1. Individual Volumetric Blood Flow (pL/s) of Arterioles Within the Proximal, Middle, and Distal Part of Control Flaps (CON), Heat Shock-Preconditioned Flaps (HSP), and Heat Shock-Preconditioned Flaps Treated With SnPP-IX (HSP-SnPP-IX) at Days 1, 3, 5, 7, and 10 After Flap Elevation

Capillary Perfusion
In controls, functional capillary density was found reduced in the middle and completely abolished in the distal part of the flap. There was no recovery over the 10-day observation period (Fig. 5). Heat shock preconditioning was capable of completely preserving the functional capillary density within all parts of the flap (Fig. 5). The additional treatment with SnPP-IX did not only blunt the heat shock-mediated protection from capillary perfusion failure within the distal part of the flap but markedly aggravated the reduction of capillary density within the proximal and middle area (Fig. 5).
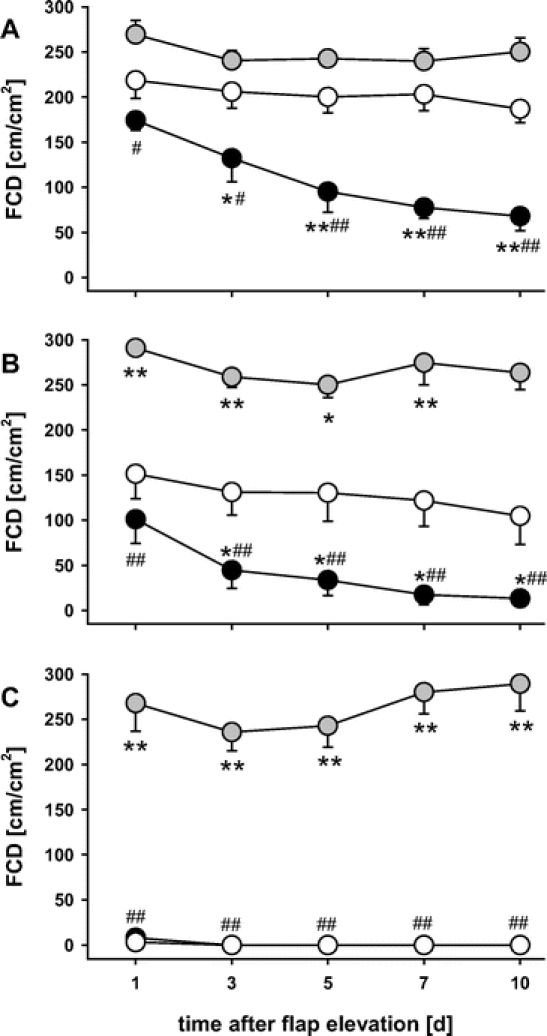
FIGURE 5. Functional capillary density (FCD) over the 10-day observation period within the proximal (A), middle (B), and distal part (C) of control flaps (white circles), heat shock-preconditioned flaps (gray circles), and heat shock-preconditioned flaps treated with SnPP-IX (black circles). Note the preservation of FCD in heat shock-preconditioned flaps. In contrast, control and in particular SnPP-IX-treated flaps show a significant decrease of FCD in the proximal and the middle area, and a complete abrogation of capillary perfusion in the distal part of the flaps. Data are mean ± SEM. *P < 0.05, **P < 0.001 versus control. #P < 0.05, ##P < 0.001 versus heat shock.
In control flaps, analysis of capillary geometry showed a massive dilation of capillaries within the middle area of the flap, which corresponds with the red zone with vascular remodeling. Within the distal part of the flaps, however, the initial capillary constriction did not recover during the 10-day postsurgery period (Fig. 6). Heat shock preconditioning normalized capillary diameters within the proximal and middle area and resulted in a marked dilation within the distal part of the flaps (Fig. 6). Functional inhibition of HSP-32 induced initial capillary constriction within all parts of the flaps, which did not recover over the entire observation period (Fig. 6).
