

Abstract
Objective:
To compare the in vitro osteogenic differentiation and in vivo ectopic bone forming capacity of human bone marrow stromal cells (BMSCs) and jaw periosteal cells (JPCs), and to identify molecular predictors of their osteogenic capacity.
Summary Background Data:
JPC could be an appealing alternative to BMSC for the engineering of cell-based osteoinductive grafts because of the relatively easy access to tissue with minimal morbidity. However, the extent of osteogenic capacity of JPC has not yet been established or compared with that of BMSC.
Methods:
BMSCs and JPCs from the same donors (N = 9), expanded for 2 passages, were cultured for 3 weeks in osteogenic medium either in monolayers (Model I) or within 3-dimensional porous ceramic scaffolds, following embedding in fibrin gel (Model II). Cell-fibrin-ceramic constructs were also implanted ectopically in nude mice for 8 weeks (Model III). Cell differentiation in vitro was assessed biochemically and by real-time RT-PCR. Bone formation in vivo was quantified by computerized histomorphometry.
Results:
JPCs had lower alkaline phosphatase activity, deposited smaller amounts of calcium (Model I), and expressed lower mRNA levels of bone sialoprotein, osteopontin, and osterix (Models I and II) than BMSCs. JPCs produced ectopic bone tissue at lower frequency and amounts (Model III) than BMSCs. Bone sialoprotein, osteopontin, and osterix mRNA levels by BMSCs or JPCs in Model II were markedly higher than in Model I and significantly more expressed by cells that generated bone tissue in Model III.
Conclusions:
Our data indicate that JPCs, although displaying features of osteogenic cells, would not be as reliable as BMSCs for cell-based bone tissue engineering, and suggest that expression of osteoblast-related markers in vitro could be used to predict whether cells would be osteoinductive in vivo.
Using 3 different model systems, we determined that human bone marrow stromal cells have a higher in vitro differentiation and in vivo bone forming capacity than jaw periosteal cells from the same individuals. Our findings are discussed in the light of cell-based bone tissue engineering approaches.
Congenital or acquired bony defects in the head and neck, as well as of long bones, can be treated using a variety of bone substitutes. If defects are small and surrounded by normal bone, materials from inorganic (ceramics), organic (polymers), or allogenic origin are typically used.1 Larger defects require either free autologous bone grafts or microvascular bone flaps harvested from different sites2. The most challenging problems associated with the latter techniques are the limited availability of free autologous bone grafts and the considerable donor site morbidity introduced by vascularized bone flaps.3 A promising approach to overcome these limitations is the use of osteogenic progenitor cells in combination with osteoconductive materials to engineer osteoinductive grafts.
It is well known that bone marrow stroma contains progenitor cells with osteogenic potential, generally referred to as mesenchymal stem cells or bone marrow stromal cells (BMSCs).4–6 Human BMSCs have been demonstrated to differentiate toward the osteoblastic lineage in vitro,7,8 to form bone tissue upon ectopic implantation in nude mice,9 and to support the repair of large segmental defects in a few clinical cases.10 However, BMSCs are found in human marrow aspirates at low frequency (approximately 0.01% of the total mononucleated cells), and their expansion by serial passaging, which is consequently required, is associated with a dramatic reduction in the differentiation capacity.11,12 In addition, a wide variability has been reported in the osteogenic ability of BMSCs from different donors13,14 or harvested using different procedures.15 Moreover, the aspiration of bone marrow has to be carried out in a sterile environment and is generally associated with considerable pain.
It is also well established that the periosteum is a source of cells with osteogenic potential, which is maintained even in elderly individuals.16 Human periosteal cells loaded into porous polymeric scaffolds have been shown to differentiate toward the osteoblastic lineage in vitro17 and to form traces of bone tissue ectopically.18 Harvest of the jaw periosteum as a source of osteogenic cells is particularly appealing because of the simplicity of the procedure and the minimal morbidity induced. Recently, Schimming and Schmelzeisen reported the clinical use of jaw periosteal cells (JPCs) in combination with a polymer fleece in the context of the maxillary sinus elevation procedure.19 Although radiologically detectable bone formation was found in 18 of 27 patients, it was not possible to prove that the newly deposited bone tissue was formed by the implanted periosteal cells and not by cells surrounding the defect. Thus, it remains unclear to which extent human JPCs are capable to induce bone formation in vivo and how this capacity is related to that of BMSCs.
