

Abstract
Objectives:
To evaluate the effects of neoadjuvant therapy with the selective cyclooxygenase-2 (COX-2) inhibitor celecoxib in vitro and in patients with esophageal adenocarcinoma on COX-2 and MET expression.
Summary Background Data:
High COX-2 and/or MET expression levels are negative prognostic factors for adenocarcinoma of the esophagus. Nonsteroidal anti-inflammatory drugs (NSAIDs) and selective COX-2 inhibitors exert anticancer mechanisms as is evident from epidemiologic studies and from experimental models for esophageal cancer. The mechanisms and the significance of these findings in patients with adenocarcinoma of the esophagus are unknown.
Methods:
Esophageal adenocarcinoma cell lines were used to asses the effects in vitro. To study the clinical effects 12 patients with esophageal adenocarcinoma were included for neoadjuvant treatment (4 weeks) with celecoxib at 400 mg twice daily. Fifteen patients not receiving NSAIDs or celecoxib were included as a control. Effects were evaluated using the MTT-cell viability test, Western blot analysis, immunohistochemistry, and RT-PCR.
Results:
In vitro celecoxib administration resulted in decreased cell viability, increased apoptosis, and decreased COX-2 and MET expression levels. In patients, neoadjuvant treatment with celecoxib significantly down-regulated COX-2 and MET expression in the tumor when compared with the nontreated control group and when compared with pretreatment measurements.
Conclusions:
This is the first study to show in vitro and in patients with esophageal adenocarcinoma that selective COX-2 inhibition down-regulates COX-2 and MET expression, both important proteins involved in cancer progression and dissemination. Therefore, (neo)adjuvant therapy with celecoxib might have clinical potential for patients with esophageal adenocarcinoma.
This study demonstrates for the first time both in vitro and in patients that neoadjuvant celecoxib administration down-regulates COX-2, MET, and VEGF in esophageal adenocarcinoma. Since COX-2 and MET are both negative prognostic factors and important oncogenes involved in cancer progression and dissemination, the role of selective COX-2 inhibition as a neoadjuvant treatment of esophageal adenocarcinoma deserves further exploration.
Adenocarcinoma of the esophagus, developing via the Barrett's metaplasia-dysplasia-carcinoma sequence, is associated with a rapidly rising incidence and a poor prognosis.1,2 The best curative option is surgical resection, but even after extensive surgery, overall survival rates rarely exceed 25%.3 Advances in careful preoperative selection, radical surgery, and conventional (neo)adjuvant chemo- and radiotherapy have only shown limited improvement of prognosis.4,5 To improve the therapeutic options for patients with esophageal cancer, current research focuses on the biologic mechanisms of cancer progression and dissemination and targets for specific chemotherapeutic treatment strategies.
Epidemiologic studies have demonstrated approximately 50% reduction in the incidence of gastrointestinal adenocarcinomas in persons regularly taking aspirin or other nonsteroidal anti-inflammatory drugs (NSAIDs).6 One of the target enzymes of NSAIDs is cyclooxygenase-2 (COX-2), a rate-limiting enzyme in prostaglandin E2 synthesis.7 The importance of COX-2 in carcinogenesis and cancer progression has been implicated in cervical, breast, prostate, and various gastrointestinal cancers, including esophageal adenocarcinoma.8 During the multistep malignant degeneration of Barrett's epithelium into esophageal adenocarcinoma, the COX-2 enzyme is increasingly expressed, suggesting an important role in this carcinogenic process.9 In addition, COX-2 expression has been identified as an independent prognostic variable for esophageal adenocarcinoma, indicating that COX-2 could be an attractive molecular target for specific chemotherapeutic treatment.10 NSAIDs and selective COX-2 inhibitors have been shown to induce apoptosis and to decrease proliferation in vitro, in animal models for esophageal cancer and in patients with adenomas of the colon.11–13 However, the exact mechanism by which NSAIDs and in particular selective COX-2 inhibitors exert their anticarcinogenic effects remains to be elucidated.
An important cellular pathway causing tumor cell survival, proliferation, and invasion is mediated by the hepatocyte growth factor (HGF).14–17 The receptor for HGF is called MET, a proto-oncogene that has been implicated in progression and dissemination of several cancer types, including esophageal cancer.18–23 In experimental models, the activation of MET causes decreased apoptosis and enhanced proliferation, angiogenesis, and invasion.24,25 Interestingly, COX-2- and MET-dependent signal transduction pathways are functionally connected in cancer. Prostaglandins have been shown to promote MET activation and subsequent oncogene transcription in colorectal cancer, causing decreased apoptosis and increased proliferation and angiogenesis.26 In addition, NSAIDs have been shown to inhibit HGF/MET-dependent signal transduction, resulting in decreased proliferation and invasiveness in experimental cancer models.14,27 Therefore, we postulated that inhibition of MET may constitute an important factor in explaining the anticarcinogenic effects of NSAIDs.
Although results from various in vitro and animal studies, and from human epidemiologic studies support COX-2 inhibition as a novel chemotherapeutic strategy for esophageal adenocarcinoma, clinical implementation in daily practice is still debated. Recent reports about the increased incidence of cardiovascular events have raised questions about the safety of long-term use of selective COX-2 inhibitors for chemoprevention in the general population. However, because of the poor prognosis of patients with esophageal cancer, these side effects play only a minor role in the (neo)adjuvant setting. Elucidating the mechanisms of selective COX-2 inhibitors in cancer provides further insight in carcinogenesis and might thus reveal targets for novel therapy. Therefore, the aim of this study was to characterize the molecular mechanisms and the potential clinical role of selective COX-2 inhibitors in the treatment of esophageal adenocarcinoma.
PATIENTS, MATERIALS, AND METHODS
Cell Cultures
To evaluate the biochemical effects of COX-2 inhibition in vitro, 2 human esophageal adenocarcinoma cell lines (OE19 and OE33) were used. In the OE19 cells, only weak expression of the COX-2 protein was observed, whereas the OE33 cells demonstrated a high COX-2 expression (data not shown).28 Both cell lines, originally derived from a human esophageal adenocarcinoma, were obtained from the European Collection of Cell Cultures. The adherent cells were cultured in Dulbecco modified Eagle medium (Sigma-Aldrich, St. Louis, MO), supplemented with 10% fetal calf serum (Integro, Leuvenheim, the Netherlands), 5 mmol/L l-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (Invitrogen Corp., Carlsbad, CA) at 37°C in a 5% CO2 atm. Cells were passaged 20 times maximally. Stock solutions of the pure compound celecoxib (kindly provided by Pfizer, New York, NY) were made in dimethyl sulfoxide. A final dimethyl sulfoxide concentration of 0.05% was used for all in vitro experiments including control experiments.
Cell Viability in Cell Culture
The cell viability was assessed by the mitochondrial function, measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenil tetrazolium bromide (MTT) reduction activity as previously reported.29 Briefly, cells were seeded in a 96-well plate and incubated with increasing concentrations of celecoxib. After 36 and 72 hours, the cells were incubated with 0.5 mg/ml MTT (Sigma-Aldrich) for 30 minutes at 37°C. Subsequently, the media were aspirated and the cells were lysed in isopropanol/0.04 mol/L HCl, where after the absorbance was read at 560 nm, using a microplate reader (Bio-Rad Laboratories, Veenendaal, The Netherlands).