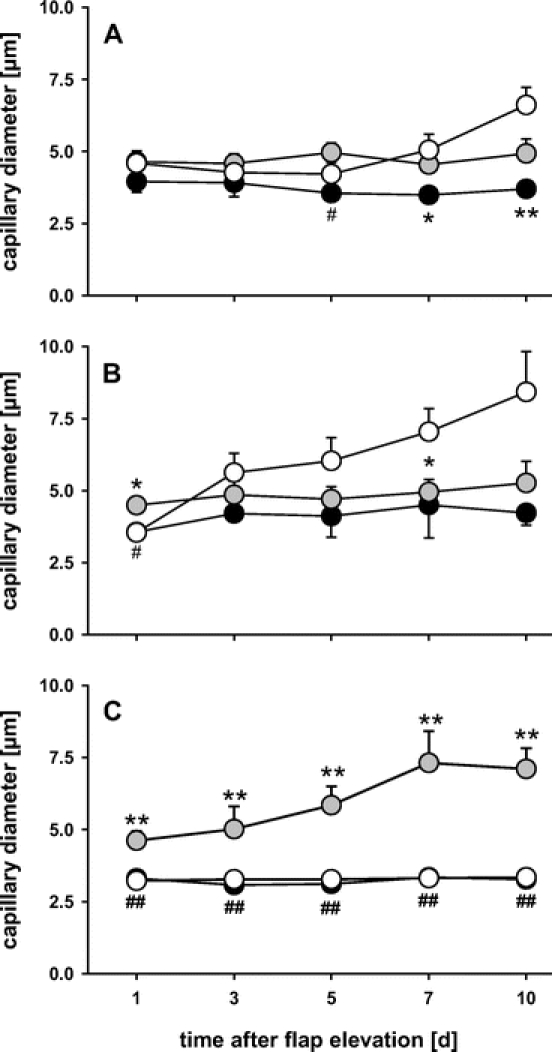
FIGURE 6. Capillary diameters over the 10-day observation period within the proximal (A), middle (B), and distal part (C) of control flaps (white circles), heat shock-preconditioned flaps (gray circles), and heat shock-preconditioned flaps treated with SnPP-IX (black circles). Note that, within the distal part of the flap, the capillaries are found dilated in heat shock-preconditioned flaps, but markedly constricted in control and SnPP-IX-treated flaps. Data are mean ± SEM. **P < 0.001 versus control. ##P < 0.001 versus heat shock.
As a consequence of capillary dilation, control animals showed a marked increase of the individual volumetric blood flow within the proximal and middle area of the flaps, while blood flow ceased completely within the distal part (Table 2). In contrast, heat shock preconditioning resulted in stable capillary perfusion in all parts of the flaps (Table 2). SnPP-IX-treatment provoked a marked reduction of the individual capillary blood flow within the proximal and middle area, and a complete cessation within the distal part of the flaps (Table 2).
TABLE 2. Individual Volumetric Blood Flow (pL/s) of Capillaries Within the Proximal, Middle, and Distal Part of Control Flaps (CON), Heat Shock-Preconditioned Flaps (HSP), and Heat Shock-Preconditioned Flaps Treated With SnPP-IX (HSP-SnPP-IX) at Days 1, 3, 5, 7, and 10 After Flap Elevation

Edema Formation
In controls, 10-day flap elevation caused massive interstitial edema within the critically perfused middle part of the flap, while the distal necrotic part obviously showed no edema formation but desiccation of tissue (Fig. 7). Heat shock preconditioning reduced edema formation in the middle part and prevented desiccation and, thus, necrosis in the distal part of the flap. Most impressively, functional blockade of HSP-32 by SnPP-IX significantly reduced the overall interstitial space in the proximal and middle, but in particular in the distal part of the flap, which indicates pronounced desiccation as the consequence of extended necrosis within all areas of the flap (Fig. 7).
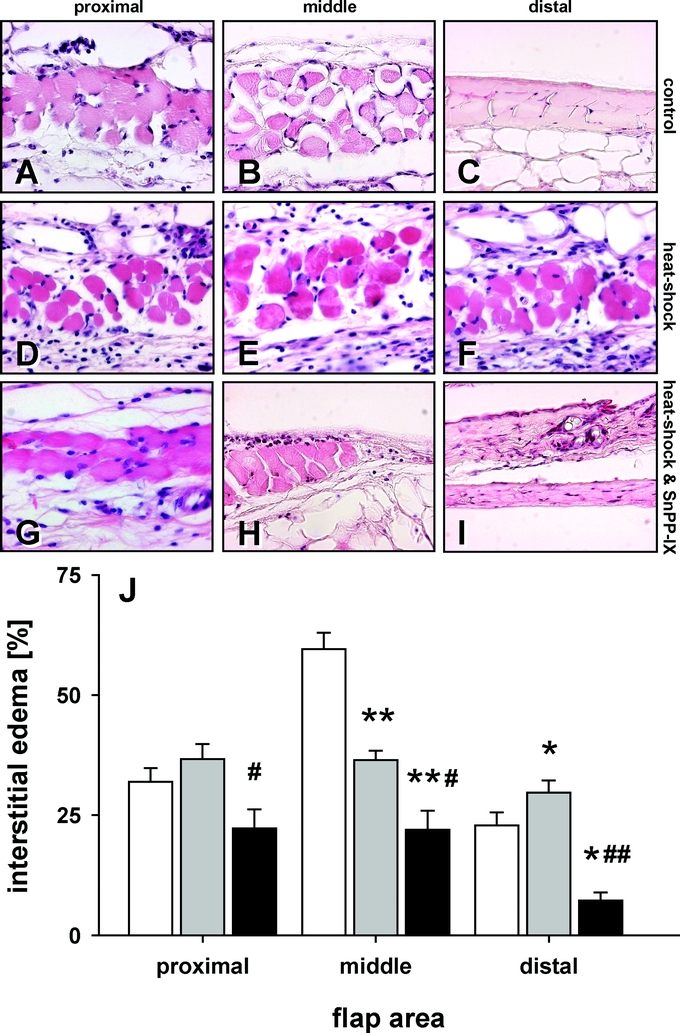
FIGURE 7. Longitudinal sections of the proximal, middle, and distal area of control flaps (white bars), heat shock-preconditioned flaps (gray bars), and heat shock-preconditioned flaps treated with SnPP-IX (black bars). Note the massive interstitial edema formation in the middle area of control flaps (B and J) and desiccation of the tissue distally, reflecting necrosis (C and J). Heat shock preconditioning reduces edema formation in the middle area (E and J) and prevents necrosis in the distal part (F and J). This protection is completely blunted by SnPP-IX teatment (H, I, and J). Data are mean ± SEM. *P < 0.05, **P < 0.001 versus control. #P < 0.05, ##P < 0.001 versus heat shock.