In this paper, we first aimed at comparing the in vitro differentiation and in vivo bone formation of BMSCs and JPCs from the same individuals, using 2-dimensional (2D) and 3-dimensional (3D) culture models and an ectopic assay in nude mice. Considering the need of reproducibility or at least of predictability in the cell osteoinductive ability for a standardized clinical use, we then investigated whether the patterns of expression of osteoblast-specific markers in 2D or 3D cultures could be predictive of the cell capacity to form bone tissue in the ectopic model.
MATERIALS AND METHODS
Human Material
Bone marrow aspirates and biopsies of jaw periosteum were obtained during maxillofacial routine intervention, involving both exposure of the periosteum of the jaw and harvesting of autologous bone from the iliac crest. Samples were obtained from 9 healthy donors (5 males and 4 females, 21–80 years of age) in accordance with the local ethical committee, following informed consent. Marrow aspirates (approximately 20 mL) were taken using a bone marrow biopsy needle inserted through the cortical bone and immediately transferred into plastic tubes containing 12,000 IU heparin. During the same surgical procedures, biopsies of jaw periosteum (approximately 10 × 5 mm) were harvested after raising a mucoperiosteal flap and immediately transferred into plastic tubes containing alpha Modified Eagle Medium (α-MEM). Care was taken not to damage the cambium layer of the periosteum, which is localized on the inner aspect of the flap, and to dissect the soft tissue from the harvested periosteum, using surgical magnification loupes.
Cell Isolation and Expansion
BMSCs
After diluting the marrow aspirates with phosphate-buffered saline (PBS) at a ratio of 1:3, nucleated cells were isolated using a density gradient solution (Histopaque, Sigma Chemical, Buchs, Switzerland). Complete medium (CM) consisted of α-MEM with 10% fetal bovine serum (GIBCO-BRL Life Technologies, Basel, Switzerland), 4.5 mg/mL d-glucose, 0.1 mmol/L nonessential amino acids, 1 mmol/L sodium pyruvate, 100 mmol/L HEPES buffer, 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.29 mg/mL l-glutamine. Nucleated cells (30–50 million from 20 mL aspirates) were plated at a density of 100,000 cells/cm2 in CM supplemented with 5 ng/mL fibroblast growth factor-2 (R&D Systems, Wiesbaden, Germany) and 10 nmol/L dexamethasone (Sigma) (FGF/Dex), previously shown to enhance osteogenic commitment of BMSCs,20 and cultured in a humidified 37°C/5% CO2 incubator. The initial number of BMSCs available from each donor, calculated considering an approximate yield of 0.01% BMSCs out of marrow-derived nucleated cells, was in the range of a few thousand cells.
JPCs
After rinsing the periosteum thoroughly with PBS containing 100 U/mL penicillin and 100 μg/mL streptomycin, the biopsies were minced in small pieces and digested in 0.5% type II collagenase (Worthington Biochemical Corporation, Lake Wood, NJ) for 4 hours at 37°C. The isolated cells were centrifuged, resuspended in CM supplemented with FGF/Dex, plated in a 56 cm2 dish, and cultured in a humidified 37°C/5% CO2 incubator. The initial number of JPCs available from each donor could not always be reliably assessed because of the limited size of the biopsy but was in the range of a few thousand cells.
Differentiation Assays
Upon reaching subconfluence, BMSCs and JPCs were detached using 0.05% trypsin/0.53 mmol/L EDTA (GIBCO-BRL) and replated at a density of 3000 cells/cm2. After getting again subconfluent, cells were used in the following 3 models.
Model I: Osteogenic Differentiation in 2D Cultures (9 Donors)
BMSCs and JPCs were seeded at a density of 3000 cells/cm2 into 6-well dishes and cultured in osteogenic medium (OM), consisting of CM supplemented with 10 nmol/L Dex, 0.1 mmol/L l-ascorbic acid-2-phosphate, and 10 mmol/L β-glycerophosphate.21 Cultures were harvested after 1, 7, 14, and 21 days and processed for biochemical and mRNA analysis as described below.