Western Blot Analysis
The esophageal cancer cell lines were seeded in 6-well plates to be at 50% confluence at the time of celecoxib administration. After the time of incubation, cells were kept on ice and washed with ice-cold phosphate-buffered saline. Cells were harvested in 300 μL of sample buffer (125 mmol/L Tris/HCl, pH 6.8; 4% sodium dodecyl sulfate (SDS); 1% β-mercaptoethanol; 20% glycerol; 1 mg of bromphenol blue). After 5 seconds of sonification and a 5-minute incubation at 95°C, 20 μL was loaded onto SDS-PAGE and subsequently transferred to a polyvinylidene difluoride membrane. The membranes were blocked with Tris-buffered saline (TBS) supplemented with 0.05% Tween-20 (wash buffer) and 1% casein and incubated with primary antibody overnight at 4°C, diluted in blocking buffer. Primary antibodies used for Western blot analysis were: anti-COX-2 (Cayman Chemical Co., Ann Arbor, MI), anti-MET, DL21 (Upstate Biotechnology, Lake Placid, NY), anticleaved-caspase-3, (Cell Signaling Technology, Beverly, MA) a marker for apoptosis, and anti-β-actin (c19; Santa Cruz Biotechnology, CA), which was used for loading control. Phospho-specific antibodies against MET and against the downstream kinases of the MET induced signal transduction were used to assess the effects of celecoxib after 30 minutes of incubation; anti-MET (DQ-13) (Upstate), anti-phospho-MET (Tyr1234/1235), anti-phospho GAB-1 (Tyr472), and anti-phospho-ERK-1 and -ERK-2 (Thr202/Tyr204) (Cell Signaling Technology). Subsequently, the membranes were washed and incubated with a horseradish peroxidase-conjugated secondary antibody in blocking buffer. The immunoreactive bands were detected using the LumiLight Western Blotting Substrate (Roche Molecular Biochemicals, Mannheim, Germany).
Patients
To evaluate the biochemical effects of selective COX-2 inhibition in vivo, celecoxib (400 mg twice daily) was administered as neoadjuvant treatment in patients with esophageal adenocarcinoma. The duration of treatment was 4 weeks until the day of surgery. The protocol was approved by the institutional Medical Ethics Committee. Written informed consent was obtained from all patients before inclusion. Within a 1-year period, 12 patients were selected for celecoxib treatment and 15 similar patients were prospectively selected as a control group. Inclusion criteria were age >18 years, histologically proven adenocarcinoma of the distal esophagus (Siewert type 1), and eligible for surgery with curative intent. Patients were excluded in case of known cardiovascular disease, a history of heart failure, hypertension, renal failure, history of gastric/duodenal ulcer or inflammatory intestinal disease, use of aspirin or NSAIDs and known hypersensitivity to these drugs. All patients were preoperatively evaluated with CT scan of chest and abdomen, whole body PET scan, endoscopic ultrasonography, external ultrasonography of the neck, indirect laryngoscopy, and a chest x-ray to adequately determine cTNM stage. None of the patients underwent neoadjuvant treatment other than celecoxib.
Tissue Collection
At the time of diagnosis, endoscopic biopsies were taken from the tumor, fixed in 10% neutral-buffered formalin, and paraffin-embedded according to standard procedures. After surgery, representative samples of the tumor were taken from the resection specimen as assigned by a GI pathologist (G.J.A.O.). The specimens were both snap-frozen at −180°C and stored in −80°C, and paraffin embedded. At a later stage, these tissue samples were analyzed immunohistochemically and with quantitative reverse transcription-PCR assay (RT-PCR).
Tissue Microarray
A tissue microarray (TMA) was made to semi-quantify the protein expression levels in the resection specimens of treated and nontreated patients. Formalin-fixed, paraffin-embedded tumor samples were used for the TMA. First, the sections were reviewed histologically and marked by a pathologist (F.J.W.K.) to select 3 morphologically representative regions for each of the tumor samples. Then from these regions, cylindrical core tissue specimens (diameter = 0.6 mm) were acquired and arrayed precisely into a new recipient paraffin block (20 × 35 mm) using a custom-built precision instrument (Beecher Instruments, Silver Spring, MD).
Immunohistochemical Analysis
Immunohistochemical analysis was performed of the endoscopic biopsies before treatment and of the resection specimens after treatment to compare the protein expression levels. The TMA was evaluated immunohistochemically to analyze protein expression levels of treated versus nontreated patients. Five-micrometer-thick sections were cut, incubated overnight at 37°C, and subsequently deparaffinized in xylene, rehydrated, and treated with 3% H2O2 in methanol for 10 minutes to block endogenous peroxidase activity. All specimens were subjected to heat-induced antigen retrieval in 10 mmol/L sodium citrate buffer (pH 6.0) for 10 minutes at 95°C. To block aspecific binding, the slides were incubated with TBS supplemented with 5% goat serum. Sections were incubated with the primary antibodies anti-COX-2 (1:100), anti-MET (1:100), anticleaved caspase-3 (1:200), anti-Ki-67 (1:100) (clone MIB1; Ylem, Rome, Italy), a marker for proliferation, anti-CD31 (1:50) (clone JC/70 A; DAKO, Glostrup, Denmark), an endothelial marker used to evaluate angiogenesis and antivascular endothelial growth factor (VEGF) (1:500) (clone C-1; Santa Cruz Biotechnology, Santa Cruz, CA) diluted in TBS with 1% bovine serum albumin overnight at 4°C. After washing, the sections were incubated with antimouse/rabbit-peroxidase polymer at 30 minutes at room temperature (Powervision; Immunovision, Inc., Daly City, CA). Diaminobenzidine chromogen (Sigma) was used for visualization, where after the sections were counterstained with hematoxylin and embedded. Specificities of the antibodies were confirmed by negative controls using irrelevant immunoglobulins instead of primary antibodies. Colon cancer tissue was included as a positive control. The analysis of all tissue sections was performed independently by 3 different investigators (J.B.T., C.J.B., and G.J.A.O.) without patient identification parameters. For COX-2 immunohistochemical staining, the following scoring criteria of tumor cells were agreed upon before the analysis: 0, no staining; 1, weak diffuse cytoplasmic staining (may contain stronger intensity in <10% of cancer cells); 2, moderate to strong granular cytoplasmic staining in 10% to 90% of cancer cells; 3, more than 90% of tumor cells stained with strong intensity; 4, as 3 with the addition of very strong granular staining. These scoring criteria have previously been described in our previous report on COX-2 expression in esophageal adenocarcinoma.10 For evaluation of MET, the same scoring criteria were used, since the staining pattern is similar except for the membranous localization of the MET receptor. Cleaved caspase-3 and Ki-67 immunopositivity were determined as the percentage of tumor cells with positive staining counted in 3 separate ×40 microscopic fields (at least 200 cells per field were counted). Subsequently samples were scored as 0 if no cells were positively stained; 1, when less than 1% of the cancer cells were positive. A score of 2 was considered if 1% to 5% immunostained cancer cells were positive, a score of 3 when 5%, a score of 4 when more than 5% of the tumor cells were positive. The evaluation of microvessel density by CD31 was performed by counting individual microvessels in 2 or 3 separate random ×40 fields and classified in 4 groups. Areas of diffuse hemorrhage or necrosis were neglected. Since the VEGF production by the tumor cells stimulates the formation of new vessels, characterized by increased endothelial CD31 expression, immunohistochemistry for VEGF was also included to evaluate the inhibitory effects of the 4-week exposure to celecoxib on angiogenesis. The following scoring criteria for VEGF expression in the cytoplasm of the tumor cells were used: 0, no staining; 1, weak diffuse cytoplasmic staining (may contain stronger intensity in <10% of cancer cells); 2, moderate to strong cytoplasmic staining in 10% to 80% of cancer cells; 3, tumor cells stained with strong intensity similar to the endothelial cells which served as a positive control (similar to score 3).