Apoptotic Cell Death
At day 1 after flap elevation, in vivo analysis of apoptotic cell death within the distal part of control flaps revealed ∼350 cells/mm2, which demonstrated chromatin condensation, fragmentation, and margination (Fig. 8). Heat shock preconditioning was capable of significantly (P < 0.05) reducing the number of apoptotic cells, while inhibition of HSP-32 by SnPP-IX almost completely abrogated the heat shock preconditioning-induced protection against the programmed cell death (Fig. 8).
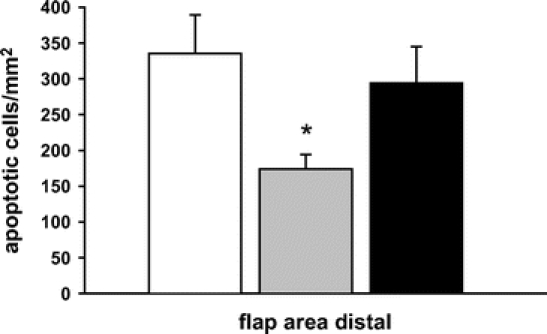
FIGURE 8. Apoptotic cell death at day 1 after flap elevation within the distal part of control flaps (white bar), heat shock-preconditioned flaps (gray bar), and heat shock-preconditioned flaps treated with SnPP-IX (black bar). Note the significant lower number of apoptotic cells in heat shock-preconditioned flaps when compared with controls, and the abrogation of protection by functional inhibition of HSP-32 with SnPP-IX. Data are mean ± SEM. *P < 0.05 versus control.
DISCUSSION
The major findings of the present study are that the induction of heat shock in randomly perfused ischemic tissue drastically reduces flap necrosis by the action of HSP-32 and that this protection is mediated by the maintenance of an adequate microvascular perfusion rather than by an induction of ischemic tolerance of the tissue.
Previous studies have shown that heat shock preconditioning is effective to prevent organ dysfunction30,31 and organ injury in a variety of disease states, including chronic inflammation,32,33 sepsis,34 ischemia-reperfusion,35,36 and graft rejection.37 Although the mechanisms of protection are not completely elucidated yet, detailed analysis in reperfused tissue revealed that carbon monoxide derived from heme degradation by HSP-32 may contribute to the amelioration of injury.23,38
After renal and myocardial reperfusion and reoxygenation, experimental studies have indicated that HSP-32 attenuates apoptotic cell death.39,40 Herein, we show for the first time that in chronic ischemia heat shock preconditioning is also capable of reducing injury, including both apoptotic cell death and final tissue necrosis. The fact that the conditioning procedure induced HSP-32 and that competitive inhibition of HSP-32 function completely abrogated the protection from necrosis indicates a major role of this molecule in the prevention of chronic ischemia-induced tissue injury.
In reperfusion-reoxygenation injury, Yang et al speculated that renal protection is the result of a HSP-70-associated increase in ischemic tolerance,41 whereas in hepatic reperfusion injury, others have shown a hyperthermia-induced improvement of the microcirculation, which was associated with the expression of both HSP-70 and HSP-32.42 In skeletal muscle, a more detailed analysis revealed that the improvement of the microcirculation is caused selectively by HSP-32 function, most probably through carbon monoxide derived from heme degradation.23
To elucidate whether in chronic ischemic skin HSPs mediate the protection from tissue necrosis by an increase of tissue tolerance or by an improvement of microvascular perfusion, we studied the microcirculation over a 10-day observation period. The data of the present study indicate that volumetric blood flow increased significantly within individual arterioles, and that this was due to arteriolar dilation rather than elevation of intravascular red blood cell velocity. Indeed, this arteriolar dilation response is mediated by HSP-32 because functional inhibition of HSP-32 by SnPP-IX was associated with a complete abrogation of the heat shock-mediated reactive arteriolar dilation.