Model II: Osteogenic Differentiation in 3D Cultures (5 Donors)
BMSCs and JPCs were resuspended in fibrin gel (Baxter, Austria) and statically loaded into porous tricalcium phosphate blocks (cubes of approximately 6 mm side; Vitoss, Orthovita, Belgium) or bovine bone-derived granulates (250–500 μm particles, for a total dry weight of 35 μg; Bio-Oss, Geistlich, Switzerland). The use of the fibrin gel was introduced to more reproducibly load cells in the ceramic scaffolds. Briefly, 1 million of expanded cells were resuspended in 30 μL of the fibrinogen component (diluted to 20 mg/mL), quickly mixed with 30 μL of the thrombin component (diluted to 6 IU/mL), and immediately loaded into and around the scaffolds, where fibrin polymerized during incubation for 15 minutes in a humidified 37°C/5% CO2 incubator. The cell-fibrin-scaffold constructs were harvested after 10 and 20 days of culture in OM and processed for mRNA analysis as described below.
Model III: Ectopic Implantation in Nude Mice (5 Donors)
BMSCs and JPCs from 5 donors (21, 28, 40, 63, and 63 years of age) were loaded using fibrin gel into Vitoss or Bio-Oss scaffolds as described for Model II and implanted subcutaneously in nude mice (CD-1 nude/nude, Charles River). After 8 weeks, mice were killed and explants were assessed histologically and by computerized histomorphometry as described below.
Analytical Methods
Proliferation Rate During Expansion
The number of doublings of BMSCs and JPCs during the second passage of expansion was determined as the logarithm in base 2 of the fold increase in the number of cells during expansion. The proliferation rate of BMSCs and JPCs was defined as the number of doublings during the second passage of expansion divided by the time required for expansion and was expressed as doublings/day. Cell counts were performed using 0.4% trypan blue solution (Sigma-Aldrich, St. Louis, MO).
Biochemical Analyses During Differentiation
DNA Assays and Alkaline Phosphatase (AP) Activity
Cultures were rinsed with PBS and the cell layers scraped in 0.01% sodium dodecyl sulfate. DNA amounts were measured in triplicate aliquots of the same sample using the CyQuant cell proliferation assay kit (Molecular Probes, Leiden, NL) according to the manufacturer's instructions. AP activity was measured in triplicate aliquots as the rate of conversion of p-nitrophenyl phosphate using Sigma kit 104. AP activity was expressed as nanomoles of p-nitrophenol/min/μg DNA.
Calcium Assays
Total calcium was measured using Sigma Kit 587 after rinsing and extracting the cell layer in 0.5 N HCl, as described elsewhere.8 The amount of deposited calcium was expressed as μg/dish.
Total RNA Extraction and cDNA Synthesis
RNA was extracted from cell layers using Trizol (Life Technologies, Basel, CH) and the standard single-step acid-phenol guanidinium method. Cell layers were first sonicated for 1 minute while in Trizol. RNA was treated with DNAse using the DNA-free Kit (Ambion). cDNA was generated from 16 μL of RNA by using 500 μg/mL random hexamers (Catalys) and 1 μL of 50 U/mL Stratascript reverse transcription (Stratagene) in the presence of dNTPs.
Real-Time Quantitative RT-PCR
PCR reactions were performed and monitored using the ABI Prism 7700 Sequence Detection System (Perkin-Elmer/Applied Biosystems, Rotkreuz, Switzerland). The PCR master mix was based on AmpliTaq Gold DNA polymerase (Applied Biosystems). In the same reaction, cDNA samples (5 μL for a total volume of 25 μL per reaction) were analyzed both for the gene of interest and the reference gene (18-S rRNA) using a multiplex approach (Perkin Elmer User Bulletin N. 2), as previously described.7 The probe for 18-S rRNA was fluorescently labeled with VIC (Perkin Elmer Corp.) and 6-carboxy-tetramethyl-rhodamin (TAMRA), whereas probes for genes of interest were labeled with 6-carboxy-fluorescein (FAM) and TAMRA. Primers and probes used for human 18-S, bone sialoprotein-I (BSP), osteopontin (OP), and core binding factor 1 (cbfa-1) were as previously described7 and those for Osterix (osx) were commercially available (Sp7 transcription factor, Applied Biosystems, Foster City, CA). Expression levels of each gene of interest were calculated by normalizing the quantified RNA amount to the 18s rRNA and by further dividing the resulting value by that previously obtained in human osteoblast cultures from cortical bone (average of 5 donors) using an identical procedure (2ΔΔCt formula, Perkin Elmer User Bulletin No. 2).7 Each sample was assessed at least in duplicate for each gene of interest.