For the evaluation of protein expression using the TMA, patients were only included if at least 2 paraffin samples of the 3 included slides on the TMA could be evaluated. Because of inadequacy of the tissue present on the TMA of 1 patient in the treated group, this patient could not be included in the comparison of the treated versus the nontreated group. The immunohistochemical comparison of endoscopic biopsies before treatment and of resection specimens after treatment was performed in 9 of the 11 patients in the treatment group. Because of insufficient tissue quality (only dysplastic, or normal epithelial cells) in the biopsies of 2 patients, these were excluded from the evaluation of the protein expression before and after treatment.
RNA Isolation and RT-PCR
Frozen resection specimens were selected to contain more than 70% tumor cells. Total RNA was isolated from the frozen resection specimens using Trizol reagent (Life Technologies Inc, Gaithersburg, MD) according to the manufacturer's protocol and quantified by spectrophotometry. Reverse transcription was performed with 5 μg of total RNA using superscript transcriptase III (Life Technologies Inc). Primers for MET and TATA-box-binding protein (TBP) mRNA (a housekeeping gene used to correct for loading) were designed to amplify across an exon/exon boundary to prevent amplification of genomic DNA. The primers used for amplification of MET were 5′-CAGATGTGTGGTCCTTTG-3′ (forward) and 5′-ATTCGGGTTGTAGGAGTCT-3′ (reverse). The PCR reaction was performed on the LightCycler instrument using the LightCycler FastStart DNA Master SYBR green 1 reaction components (Roche Diagnostics). The cycling conditions were as follows: initial denaturation at 95°C for 10 minutes, followed by 40 cycles of 95°C for 5 seconds, 60°C for 10 seconds, and 72°C for 15 seconds. All runs included a negative water control. The samples were quantified using Roche Lightcycler software, version 3.5 (Roche Diagnostics) and linregPCR software, version 7.4 as described.30
Statistical Analysis
Data obtained from Western blot analysis, immunohistochemistry, and RT-PCR were analyzed using the Student t test (2 sided) with a level of significance at P value < 0.05. Graphically, data are shown as dots, line or bar graphs depicting the mean values and the standard errors of mean. All statistical analyses were performed using the Statistical Software Package version 11.5 (SPSS Inc., Chicago, IL).
RESULTS
Effects of Celecoxib on Cell Viability and Apoptosis In Vitro
Before the effects of COX-2 inhibition in patients with esophageal cancer were investigated, it was first analyzed whether esophageal adenocarcinoma cell lines react to the COX-2 inhibitor celecoxib by a decrease in cell viability and by induction of apoptosis. Celecoxib administration reduced cell viability in both cell lines at 40 μmol/L and more, as detected by the incorporation of tryptan blue by the MTT assay after 36 hours (Fig. 1A). At 72 hours, celecoxib is more effective, especially at concentrations of 20 and 40 μmol/L, to decrease viability in both cell lines. In agreement, an increase in the cleavage product of caspase-3, an unequivocal marker for cellular apoptosis, was observed as detected by Western blot analysis after 72 hours in both cell lines (Fig. 1B).
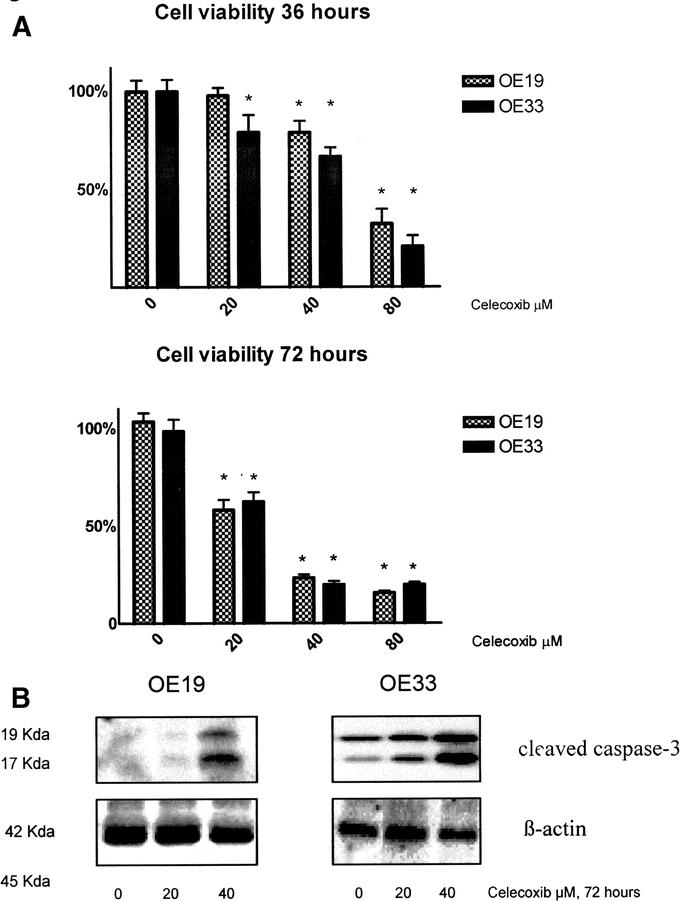
FIGURE 1. Celecoxib reduces cell viability and induces apoptosis in vitro. A, The viability of esophageal cancer cells (OE19 and OE33) was measured by a MTT assay. After 36 hours of exposure to celecoxib, the viability was significantly decreased at concentrations of celecoxib of 20 μmol/L and above in OE33 cells and at 40 μmol/L and above in OE19 cells. After 72 hours of exposure to celecoxib, the viability was decreased already at 20 μmol/L and above for both cell lines. *Significant decrease compared with the control (P < 0.05). B, Western blot analysis of OE19 and OE33 cell lysates after 72 hours incubation with celecoxib. The cleaved caspase-3 products of 17 and 19 kDa are shown as markers for apoptosis. Celecoxib progressively induces apoptosis at increasing concentrations. The level of β-actin is shown as a loading control.
Effects of Celecoxib on COX-2 and MET Protein Expression In Vitro
Subsequently, the effects of celecoxib in vitro on the expression of COX-2 and MET protein levels were investigated in the cell line OE33. The expression levels of the proteins were determined after 36 hours of treatment, at a time point without significant cell death. Decreased COX-2 protein levels were observed after celecoxib administration. Also, the levels of MET protein were decreased when compared with the levels of β-actin, albeit at relatively high concentrations of celecoxib (Fig. 2).
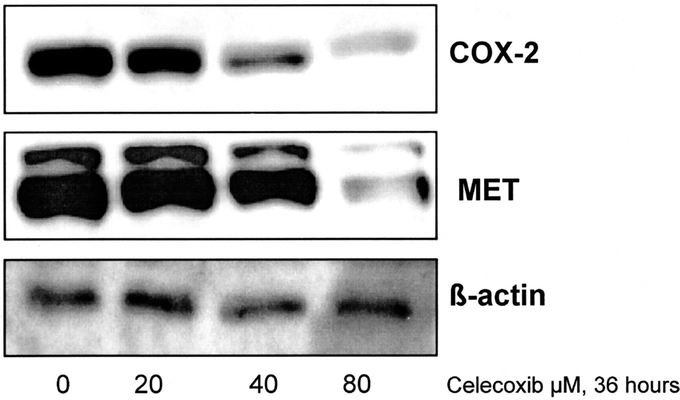
FIGURE 2. Celecoxib down-regulates both COX-2 and MET protein levels in vitro. Western blot analysis of OE33 cell lysates after 36 hours incubation with celecoxib. COX-2 protein levels are already decreased at low concentrations of celecoxib, whereas MET protein is down-regulated at higher concentrations. The level of β-actin is shown as a loading control.