HSP-32, which has been identified as HO-1, is the rate-limiting enzyme in the degradation of heme to biliverdin, free iron, and carbon monoxide.43 Carbon monoxide is known to be a potent endogenous vasodilator,44 acting analogous to nitric oxide.45 Although discussed controversially, there is evidence that vasodilation after heat shock is not mediated by nitric oxide.46 Given that the expression of HSP-32 was evident only after local heat shock preconditioning when compared with controls, our data suggest that HSP-32 via its degradation product carbon monoxide is crucially involved in chronic ischemia-induced arteriolar dilation. This view is in line with the results of a report of Kozma et al, demonstrating that inhibition of HSP-32 by metalloporphyrins causes a rapid and sustained constriction of gracilis muscle arterioles in rats.47
During the last decade, a variety of studies have underlined the biologic relevance of carbon monoxide in the regulation of vascular tone. Hypoxia, shear stress, and stretch have been shown to induce HSP-32 and to result in elevated production of carbon monoxide, especially by smooth muscle cells.48 Carbon monoxide, in turn, may regulate vascular tone by stimulation of intracellular cyclic guanosine monophosphate levels.49 Furthermore, it produces a paracrine suppression of mRNA for endothelin-1 and platelet-derived growth factor in endothelial cells.50 Stanford et al51 could demonstrate in human pulmonary artery smooth muscle cells that a carbon monoxide-releasing molecule from heme degradation inhibits endothelin-1 release. These authors concluded from their in vitro study that under certain pathophysiologic conditions, such as chronic hypoxia, carbon monoxide may act as an endogenous break on endothelin-1.51 The functional balance between carbon monoxide and endothelin-1 was also studied by Zhang et al in pulmonary interlobar arteries.52 These studies indicated that carbon monoxide derived from heme degradation inhibits hypoxia-induced pulmonary vasoconstriction by reducing both the absolute endothelin-1 vascular levels and its sensitivity to endothelial cells.52 Thus, the heat shock-induced arteriolar dilation in chronic ischemia, observed in the present study, may be caused by the action of HSP-32-derived carbon monoxide, including both its direct vasodilative potential and its endothelin-1 inhibiting function.
The heat shock-mediated arteriolar dilation was associated with a maintained capillary density and a drastic increase in volumetric blood flow in individual capillaries. Comparable to the arteriolar hemodynamics, the increased capillary volume flow was caused by capillary dilation rather than increased red blood cell velocity. This capillary dilation may represent just a passive response to the total increase of arteriolar blood flow. On the other hand, carbon monoxide may also have been capable of inducing active dilation of the nutritive capillaries by stimulating pericyte function, as has been reported for heat shock-mediated sinusoidal dilation in hemorrhagic shock livers.53 In final consequence, the dilation of the capillaries after heat shock preconditioning guaranteed the maintenance of an adequate functional capillary density, which must be considered as the major prerequisite in preventing the chronic ischemia-induced tissue necrosis.
It remains to be clarified whether the improvement of tissue survival was achieved by an increase of ischemic tolerance. By definition, an increase of ischemic tolerance would have resulted in tissue survival despite persistence of significant microcirculatory perfusion failure and thus decreased oxygen supply. Although we cannot exclude that heat shock preconditioning additionally affected ischemic tolerance, the effect on nutritive perfusion was sufficient to guarantee tissue survival.
Of interest, blockade of HSP-32 by SnPP-IX did not only blunt the protective action of the heat shock preconditioning procedure but additionally increased tissue injury and necrosis when compared with nontreated controls. This aggravation of injury may be due to the fact that SnPP-IX did not only block the HSPs induced by the conditioning procedure but also those triggered by the flap elevation surgery. In nontreated controls, the microvascular remodeling within the middle area of the flap is probably caused by the endogenous heat shock response and resulted in some protection from necrosis. The lack of microvascular remodeling in the demarcation zone of the middle area after SnPP-IX treatment may be due to the inhibition of the surgery-induced endogenous heat shock response, and may indeed explain the aggravation of necrosis when compared with that of nontreated controls.
CONCLUSION
The present study demonstrates for the first time, in an in vivo murine model of chronic ischemia, that the induction of HSP-32 after repetitive local heat preconditioning of the skin significantly preserves endangered myocutaneous ischemic tissue from apoptotic and necrotic cell death. This protection from ischemic tissue loss seems to be the result of the HSP-32-mediated vasoactive action of carbon monoxide, improving microcirculatory blood flow on an arteriolar and capillary level rather than increasing ischemic tolerance of the tissue.
Discussions
Dr. Ploeg: Thank you very much. First, I would like to thank the Program Committee for the privilege to comment on this presentation as well as thanking Dr. Harder for sending me the manuscript.
I do compliment Dr. Harder and his team for this very elegant study indeed. On first sight, it may look like we are drawn into a most secluded outskirt of experimental plastic surgery. But in fact and on the contrary the issue is a major topic in our daily surgical routine: how does tissue respond to our sometimes brutal, sometimes respectful handling? In my view, this does lead us to a most relevant topic in surgery: which critical factors do affect the balance of injury and repair?