Bone Quantification
Explants were fixed in 4% buffered formalin for 24 hours, decalcified with 0.5 mol/L ethylenediaminetetraacetic acid, pH 8, for 7 to 10 days, paraffin embedded, and cross-sectioned (5 μm thick) at 3 different levels. Sections were stained with hematoxylin/eosin and Masson/Trichrome and assessed both qualitatively for the appearance of bone tissue and quantitatively by computerized bone histomorphometry as previously described.22 Briefly, for each cross section, stained by hematoxylin/eosin, 3 or 4 images (sufficient to cover the construct cross sections) were acquired and used to measure the area covered with bone tissue and the total area available for tissue ingrowth (total implant area minus the undegraded scaffold area) by computerized image analysis (Scion Image, Scion Corp., Frederick, MD). The amount of bone tissue was then calculated as a percentage of the total implant area (% area) or of the total space available for tissue ingrowth (% tissue).
Statistical Analysis
Each assessment was performed on 2 or 3 independent cultures or constructs for each donor, cell source, and experimental group. All values are presented as mean ± SD. Differences between experimental groups were statistically assessed using Wilcoxon's nonparametric tests for paired samples, with P < 0.01 considered to be statistically significant. Correlation between in vitro mRNA expression levels of osteoblast-related genes of different primary cultures was assessed by Pearson's correlation tests on the logarithm of the values, with P < 0.01 considered to be statistically significant.
RESULTS
Expansion of BMSC and JPC
BMSCs and JPCs were expanded for 2 passages and a total number of up to 12 doublings. During the second passage of the expansion phase, JPCs proliferated at a significantly higher rate (0.70 ± 0.18 doublings/day) than BMSCs (0.50 ± 0.14 doublings/days). After a total time of 14 to 17 days, it was possible to obtain at least 10 million cells of either cell type, sufficient for use in the 3 different models.
Model I: Osteogenic Differentiation in 2D Cultures
Biochemical Analyses
During osteogenic differentiation, DNA amounts in JPC cultures progressively increased with time and were always significantly higher than in BMSC cultures, likely indicating that JPCs continued to proliferate faster than BMSCs (Fig. 1A). AP activity by BMSCs, normalized to the DNA amount, reached a typical peak between 7 and 10 days of culture,7 followed by a slight decrease (Fig. 1B). AP activity by JPC was significantly lower at each time point and slowly increased with time, without displaying peak levels and reaching stable values after 14 days. Accumulation of calcium in the extracellular matrix was, for both cell types, detectable only after the first week in OM, and then steadily increased. BMSCs deposited significantly more calcium than JPCs during the first 2 weeks of culture in OM (Fig. 1C).