Short-term Effects on MET Signal Transduction of Celecoxib In Vitro
To provide insight in the short-term effects of COX-2 inhibition, we have investigated the effects on MET phosphorylation and its induced activation of signal transduction employing short time (30 minutes) incubation with celecoxib. This is an early time point at which the protein levels cannot yet have changed significantly. However, the phosphorylation status of a protein, reflecting its activation level, can change within short time frames.
The activation of the MET protein is decreased after celecoxib administration, which is demonstrated by the decrease of the phosphorylation of MET (p-MET), whereas the total MET protein is unchanged (Fig. 3). The downstream targets of MET induced signal transduction are GAB 1 and ERK-1 and -2. Upon activation by phosphorylation, these kinases induce increased expression of COX-2 and MET (positive feedback loop).25 In agreement with the decline in MET activation, decreased levels of phosphorylated GAB 1 and ERK-1 and -2 were observed after 30 minutes of celecoxib administration at both concentrations of 25 and 50 μmol/L (Fig. 3).
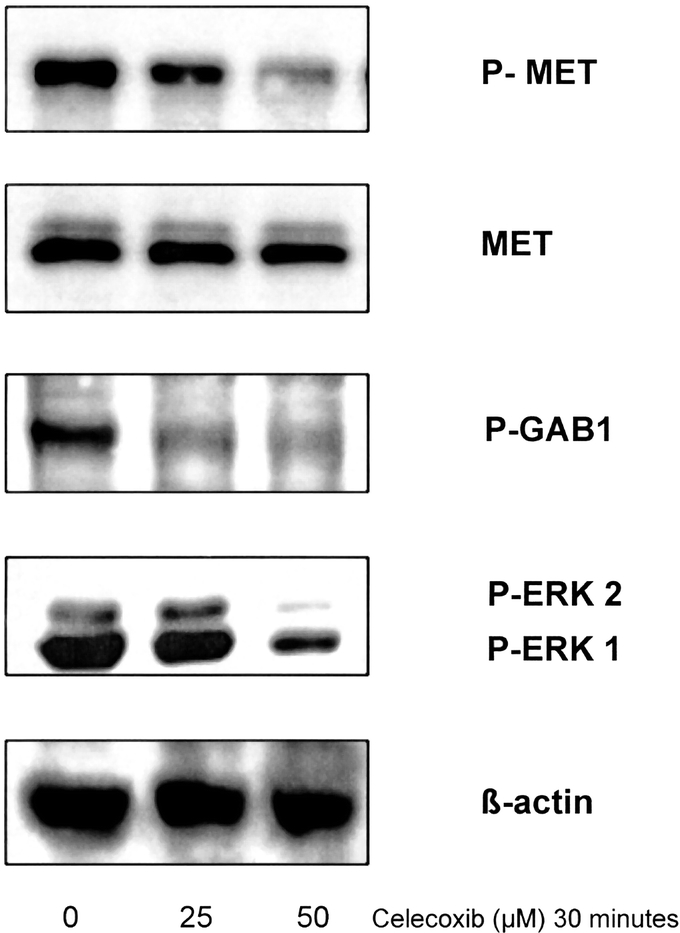
FIGURE 3. Celecoxib targets MET activation and downstream signal transduction in vitro. Western blot analysis of OE33 cell lysates after 30-minute incubation with celecoxib. The levels for phospho-MET, MET, phospho-GAB1, phospho-ERK-1 and ERK-2, and β-actin protein levels are shown. After 30 minutes, celecoxib induces a decrease in the phosphorylated form of MET. The total MET protein is shown as a control (unchanged). This is accompanied by a decrease of activation of the downstream targets GAB1, ERK-1, and ERK-2. The level of β-actin is shown as a loading control.
Comparison Between Tumoral COX-2 and MET Expression in Patients After Celecoxib Treatment and Control Patients
To investigate the potential clinical significance of the biochemical results obtained in vitro, 12 patients with an adenocarcinoma of the distal esophagus underwent neoadjuvant treatment with celecoxib. One adverse event of celecoxib treatment was registered in a patient experiencing a mild cutaneous rash that spontaneously resolved after stopping celecoxib treatment. This patient was excluded from further analysis. No other adverse events (including cardiovascular events) were identified during treatment. The median treatment duration of the remaining 11 patients was 4 weeks with a compliance rate of 100%, as explicitly indicated by the patients. The clinicopathologic parameters of both groups are described in Table 1. The study groups were comparable in age, sex, and pTNM staging. There was no significant difference in the uTNM staging before celecoxib treatment versus after treatment. It should be noticed, however, that both scores cannot be compared objectively because of the limited accuracy of the preoperative ultrasound compared with histology.31
TABLE 1. Clinicopathologic Parameters of Patients Treated With Celecoxib in Comparison With the Nontreated Control Group
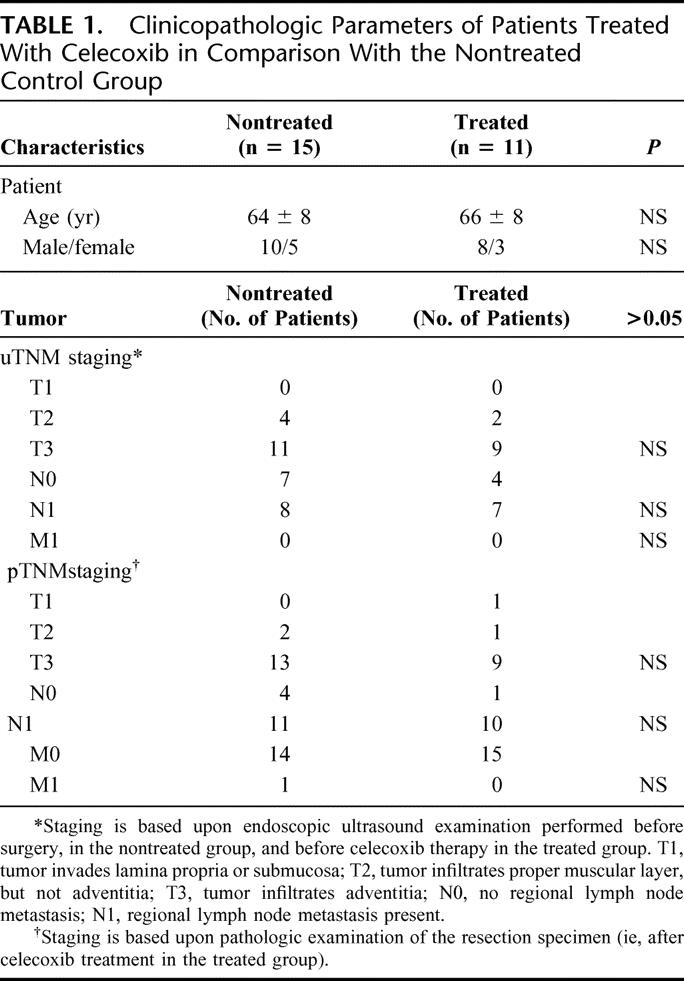
Figure 4 shows examples of immunohistochemical stainings of tissue spots presented on the TMA. Of 1 patient in the treated group, no adequate tissue was present on the TMA; therefore, this patient was excluded in the comparison of the treated versus the nontreated group. In agreement with the results obtained in vitro, the level of COX-2 protein was significantly lower in patients treated with celecoxib when compared with the nontreated control group (P < 0.01, Fig. 5). Also, a significant decrease in MET expression was observed in the celecoxib-treated group (P < 0.01). No significant differences in the immunohistochemically determined levels of apoptosis (cleaved caspase-3) or proliferation (Ki-67) were detected. Nor were any differences observed in the quantity of endothelial cells as displayed by the expression of CD31. However, the VEGF expression localized in the cytoplasm of tumor cells was significantly decreased in the treated patients versus the nontreated patients (P = 0.02).