In this study, you have asked yourself 3 questions: first, do repetitive heat applications induce HO-1 (HSP 32)? Your answer is yes as you are able to selectively block HO-1 with tin-protoporphyrin, which annihilates HO-1 appearance. I do not understand, however, that you see no HO-1 induction in the unconditioned control after inducing ischemia. In our experiments in rats and in humans, we have observed a definite ischemic-stress related HO-1 response in kidney, liver, and intestine. Unless, of course, I misunderstand your type of sham control and there was no ischemia applied. But then, I am afraid, I have to say, you have chosen the wrong control.
Next, you attempt to prove that heat induces protective HO-1 and is responsible for better survival. I still wonder whether HO-1 is a strong outcry for help, and thus a very helpful biomarker, or indeed a cytoprotective agent. In this context, I have a second question. Explain to me, please, how is it possible to precondition 24 hours before the event, induce HO-1, and with it the short-lived CO, which then for days will dilate the vessels and achieve these results. Also, you found awful results in your tin-protoporphyrin controls, even worse than in the sham controls. I think, as we know that tin-protoporphyrin is toxic, that you should have added an extra group just looking at the effect of tin-protoporphyrin in controls.
Finally, a last topic: You ask yourself the question, whether the beneficial effect of HO-1 is due to dilatation, and pedronitro perfusion or due to as you call it, and I love the term, a cross ischemic tolerance. Let us not forget that ischemic stress induces a multitude of cytokines, endothelial activation, up-regulation of von Willebrand factor, selectins, and adhesion molecules. This pronounced pro-inflammatory and pro-coagulatory response will affect the vasculature of your flap. Now, you conclude that HO-1-induced dilatation is the clue with better perfusion, and I really doubt that. CO does give a vascular dilatation, but do not forget that, at the same time, HO-1 products, such as free iron and the biliverdin, are very strong antioxidants. So, to distinguish between the dilatation effect or the, let us say, inflammatory effect, should you not have added another group using CO particles in the inhalation anesthetic of the mouse model or even added selective antioxidants or dexamethasone to down-regulate the pro-inflammatory sparks?
I am looking forward to your reply, and thank again the Program Committee for the opportunity to discuss this paper.
Dr. Harder: Thank you for these constructive comments.
The first question, if you remember the final necrosis in the SnPP-1X group comprised approximately 70% of the flap surface when compared to about 50% in the control group, which was just a nontreated chronic ischemic flap.
There is a difference of 20%, and this difference has to do with the blockage by SnPP-1X, not only from the heat shock-induced molecule HSP 32 but also with the endogenous HSP 32, which is induced by surgery and ischemia alone.
Concerning the control group we have data of only the control or sham operated group as you mention it, namely untreated ischemic flap. Due to extensive amount of data, the skinfold chamber without any flap has not been shown. Though, I can say that within all 3 corresponding areas of the nonischemic tissue within the chamber, over the 10 days we observe no changes concerning blood flow, be it on an arterial, capillary and/or venous level. Concerning the microvascular dilation over these 10 days, it is so that SnPP-IX was administered until necrosis has fully developed, which is approximately day 3. At that point, you cannot change anything any more.
Why does carbon monoxide save this ischemic tissue in the flap? We think that the first 24 hours is the crucial time span. If within this time you can reestablish or maintain a perfusion in this critically ischemic tissue, you can save the endangered tissue over days. Concerning a possible cross tolerance, or ischemic tolerance, I think it is very difficult to see whether it is only due to the maintenance or an amelioration of the microperfusion or also just due to an amelioration of the ischemic tolerance of the tissue, which as I said would imply that the tissue survives despite a failure of the microperfusion. The data we have, clearly shows that the amelioration of flap survival is due to the maintenance of the microcirculation, which I think is the main reason for survival.
SnPP-IX is toxic, this is true. It is a significant problem when running the experiments, because we lose quite an amount of animals. Of course, we also blocked biliverdin especially, another product of the HSP-32 metabolism. While I think that the accumulated free iron is not a big problem in this pathway, biliverdin acts as a strong antioxidant. Previous experiments done in the lab in skin flaps as well as in myocutaneous flaps have shown the following when differentiating between the effect of biliverdin and carbon monoxide. If there was any antioxidant effect in the survival of this chronic ischemic issue, it was in the true skin flap, which accounted for about 10% when compared to 90% CO-mediated improvement of the microvasculature. These figures were reproduced in the myocutaneous flap, which mimicks an ischemia reperfusion model. To protect microcirculation, the antioxidative effect of biliverdin represented about 10% to 15% when compared to 85% to 90% CO-mediated effect.
Dr. Tukiainen: Dr. Harder, thank you for this presentation. Could you postulate, what would happen, if you would use cooling or ischemic preconditioning, instead of using heat? Would the mechanism be the same?
Dr. Harder: As I said, the supraphysiological stressors are nonspecific. I would assume, and I know from literature, that cooling would induce the same mechanism, and so probably the same protection. However, our rationale for using heat is not because I would like to come to Scandinavia because you prefer the sauna to the snow. But it is just a practical point of view. To precondition a superficial organ or surface, such as skin or muscle, I believe that heat of 43°C is much better tolerated than cooling, which requires a temperature of about 4°C.