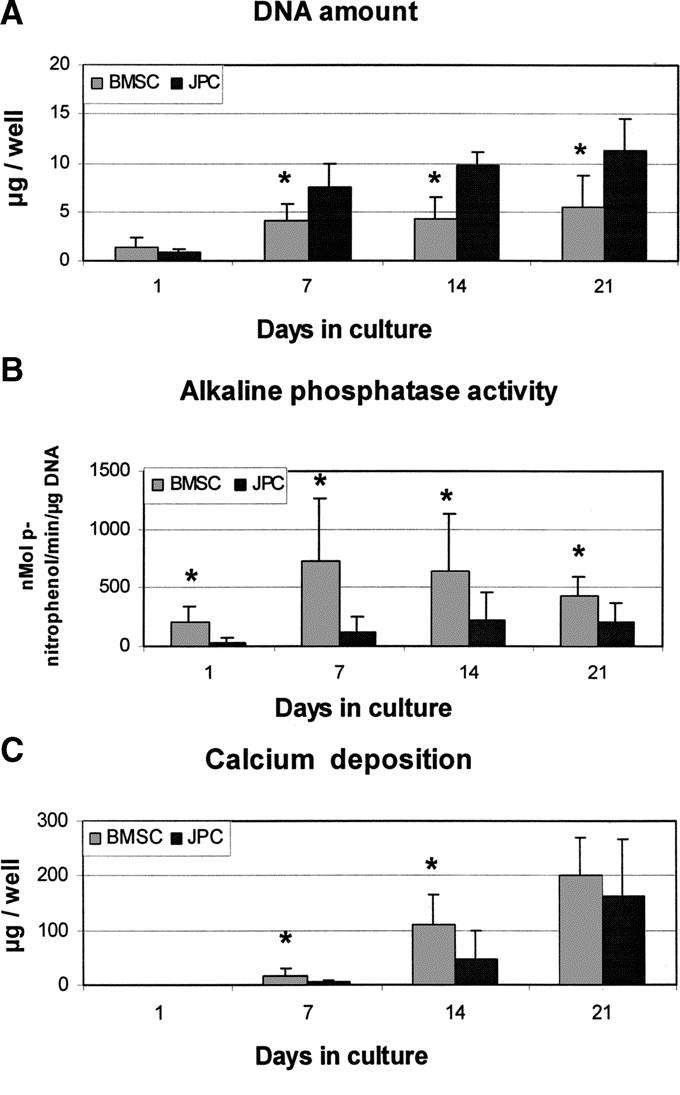
FIGURE 1. Biochemical analyses during cell differentiation in Model I. Human bone marrow stromal cells (BMSC) and jaw periosteal cells (JPC) were cultured in Model I (2D cultures) and assessed at different time points for the amount of DNA (A), alkaline phosphatase activity (B), and deposition of calcium (C). *Statistically significant difference (P < 0.01) between BMSC and JPC cultures.
Real-Time RT-PCR
During culture in OM, BMSCs and JPCs were assessed for the mRNA expression of genes encoding 1) extracellular matrix proteins reported to reflect the extent of cell differentiation into an osteoblast-like phenotype (ie, BSP and OP)7 and 2) transcription factors reported to be required for bone formation (ie, cbfa-1 and osx).23 Expression of BSP and OP by BMSC constantly increased with time, reaching levels around 10-fold higher than those measured in human osteoblasts (Fig. 2A, B). BSP mRNA expression by JPCs remained negligible throughout the duration of the culture. OP mRNA expression levels in JPC cultures became detectable only after 14 days and remained significantly lower than those measured in BMSC cultures. The mRNA expression levels of cbfa-1 remained constant with time and were similar for the 2 cell types (Fig. 2C). The mRNA expression of osx in BMSC cultures increased with time and approached levels measured in human osteoblasts after 21 days, whereas it remained negligible in JPC cultures (Fig. 2D).