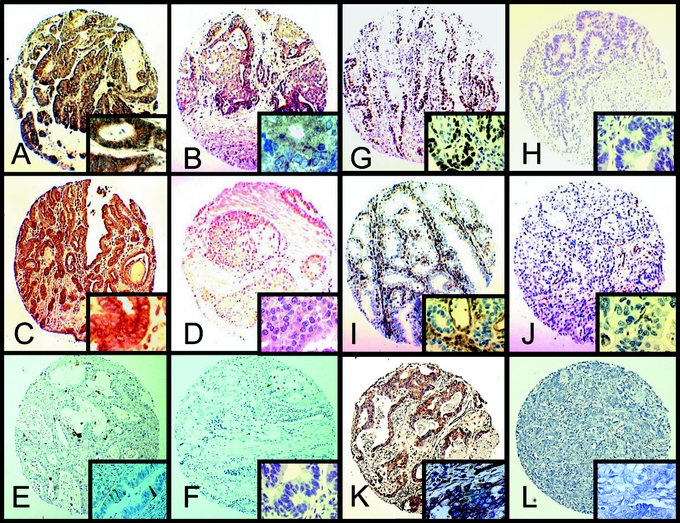
FIGURE 4. Examples of immunohistochemical analysis of resection specimens of patients plotted in the tissue microarray. A, High COX-2 expression. B, Low COX-2 expression. C, High MET expression. D, Low MET expression. E, High cleaved caspase-3 staining. F, Low cleaved caspase-3 staining. G, High Ki-67 expression. H, Low Ki-67 expression. I, High CD31 expression. J, Low CD31 expression. K, High VEGF expression. L, Low VEGF expression (original magnification ×10). The squares at the lower right corner represent the same tissue at original magnification ×50.

FIGURE 5. Significant down-regulation of COX-2, MET, and VEGF protein levels in patients treated with celecoxib compared with a nontreated control group. Semi-quantitative immunohistochemical analysis of resection specimens. The COX-2, MET, and tumor-specific VEGF protein levels were significantly decreased (P < 0.01, P < 0.01, and P = 0.02, respectively) in the resection specimens of treated patients operated on for adenocarcinoma of the esophagus as compared with the nontreated control group. A significant increase in apoptosis as analyzed by cleaved caspase-3 staining or decrease in the quantity of endothelial cells (CD31 staining) was not observed. Proliferation as measured by Ki-67 staining tended to decrease, although not significantly (P = 0.14).
To validate the decline of MET observed in the semi-quantitative immunohistochemical analysis, the RNA levels of MET in the resection specimens of patients treated with celecoxib were compared with those of the control patients. The amount of MET, relative to the amount of the housekeeping gene TBP, was decreased in patients treated with celecoxib as compared with control patients, although this did not reach statistical significance (P = 0.16, Fig. 6).
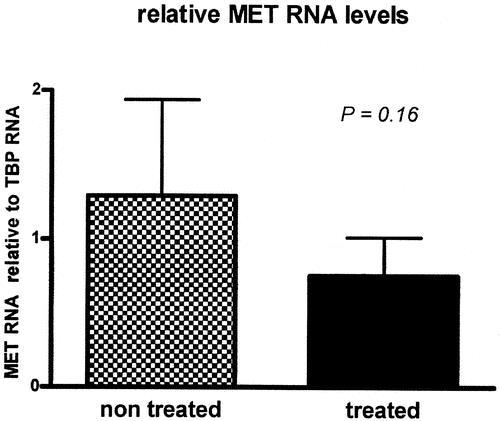
FIGURE 6. MET RNA tends to be decreased in patients treated with celecoxib compared with a nontreated control group. Quantification of MET RNA in frozen resection specimens from treated patients and nontreated patients. Total RNA was extracted from tissue regions containing at least 70% tumor tissue. After correction for the housekeeping gene TBP levels, the MET RNA tended to be decreased, although not significantly (P = 0.16).
Comparison Between Tumoral COX-2 and MET Expression Before and After Celecoxib Treatment
To confirm the effects of celecoxib on biochemical parameters observed both in vitro and in patients, the expression patterns of COX-2 and MET protein levels were also measured in tissue samples from the same patient taken before and after treatment. Endoscopic biopsies, taken before treatment, contained adequate tumor tissue in 9 of 11 patients. In these 9 patients, protein levels of COX-2 and MET were evaluated by semi-quantitative immunohistochemistry. Again, a significant decline in COX-2 and MET expression was detected (P = 0.002 and P < 0.001, respectively). In all but 1 patient, a decreased COX-2 expression was observed after celecoxib treatment. A decrease in MET expression was demonstrated in all 9 patients (Fig. 7).
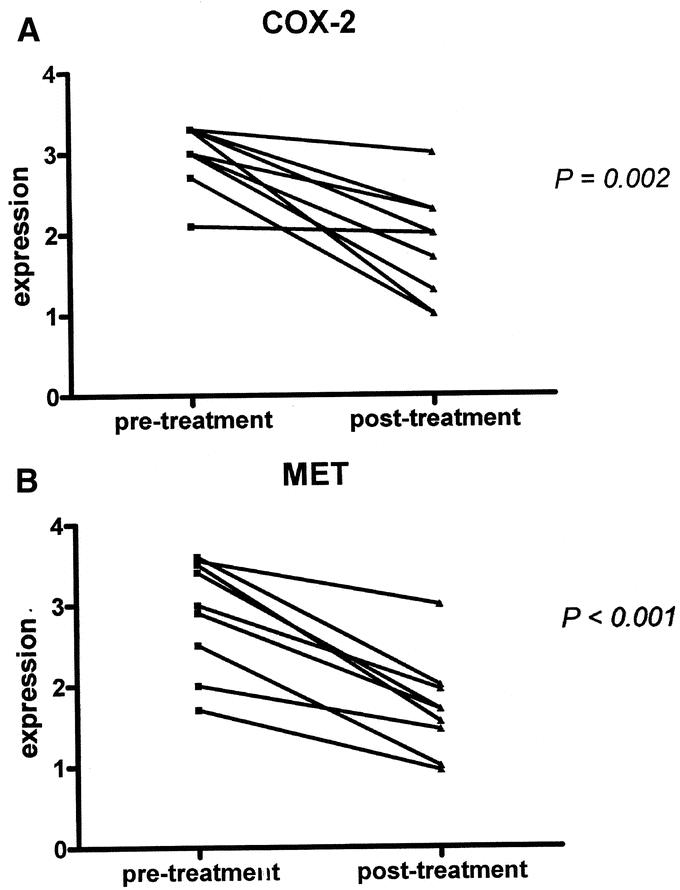
FIGURE 7. Celecoxib treatment results in down-regulation of COX-2 and MET protein expression in tissue after treatment compared with endoscopic biopsies derived before treatment. Semiquantitative immunohistochemical analysis in endoscopic biopsies before treatment compared with resection specimens after neoadjuvant celecoxib treatment. A, In all but 1 patient, decreased COX-2 protein levels were observed after treatment. B, A decline in MET protein levels was demonstrated in all patients.