Dr. Clavien: Dr. Harder, I would like to congratulate you for these convincing sets of experiments using a myocutaneous flap model. It is very nice to see how much we can learn from a flap model, as similar endpoints are much more difficult to evaluate in a solid organ model such as the liver or the kidney. I have 2 questions. The main strategy to trigger the protective effects of preconditioning in solid organ is a short period of ischemia rather than heat. Do you have any evidence that similar protection can be achieved with ischemic preconditioning. Second, a major target of protection from preconditioning in vivo is the endothelial cells. You looked mostly at vasodilatation and tissue injury. Did you also look more specifically at endothelial cell injury, for example, in terms of cell death, expression of adhesion molecules, or whether they may express some pro-coagulant activity?
Dr. Harder: Although we did not study in the present model ischemic preconditioning, others have demonstrated in various models that this procedure is also capable of improving flap survival. Nonetheless, in elective flap surgery, heat preconditioning may have some advantages over ischemic preconditioning due to its ease in application without side effects. Ischemic preconditioning would require manipulation and clamping of the soft tissue pedicle (random pattern flap) or the vascular pedicle (island flap) including the nourishing artery, thus endangering this small, sensible vessel for iatrogenic injury.
From our experiments, we do not have any indication for a specific protection of endothelial cells by heat shock preconditioning. However, in the model presented, there is neither evidence for distinct microvascular thrombosis nor indication for preferential apoptotic cell death of endothelial cells. The intravital microscopy shows leakage of the microvasculature, reflecting an increase in vascular permeability in ischemic tissue areas. However, leakage is neither prevented nor reduced by heat shock, indicating that protection is mediated by a mechanism different from inhibition of permeability increase.
Footnotes
Y. H. is a recipient of a fellowship of the Swiss National Science Foundation (SNF No. PBBSB-102601), the “Freiwillige Akademische Gesellschaft,” and the “Margarete und Walter Lichtenstein Stiftung, Medical Department” in Basel, Switzerland.
Reprints: Yves Harder, MD, Institute for Clinical & Experimental Surgery, University of Saarland, D-66421 Homburg/Saar, Germany. E-mail: yvesharder@bluewin.ch.
REFERENCES
- 1.Kearns SR, Moneley D, Murray P, et al. Oral vitamin C attenuates acute ischaemia-reperfusion injury in skeletal muscle. J Bone Joint Surg Br. 2001;83:1202–1206. [DOI] [PubMed] [Google Scholar]
- 2.van Uchelen JH, Werker PM, Kon M. Complications of abdominoplasty in 86 patients. Plast Reconstr Surg. 2001;107:1869–1873. [DOI] [PubMed] [Google Scholar]
- 3.Erni D, Harder YD. The dissection of the rectus abdominis myocutaneous flap with complete preservation of the anterior rectus sheath. Br J Plast Surg. 2003;56:395–400. [DOI] [PubMed] [Google Scholar]
- 4.Angrisani L, Favretti F, Furbetta F, et al. Italian Group for Lap-Band System: results of multicenter study on patients with BMI < or =35 kg/m2. Obes Surg. 2004;14:415–418. [DOI] [PubMed] [Google Scholar]
- 5.Berghmans T, Tragas G, Sculier JP. Age and treatment of non-small-cell lung cancer: a database analysis in elderly patients. Support Care Cancer. 2002;10:619–623. [DOI] [PubMed] [Google Scholar]
- 6.Finseth F, Cutting C. An experimental neurovascular island skin flap for the study of the delay phenomenon. Plast Reconstr Surg. 1978;61:412–420. [DOI] [PubMed] [Google Scholar]
- 7.Menger MD, Barker JH, Messmer K. Capillary blood perfusion during postischemic reperfusion in striated muscle. Plast Reconstr Surg. 1992;89:1104–1114. [DOI] [PubMed] [Google Scholar]
- 8.Vollmar B, Menger MD. Assessment of microvascular oxygen supply and tissue oxygenation in hepatic ischemia/reperfusion. Adv Exp Med Biol. 1997;428:403–408. [DOI] [PubMed] [Google Scholar]
- 9.Amon M, Menger MD, Vollmar B. Heme oxygenase and nitric oxide synthase mediate cooling-associated protection against TNF-alpha-induced microcirculatory dysfunction and apoptotic cell death. FASEB J. 2003;17:175–185. [DOI] [PubMed] [Google Scholar]