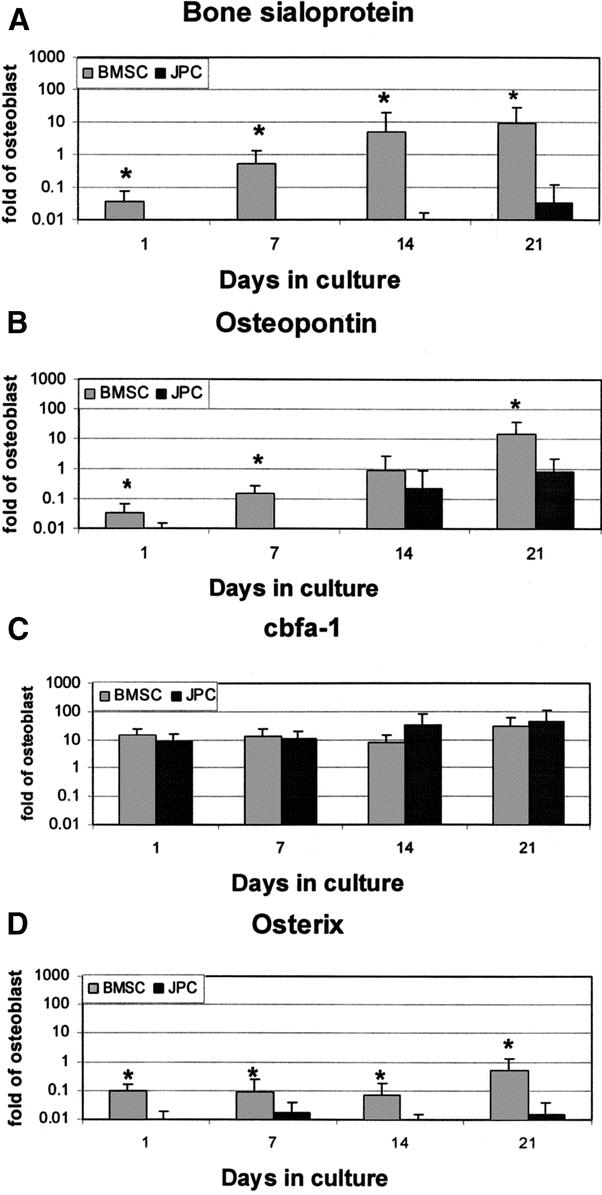
FIGURE 2. Expression of osteoblast-related genes in Model I. Human bone marrow stromal cells (BMSC) and jaw periosteal cells (JPC) were cultured in Model I (2D cultures) and assessed at different time points for the mRNA expression of bone sialoprotein-I (A), osteopontin (B), cbfa-1 (C), and osterix (D). Values, normalized to 18 s, were expressed as fold difference from those previously measured in human osteoblasts. *Statistically significant differences (P < 0.01) between BMSC and JPC cultures.
Model II: Osteogenic Differentiation in 3D Cultures
Real-Time RT-PCR
Cell-fibrin-scaffold constructs were assessed for the mRNA expression of osteoblast-related genes after 10 or 20 days of culture in OM. In general, results obtained using Bio-Oss or Vitoss were very similar (Fig. 3). The mRNA expression levels of BSP and OP by BMSCs were markedly higher (respectively 15- and 13-fold after 20 days) than those measured in Model I (Fig. 3A, B). The same trend was observed for BSP and OP levels expressed by JPCs, which however remained significantly lower than those expressed by BMSC. Similar to our observations in Model I, both cell types expressed cbfa-1 at comparable levels at all assessed time points (Fig. 3C). The mRNA expression levels of osx by BMSC and JPC were markedly higher in Model II than in Model I (respectively, 8- and 16-fold after 20 days) but remained significantly lower in JPCs than in BMSCs (Fig. 3D).

FIGURE 3. Expression of osteoblast-related genes in Model II. Human bone marrow stromal cells (BMSC) and jaw periosteal cells (JPC) were cultured in Model II (3D cultures) using Bio-Oss or Vitoss scaffolds and assessed at 10 or 20 days for the mRNA expression of bone sialoprotein-I (A), osteopontin (B), cbfa-1 (C), and osterix (D). Values, normalized to 18 s, were expressed as fold difference from those previously measured in human osteoblasts. *Statistically significant differences (P < 0.01) between BMSC and JPC cultures.
Model III: Ectopic Implantation in Nude Mice
Frequency of Bone Formation
For each cell type, a total of 49 cell-fibrin-scaffold constructs (24 based on Bio-Oss, 25 based on Vitoss) were implanted subcutaneously into nude mice to assess the intrinsic cell osteoinductive capacity. Bone formation could be detected in 29 of 49 (59%) constructs loaded with BMSCs and only in 9 of 49 (18%) constructs loaded with JPCs. BMSCs from all 5 donors tested could generate bone tissue in at least one construct, whereas JPCs from the 2 youngest donors only (21 and 28 years of age) were capable to form bone tissue. Considering both cell sources and all donors together, the frequency of bone formation was almost identical using the 2 scaffolds (37.5% for Bio-Oss and 38% for Vitoss). As expected, scaffolds not loaded with cells did not show any bone formation.