DISCUSSION
This study demonstrates that the selective COX-2 inhibitor celecoxib down-regulates the expression of COX-2 and MET protein levels in esophageal adenocarcinoma cell lines in vitro. This is associated with significantly decreased cell viability and increased apoptosis. These findings led to the initiation of a clinical phase II study with selective COX-2 inhibition as neoadjuvant therapy for patients with an adenocarcinoma of the esophagus. The results of this study show that 4 weeks of celecoxib treatment significantly down-regulates the expression of COX-2 and MET in vivo when comparing the same patients before and after therapy and when comparing patients with and without neoadjuvant celecoxib treatment. In addition, a significantly decreased expression of tumor-specific VEGF was observed in treated patients compared with nontreated patients. These results suggest that selective COX-2 inhibition in the (neo)adjuvant setting may significantly decrease the neoplastic aggressiveness of esophageal adenocarcinoma.
A reduction of proliferation and induction of apoptosis in esophageal cancer cell lines has previously been observed for other COX-2 inhibitors.12,32–34 However, the mechanisms explaining these phenomena as well as the significance of these findings in patients with esophageal adenocarcinoma have not been described before. This study shows that celecoxib is effective in reducing proliferation and inducing apoptosis in esophageal adenocarcinoma cell lines. More importantly, the present study shows both in vitro and in patients that celecoxib down-regulates COX-2 and MET protein expression, both prognostic markers for esophageal adenocarcinoma. This down-regulation was demonstrated in patients with esophageal adenocarcinoma treated with celecoxib in comparison with nontreated patients and in comparison of pretreatment and post-treatment levels in the treated patients.
The decrease of the COX-2 protein levels upon 4 weeks of celecoxib treatment in vivo is in agreement with the decrease of COX-2 protein levels and cell proliferation demonstrated in individuals with premalignant Barrett epithelium after 10 days of rofecoxib, also a selective COX-2 inhibitor.35 However, the present study demonstrates the effect of neoadjuvant COX-2 inhibition in patients with esophageal adenocarcinoma. This is relevant since we focused on (neo)adjuvant chemotherapy in cancer patients and not on chemoprevention in individuals with Barrett's lesions.
The observed decrease in MET signaling and decrease in MET expression are relevant findings that provide a possible explanation for the observed anticarcinogenic potential of celecoxib. Activation of MET is one of the main mechanisms involved in dissemination, and MET expression is a negative prognostic factor for survival of patients with esophageal adenocarcinoma (preliminary data), as has also been shown for other types of adenocarcinoma.22,24 The results show that the rapid effects (after 30 minutes) of celecoxib include inhibition of MET phosphorylation together with a decreased phosphorylation of GAB 1 and ERK. These proteins are important kinases downstream of MET, which regulate the transcription of both COX-2 and MET (positive feedback). Therefore, these rapid effects partly explain the observed down-regulation of COX-2 and MET.25,36
The observed decrease in VEGF expression in the tumor cells is in agreement with the decrease in COX-2 and MET. It has previously been shown that VEGF transcription is enhanced by MET signaling. Therefore, inhibition of MET by celecoxib can at least partly explain the observed decrease of VEGF expression.37
A discrepancy between tumoral VEGF and CD31, a marker for endothelial cells, was observed. Since the production of tumoral VEGF stimulates the formation of new capillaries, the inhibition of VEGF by celecoxib might finally result in a decrease of the microvessel density within the tumor. However, it is questionable if the 4-week celecoxib treatment period is sufficient to observe a decline in CD31 expression.
Although no definitive conclusions can be drawn about the exact signal transduction pathways, the significant concomitant decrease in MET phosphorylation and downstream target phosphorylation of GAB 1 and ERK, together with a decrease in COX-2, MET, and VEGF expression, suggests that MET is indeed an important target of selective COX-2 inhibition. This is in agreement with earlier the observations that prostaglandins enhance the signal transduction mediated by MET and other growth factors, resulting in increased oncogene transcription.25,26 It remains unclear, however, whether the effect of celecoxib is explained by inhibition of prostaglandin E2 production or by (additional) other direct COX-2 independent mechanisms. In vivo evidence of MET as a target of selective COX-2 inhibition was found in a rat model for esophageal ulcers. In that study, it was shown that celecoxib delayed wound healing by down-regulation of the HGF/MET signaling pathway.38 The observed decrease of MET by 4-week celecoxib administration in vivo in this study provides a rationale to analyze its potential to inhibit cancer progression and dissemination.
It is conceivable that COX-2 inhibition might eradicate undetectable micrometastases, which are frequently present at the time of surgery with curative intent, and might thus create a postoperative survival advantage. Although a significant decrease in COX-2, MET, and VEGF was observed, no significant decrease in proliferation or increase in apoptosis was demonstrated. The present study may have lacked the power to clearly show a decreased proliferation and/or increased apoptosis in vivo. However, it is important to emphasize that this is a phase II trial designed to gain insight in the biologic mechanisms underlying selective COX-2 inhibition in vivo by comparing pretreatment and post-treatment histology. No significant changes in TNM stage were observed due to the celecoxib treatment. It should be noticed, however, that the pretreatment endosonographic staging cannot be compared objectively to the post-treatment pathologic staging because of the limited accuracy of the endosonography. Moreover, the number of patients is too small to draw conclusions regarding clinical outcome. Whether selective COX-2 inhibition will truly improve patient survival has to be demonstrated in a randomized controlled trial with long-term survival data.
So far, most oncologic studies with NSAIDs and selective COX-2 inhibitors have predominantly focused on their potential chemopreventive effect. Recently, it has been shown that the long-term usage of the selective COX-2 inhibitors rofecoxib and valdecoxib is accompanied by an increased risk of serious cardiovascular events.39 This has led pharmaceutical companies to withdraw these drugs from the market. For at least 2 reasons, this observation hardly affects the presently suggested application of celecoxib in patients with esophageal adenocarcinoma. First, it should be underlined that the acceptance of toxicity is unacceptable in chemopreventive strategies for unaffected individuals, which is in sharp contrast to the chemotherapeutic setting in affected cancer patients. Second, the short-term administration as (neo)adjuvant therapy during several weeks is not likely to cause cardiovascular side effects, as opposed to the long-term chemopreventive application during many years. For safety reasons, patients in this study were excluded when at risk for cardiovascular events as determined by (family) history.
CONCLUSION
This study demonstrates for the first time both in vitro and in patients that neoadjuvant celecoxib administration down-regulates COX-2, MET, and VEGF in esophageal adenocarcinoma. Since COX-2 and MET are both negative prognostic factors and important oncogenes involved in cancer progression and dissemination, the role of selective COX-2 inhibition as a neoadjuvant treatment of esophageal adenocarcinoma deserves further exploration.
ACKNOWLEDGMENTS
The authors thank Pfizer (New York) for the provision of the celecoxib compound, which was used for in vitro experiments. The authors also wish to thank Marinke Westerterp for the assistance with the patients’ information and patients’ accrual and Chris van der Loos for his expertise and help with the immunohistochemistry for VEGF.
Discussions
Dr. Lerut: I want to congratulate you and your group on this very elegant study on what is becoming, I think, a very hot topic, given our frustration with the results of the classic chemoradiotherapy as an induction modality. In this study, you clearly show that the neoadjuvant COX-2 inhibition down-regulates important oncogenic pathways. I would like to thank you for sending me the full manuscript well ahead so that I could study this well in advance.
I have a couple of remarks and questions.
You state in your discussion part that it is unlikely or less likely that the results have been substantially influenced by biopsy sampling, due to the heterogeneity of the tumors. How sure are you of this statement? In my mind, that would mean that you take your biopsies in the pre- and post-treatment endoscopy at exactly the same place. If not, I would be a bit doubtful of your statements. I would like your comment on this part.
Secondly, it is not clear to me whether your TNM classification is the clinical pretreatment one or, what I suppose, the pathologic staging. If this is so, if you did restaging after your neoadjuvant treatment, just as we do with the classic induction or chemo and chemoradiotherapy, did you see a “response” in some of your patients on your restaging? And in the same thinking line, I would like you to speculate whether this increased apoptosis and decreased proliferation and angiogenesis as you state would result in a visible response, for instance, on PET scan. Because that is, I think, a relevant clinical issue here.