- 10.Harder Y, Contaldo C, Klenk J, et al. Improved skin flap survival after local heat preconditioning in pigs. J Surg Res. 2004;119:100–105. [DOI] [PubMed] [Google Scholar]
- 11.Inoue K, Ando S, Gyuan F, et al. A study of the myocardial protective effect of rapid cooling based on intracellular Ca, intracellular pH, and HSP70. Ann Thorac Cardiovasc Surg. 2003;9:301–306. [PubMed] [Google Scholar]
- 12.Nayeem MA, Elliott GT, Shah MR, et al. Monophosphoryl lipid A protects adult rat cardiac myocytes with induction of the 72-kD heat shock protein: a cellular model of pharmacologic preconditioning. J Mol Cell Cardiol. 1997;29:2305–2310. [DOI] [PubMed] [Google Scholar]
- 13.Reed RC, Nicchitta CV. Chaperone-mediated cross-priming: a hitchhiker's guide to vesicle transport [Review]. Int J Mol Med. 2000;6:259–264. [DOI] [PubMed] [Google Scholar]
- 14.De Maio A. The heat shock response. New Horiz. 1995;3:198–207. [PubMed] [Google Scholar]
- 15.Wynn RM, Davie JR, Cox RP, et al. Molecular chaperones: heat shock proteins, foldases, and matchmakers. J Lab Clin Med. 1994;124:31–36. [PubMed] [Google Scholar]
- 16.Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2:2557–2568. [PubMed] [Google Scholar]
- 17.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. [DOI] [PubMed] [Google Scholar]
- 18.Goda N, Suzuki K, Naito M, et al. Distribution of heme oxygenase isoforms in rat liver: topographic basis for carbon monoxide-mediated microvascular relaxation. J Clin Invest. 1998;101:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Latchman DS. Heat shock proteins and cardiac protection. Cardiovasc Res. 2001;51:637–646. [DOI] [PubMed] [Google Scholar]
- 20.Lepore DA, Knight KR, Anderson RL, et al. Role of priming stresses and Hsp70 in protection from ischemia-reperfusion injury in cardiac and skeletal muscle. Cell Stress Chaperones. 2001;6:93–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirino T. Ischemic tolerance. J Cereb Blood Flow Metab. 2002;22:1283–1296. [DOI] [PubMed] [Google Scholar]
- 22.Shimizu H, Takahashi T, Suzuki T, et al. Protective effect of heme oxygenase induction in ischemic acute renal failure. Crit Care Med. 2000;28:809–817. [DOI] [PubMed] [Google Scholar]
- 23.Rücker M, Schäfer T, Roesken F, et al. Local heat shock priming-induced improvement in microvascular perfusion in osteomyocutaneous flaps is mediated by heat shock protein 32. Br J Surg. 2001;88:450–457. [DOI] [PubMed] [Google Scholar]
- 24.Harder Y, Amon M, Erni D, et al. Evolution of ischemic tissue injury in a random pattern flap: a new mouse model using intravital microscopy. J Surg Res. 2004;121:197–205. [DOI] [PubMed] [Google Scholar]
- 25.Lehr HA, Leunig M, Menger MD, et al. Dorsal skinfold chamber technique for intravital microscopy in nude mice. Am J Pathol. 1993;143:1055–1062. [PMC free article] [PubMed] [Google Scholar]
- 26.Klyscz T, Junger M, Jung F, et al. Cap image: a new kind of computer-assisted video image analysis system for dynamic capillary microscopy. Biomed Tech. 1997;42:168–175. [DOI] [PubMed] [Google Scholar]
- 27.Gross JF, Aroesty J. Mathematical models of capillary flow: a critical review. Biorheology. 1972;9:225–264. [DOI] [PubMed] [Google Scholar]
- 28.Anderson KE, Simionatto CS, Drummond GS, et al. Tissue distribution and disposition of tin-protoporphyrin, a potent competitive inhibitor of heme oxygenase. J Pharmacol Exp Ther. 1984;228:327–333. [PubMed] [Google Scholar]
- 29.Vreman HJ, Ekstrand BC, Stevenson DK. Selection of metalloporphyrin heme oxygenase inhibitors based on potency and photoreactivity. Pediatr Res. 1993;33:195–200. [DOI] [PubMed] [Google Scholar]
- 30.Mokuno Y, Berthiaume F, Tompkins RG, et al. Technique for expanding the donor liver pool: heat shock preconditioning in a rat fatty liver model. Liver Transpl. 2004;10:264–272. [DOI] [PubMed] [Google Scholar]
- 31.Raeburn CD, Cleveland JC Jr, Zimmerman MA, et al. Organ preconditioning. Arch Surg. 2001;136:1263–1266. [DOI] [PubMed] [Google Scholar]
- 32.Healy C, Mulhall KJ, Nelligan M, et al. Postoperative stiffness and adhesion formation around repaired and immobilized Achilles tenotomies are prevented using a model of heat shock protein induction. J Surg Res. 2004;120:225–229. [DOI] [PubMed] [Google Scholar]
- 33.Willis D, Moore AR, Frederick R, et al. Heme oxygenase: a novel target for the modulation of the inflammatory response. Nat Med. 1996;2:87–90. [DOI] [PubMed] [Google Scholar]