Finally, you choose a treatment period of 4 weeks, on an arbitrary basis. Perhaps you could speculate whether you would expect a benefit of prolonging the treatment, let us say, for 6 or 8 weeks.
Dr. Tuynman: Thank you, Prof. Lerut, for your very relevant phrases, kind remarks, and good questions.
Your first question was: How can we be sure that biopsies before treatment are comparable with biopsies from the resection material? We evaluated this possible sampling error by first comparing endoscopic biopsies with resection material from untreated patients. We observed no significant alteration in the protein expression levels of COX-2 and MET. Although COX-2 tended to be lower in the resection, this was not significant. Therefore, we think that we can conclude that a sampling error is not a confounder in this study.
Your second question was about the pathologic staging: We staged the patients clinically before treatment, classified as an uTNM staging. Results are available demonstrating the staging after treatment, but because of the relatively small number of patients, we did not include this in the manuscript. We do not have the power to show clinical response. We also have survival data of the 15 treated patients versus the 11 nontreated patients, but we would not like to speculate on the clinical outcome.
The treatment period of 4 weeks was chosen because we did not want to interfere with the normal procedure. Our center is a tertiary referral center in which a strict schedule is applied. After the diagnostic biopsies, the patients received celecoxib therapy upon inclusion. During the treatment, patients were further evaluated to assess the curability. The waiting time between the first biopsies and the resection is 4 weeks. This period was the main reason to choose the treatment period. Perhaps had the patients been treated longer, we might have observed significant differences in proliferation or apoptosis. However, so far, these markers have not been shown to be of clinical significance. It has only been shown for COX-2 and MET that these are important markers for survival in esophageal adenocarcinoma. Apoptosis, for example, in the tumor is not a predictor of prognosis because tumors with a high apoptotic rate can also be aggressive. Moreover, apoptotic tumor cells are removed after 1 or 2 days by immune cells. Therefore, we think that the observed decrease in COX-2 and MET is important. Whether this can result in a decrease in proliferation rate and/or increased apoptosis, we were not able to show.
Dr. Eggermont: We want to congratulate you on what I think is an excellent example of how translational research should be conducted. Translation research in this type of setting is often associated with new adjuvant therapies and sequential biopsies, and it is a shortcut to get mechanistic insights in questions that otherwise may take much longer.
There is one point that I would like to ask you and where I think you may underestimate the potential and the antiangiogenic effect of the COX-2 inhibitor. It may berelated to 2 aspects of your study: on the one hand that the exposure to the agent might be too short and secondly that the evaluation that you have done by the CD31 marker may not be the ideal endpoint to evaluate this. Since you would expect this phenomenon to be independent of tumor type, I would suggest that you might consider doing some additional stainings for VEGF2 and VEGF3 receptor expression, to get a better insight in the antiangiogenic and antilymphangiogenic effects. If these are down-regulated, it may not have translated yet in the total drop measured and picked up by your CD31 staining, but it would have actually indicated then that the antiangiogenic effects will be more pronounced at a later point in time, and in that case it might further enhance the therapeutic insight of this agent, also for these types of tumors.
Dr. Tuynman: The discussion about exposure time to celecoxib: this can indeed be of significance, but in this study we did not want to interfere with the normal diagnostic and therapeutical timeframe in our patient group. Regarding your second remark concerning the stainings with CD31, I agree. The CD31 is a marker for endothelial cells. Indeed, the vessel forms a relatively late process, but this score has been validated very well by many others. We agree with your idea to investigate other mechanisms, like VEGF expression, although the staining for VEGF seems to be difficult. At the moment, we are also investigating immunohistochemistry using specific phosphoprotein antibodies, which might give more insight in the direct mechanisms of COX-2 inhibition. Your suggestion is indeed a good idea.
Dr. Lundell: An absolutely fascinating trial, which really opens up new approaches to the management of the disease. First, I have a slightly depressing comment, because we have just been through the phase of running a trial comparing COX-2 inhibition after radical gastric surgery with no additional treatment, where the authorities prevented us from continuing with the COX-2, due to the side effect profile. Hopefully, the Dutch authority is not that strict and rigid as the Swedish apparently is. This might be a difficult process, so prepare yourself well when you start to argue for this because a similar trial has to be conducted.
Now I will come to the actual question that is based on some sort of lateral thinking, relating the idea of using COX-2 inhibition as adjuvant or neoadjuvant therapy in GI cancer. This is based on the experiences in, for example, bronchial carcinoma and in prostatic carcinoma, in which situations data have indicated that the greatest potential for COX-2 inhibition is to act as an adjuvant to radiotherapy. In fact, COX-2 inhibitors may act as radiosensitizers. My question to you is whether you already at this stage have considered a similar approach in the next phase of evaluation the use of COX-2 inhibition in the preoperative treatment of those patients. The bottom line is that chemo or radiochemotherapy is considered by many institutions to be standard preoperative therapy already today.
Dr. Tuynman: Regarding the side effects, it is disappointing that your Swedish trial has stopped because, compared with chemotherapy for cancer patients, the side effects of COX-2 inhibition are much smaller. In addition, these are only present after 18 months, although these effects remain relevant. I completely agree with the fact that COX-2 inhibition as a single agent is interesting, but the combination with radiotherapy is even more promising. We hope to investigate this in the near future. At the moment, we are participating in a large trial in colorectal cancer, evaluating COX-2 inhibition in a randomized setting when combined with the standard adjuvant chemotherapy.
Footnotes
For the purpose of this study, the pure compound celecoxib used in vitro was kindly provided by Pfizer (New York).
Reprints: Jurriaan B. Tuynman, MD, G2-132, Academic Medical Center, Meibergdreef 9, 1105 AZ The Netherlands. E-mail: J.B.Tuynman@amc.uva.nl.