- 34.Mikami KI, Otaka M, Goto T, et al. Induction of a 72-kDa heat shock protein and protection against lipopolysaccharide-induced liver injury in cirrhotic rats. J Gastroenterol Hepatol. 2004;19:884–890. [DOI] [PubMed] [Google Scholar]
- 35.Uchinami H, Yamamoto Y, Kume M, et al. Effect of heat shock preconditioning on NF-kappaB/I-kappaB pathway during I/R injury of the rat liver. Am J Physiol Gastrointest Liver Physiol. 2002;282:G962–G971. [DOI] [PubMed] [Google Scholar]
- 36.Yamagami K, Enders G, Schauer RJ, et al. Heat-shock preconditioning protects fatty livers in genetically obese Zucker rats from microvascular perfusion failure after ischemia reperfusion. Transpl Int. 2003;16:456–463. [DOI] [PubMed] [Google Scholar]
- 37.Redaelli CA, Wagner M, Kulli C, et al. Hyperthermia-induced HSP expression correlates with improved rat renal isograft viability and survival in kidneys harvested from non-heart-beating donors. Transpl Int. 2001;14:351–360. [DOI] [PubMed] [Google Scholar]
- 38.Akamatsu J, Ueda K, Tajima S, et al. Sulfatide elongates dorsal skin flap survival in rats. J Surg Res. 2000;92:36–39. [DOI] [PubMed] [Google Scholar]
- 39.Katori M, Buelow R, Ke B, et al. Heme oxygenase-1 overexpression protects rat hearts from cold ischemia/reperfusion injury via an antiapoptotic pathway. Transplantation. 2002;73:287–292. [DOI] [PubMed] [Google Scholar]
- 40.Vulapalli SR, Chen Z, Chua BH, et al. Cardioselective overexpression of HO-1 prevents I/R-induced cardiac dysfunction and apoptosis. Am J Physiol Heart Circ Physiol. 2002;283:H688–H694. [DOI] [PubMed] [Google Scholar]
- 41.Yang CW, Kim BS, Kim J, et al. Preconditioning with sodium arsenite inhibits apoptotic cell death in rat kidney with ischemia/reperfusion or cyclosporine-induced injuries: the possible role of heat shock protein 70 as a mediator of ischemic tolerance. Exp Nephrol. 2001;9:284–294. [DOI] [PubMed] [Google Scholar]
- 42.Terajima H, Kondo T, Enders G, et al. Reduction of hepatic microcirculatory failure caused by normothermic ischemia/reperfusion-induced injury by means of heat shock preconditioning. Shock. 1999;12:329–334. [DOI] [PubMed] [Google Scholar]
- 43.Choi AM, Sylvester S, Otterbein L, et al. Molecular responses to hyperoxia in vivo: relationship to increased tolerance in aged rats. Am J Respir Cell Mol Biol. 1995;13:74–82. [DOI] [PubMed] [Google Scholar]
- 44.Stone JR, Marletta MA. Soluble guanylate cyclase from bovine lung: activation with nitric oxide and carbon monoxide and spectral characterization of the ferrous and ferric states. Biochemistry. 1994;33:5636–5640. [DOI] [PubMed] [Google Scholar]
- 45.Wang R. Resurgence of carbon monoxide: an endogenous gaseous vasorelaxing factor. Can J Physiol Pharmacol. 1998;76:1–15. [DOI] [PubMed] [Google Scholar]
- 46.Lubbe AS. Heat shock attenuates endothelium-dependent vasodilation in skeletal muscle microcirculation. Shock. 1994;2:179–184. [DOI] [PubMed] [Google Scholar]
- 47.Kozma F, Johnson RA, Zhang F, et al. Contribution of endogenous carbon monoxide to regulation of diameter in resistance vessels. Am J Physiol. 1999;276(4 Pt 2):R1087–R1094. [DOI] [PubMed] [Google Scholar]
- 48.Wagner CT, Durante W, Christodoulides N, et al. Hemodynamic forces induce the expression of heme oxygenase in cultured vascular smooth muscle cells. J Clin Invest. 1997;100:589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morita T, Perrella MA, Lee ME, et al. Smooth muscle cell-derived carbon monoxide is a regulator of vascular cGMP. Proc Natl Acad Sci USA. 1995;92:1475–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morita T, Kourembanas S. Endothelial cell expression of vasoconstrictors and growth factors is regulated by smooth muscle cell-derived carbon monoxide. J Clin Invest. 1995;96:2676–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stanford SJ, Walters MJ, Mitchell JA. Carbon monoxide inhibits endothelin-1 release by human pulmonary artery smooth muscle cells. Eur J Pharmacol. 2004;486:349–352. [DOI] [PubMed] [Google Scholar]
- 52.Zhang F, Kaide JI, Yang L, et al. CO modulates pulmonary vascular response to acute hypoxia: relation to endothelin. Am J Physiol Heart Circ Physiol. 2004;286:H137–H144. [DOI] [PubMed] [Google Scholar]
- 53.Rensing H, Bauer I, Zhang JX, et al. Endothelin-1 and heme oxygenase-1 as modulators of sinusoidal tone in the stress-exposed rat liver. Hepatology. 2002;36:1453–1465. [DOI] [PubMed] [Google Scholar]