REFERENCES
- 1.Ruol A, Zaninotto G, Costantini M, et al. Barrett's esophagus: management of high-grade dysplasia and cancer. J Surg Res. 2004;117:44–51. [DOI] [PubMed] [Google Scholar]
- 2.Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349:2241–2252. [DOI] [PubMed] [Google Scholar]
- 3.Daly JM, Fry WA, Little AG, et al. Esophageal cancer: results of an American College of Surgeons Patient Care Evaluation Study. J Am Coll Surg. 2000;190:562–572. [DOI] [PubMed] [Google Scholar]
- 4.Geh JI, Crellin AM, Glynne-Jones R. Preoperative (neoadjuvant) chemoradiotherapy in oesophageal cancer. Br J Surg. 2001;88:338–356. [DOI] [PubMed] [Google Scholar]
- 5.Hulscher JB, van Sandick JW, de Boer AG, et al. Extended transthoracic resection compared with limited transhiatal resection for adenocarcinoma of the esophagus. N Engl J Med. 2002;347:1662–1669. [DOI] [PubMed] [Google Scholar]
- 6.Thun MJ. NSAIDs and esophageal cancer: ready for trials but not yet broad clinical application. Gastroenterology. 2003;124:246–248. [DOI] [PubMed] [Google Scholar]
- 7.Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. [DOI] [PubMed] [Google Scholar]
- 8.Tuynman JB, Peppelenbosch MP, Richel DJ. COX-2 inhibition as a tool to treat and prevent colorectal cancer. Crit Rev Oncol Hematol. 2004;52:81–101. [DOI] [PubMed] [Google Scholar]
- 9.Morris CD, Armstrong GR, Bigley G, et al. Cyclooxygenase-2 expression in the Barrett's metaplasia-dysplasia-adenocarcinoma sequence. Am J Gastroenterol. 2001;96:990–996. [DOI] [PubMed] [Google Scholar]
- 10.Buskens CJ, van Rees BP, Sivula A, et al. Prognostic significance of elevated cyclooxygenase 2 expression in patients with adenocarcinoma of the esophagus. Gastroenterology. 2002;122:1800–1807. [DOI] [PubMed] [Google Scholar]
- 11.Oyama K, Fujimura T, Ninomiya I, et al. A COX-2 inhibitor prevents esophageal inflammation-metaplasia-adenocarcinoma sequence in rats. Carcinogenesis. 2004;26:565–570. [DOI] [PubMed] [Google Scholar]
- 12.Vona-Davis L, Riggs DR, Jackson BJ, et al. Antiproliferative and apoptotic effects of rofecoxib on esophageal cancer in vitro. J Surg Res. 2004;119:143–148. [DOI] [PubMed] [Google Scholar]
- 13.Keller JJ, Offerhaus GJ, Polak M, et al. Rectal epithelial apoptosis in familial adenomatous polyposis patients treated with sulindac. Gut. 1999;45:822–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sengupta S, Sellers LA, Cindrova T, et al. Cyclooxygenase-2-selective nonsteroidal anti-inflammatory drugs inhibit hepatocyte growth factor/scatter factor-induced angiogenesis. Cancer Res. 2003;63:8351–8359. [PubMed] [Google Scholar]
- 15.Michieli P, Mazzone M, Basilico C, et al. Targeting the tumor and its microenvironment by a dual-function decoy Met receptor. Cancer Cell. 2004;6:61–73. [DOI] [PubMed] [Google Scholar]
- 16.Shinomiya N, Gao CF, Xie Q, et al. RNA interference reveals that ligand-independent met activity is required for tumor cell signaling and survival. Cancer Res. 2004;64:7962–7970. [DOI] [PubMed] [Google Scholar]
- 17.Jiang WG, Martin TA, Parr C, et al. Hepatocyte growth factor, its receptor, and their potential value in cancer therapies. Crit Rev Oncol Hematol. 2005;53:35–69. [DOI] [PubMed] [Google Scholar]
- 18.Hu YC, Lam KY, Law S, et al. Profiling of differentially expressed cancer-related genes in esophageal squamous cell carcinoma (ESCC) using human cancer cDNA arrays: overexpression of oncogene MET correlates with tumor differentiation in ESCC. Clin Cancer Res. 2001;7:3519–3525. [PubMed] [Google Scholar]
- 19.Saeki H, Oda S, Kawaguchi H, et al. Concurrent overexpression of Ets-1 and c-Met correlates with a phenotype of high cellular motility in human esophageal cancer. Int J Cancer. 2002;98:8–13. [DOI] [PubMed] [Google Scholar]
- 20.Lengyel E, Prechtel D, Resau JH, et al. C-Met overexpression in node-positive breast cancer identifies patients with poor clinical outcome independent of Her2/neu. Int J Cancer. 2005;113:678–682. [DOI] [PubMed] [Google Scholar]
- 21.Zhang YW, Graveel C, Shinomiya N, et al. Met decoys: will cancer take the bait? Cancer Cell. 2004;6:5–6. [DOI] [PubMed] [Google Scholar]
- 22.Dong G, Lee TL, Yeh NT, et al. Metastatic squamous cell carcinoma cells that overexpress c-Met exhibit enhanced angiogenesis factor expression, scattering and metastasis in response to hepatocyte growth factor. Oncogene. 2004;23:6199–6208. [DOI] [PubMed] [Google Scholar]
- 23.Resnick MB, Routhier J, Konkin T, et al. Epidermal growth factor receptor, c-MET, beta-catenin, and p53 expression as prognostic indicators in stage II colon cancer: a tissue microarray study. Clin Cancer Res. 2004;10:3069–3075. [DOI] [PubMed] [Google Scholar]
- 24.Birchmeier C, Birchmeier W, Gherardi E, et al. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–925. [DOI] [PubMed] [Google Scholar]
- 25.Boccaccio C, Sabatino G, Medico E, et al. The MET oncogene drives a genetic programme linking cancer to haemostasis. Nature. 2005;434:396–400. [DOI] [PubMed] [Google Scholar]
- 26.Pai R, Nakamura T, Moon WS, et al. Prostaglandins promote colon cancer cell invasion: signaling by cross-talk between two distinct growth factor receptors. FASEB J. 2003;17:1640–1647. [DOI] [PubMed] [Google Scholar]
- 27.Abiru S, Nakao K, Ichikawa T, et al. Aspirin and NS-398 inhibit hepatocyte growth factor-induced invasiveness of human hepatoma cells. Hepatology. 2002;35:1117–1124. [DOI] [PubMed] [Google Scholar]
- 28.Rockett JC, Larkin K, Darnton SJ, et al. Five newly established oesophageal carcinoma cell lines: phenotypic and immunological characterization. Br J Cancer. 1997;75:258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. [DOI] [PubMed] [Google Scholar]
- 30.Ramakers C, Ruijter JM, Deprez RH, et al. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett. 2003;339:62–66. [DOI] [PubMed] [Google Scholar]
- 31.Westerterp M, Westreenen EHL, Reitsma JB, et al. Computed tomography, endosonography and FDG-positron emission tomography to assess neoadjuvant therapy response in esophageal cancer: a systematic review. Radiology. In press.
- 32.Cheong E, Ivory K, Doleman J, et al. Synthetic and naturally occurring COX-2 inhibitors suppress proliferation in a human oesophageal adenocarcinoma cell line (OE33) by inducing apoptosis and cell cycle arrest. Carcinogenesis. 2004;25:1945–1952. [DOI] [PubMed] [Google Scholar]
- 33.Souza RF, Shewmake K, Beer DG, et al. Selective inhibition of cyclooxygenase-2 suppresses growth and induces apoptosis in human esophageal adenocarcinoma cells. Cancer Res. 2000;60:5767–5772. [PubMed] [Google Scholar]
- 34.Yu HG, Huang JA, Yang YN, et al. The effects of acetylsalicylic acid on proliferation, apoptosis, and invasion of cyclooxygenase-2 negative colon cancer cells. Eur J Clin Invest. 2002;32:838–846. [DOI] [PubMed] [Google Scholar]
- 35.Kaur BS, Khamnehei N, Iravani M, et al. Rofecoxib inhibits cyclooxygenase 2 expression and activity and reduces cell proliferation in Barrett's esophagus. Gastroenterology. 2002;123:60–67. [DOI] [PubMed] [Google Scholar]
- 36.Trusolino L, Comoglio PM. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nat Rev Cancer. 2002;2:289–300. [DOI] [PubMed] [Google Scholar]
- 37.Dong G, Chen Z, Li ZY, et al. Hepatocyte growth factor/scatter factor-induced activation of MEK and PI3K signal pathways contributes to expression of proangiogenic cytokines interleukin-8 and vascular endothelial growth factor in head and neck squamous cell carcinoma. Cancer Res. 2001;61:5911–5918. [PubMed] [Google Scholar]
- 38.Baatar D, Jones MK, Pai R, et al. Selective cyclooxygenase-2 blocker delays healing of esophageal ulcers in rats and inhibits ulceration-triggered c-Met/hepatocyte growth factor receptor induction and extracellular signal-regulated kinase 2 activation. Am J Pathol. 2002;160:963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Juni P, Nartey L, Reichenbach S, et al. Risk of cardiovascular events and rofecoxib: cumulative meta-analysis. Lancet. 2004;364:2021–2029. [DOI] [PubMed] [Google Scholar]