Characteristics of Bone Formation
Morphologic characteristics of the bone tissue formed ectopically did not appear to depend upon the use of BMSCs or JPCs. Instead, rather distinct patterns of bone formation were observed using the 2 different scaffolds. Bovine bone-derived ceramic (Bio-Oss), composed of 100% hydroxyapatite, is characterized by a very slow degradation rate, such that negligible resorption occurred during the 8 weeks of implantation. Thus, the newly formed bone tissue was deposited, starting from the “priming surface” of the ceramic,20 only within the scaffold pores (Fig. 4A). Instead, synthetically produced ceramic (Vitoss, Orthovita), consisting of pure tricalcium phosphate, was almost completely degraded after 8 weeks in vivo, such that bone or fibrous tissues were covering the whole construct cross sections (Fig. 4B). Most of the bone tissue generated within Bio-Oss scaffolds was already remodeled (red color using the Masson-Trichrome stain) and lamellar (Fig. 4C). Instead, using Vitoss scaffolds, in addition to lamellar bone tissue found in the original ceramic voids, large amounts of freshly deposited and immature bone tissue (blue color using the Masson-Trichrome stain) were found in the areas where ceramic was being resorbed (Fig. 4D).
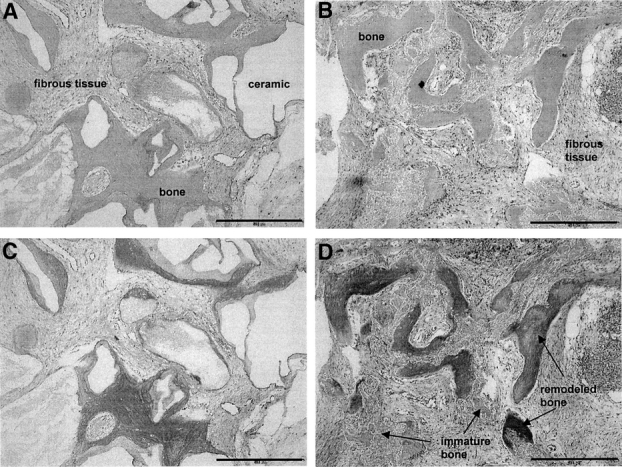
FIGURE 4. Histology of bone tissue formation in Model III. Representative cross sections of constructs generated in Model III by bone marrow stromal cells using Bio-Oss (A, C) or Vitoss (B, D) scaffolds, stained by hematoxylin/eosin (A, B) or Masson/Trichrom (C, D). Empty spaces correspond to decalcified Bio-Oss. Vitoss was almost completely resorbed at this stage. In sections stained by Masson/Trichrome, a red color indicates remodeled and lamellar bone, whereas a blue color indicates immature and freshly deposited bone. Bone formation by jaw periosteal cells displayed similar histologic patterns. Bar = 0.5 mm.
Quantification of Bone Formation
Considering all 5 donors, the average amounts of bone tissue formed by BMSCs were significantly higher than those generated by JPCs (Fig. 5). However, taking into account only the 2 youngest donors, whose JPCs were osteoinductive, the average percentages of bone tissue were similar in BMSCs and JPCs (data not shown), reaching up to 9.2% of the total tissue formed.
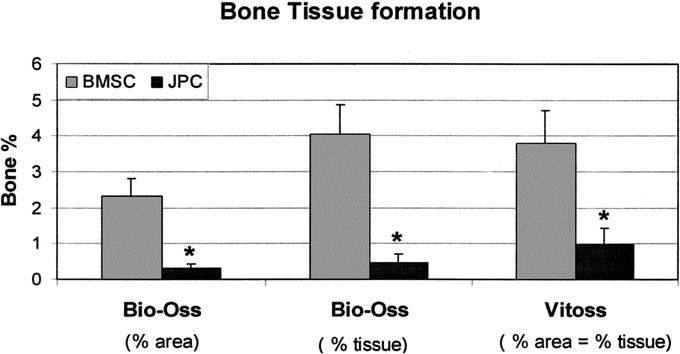
FIGURE 5. Quantification of bone tissue formation in Model III. Bone tissue formation by human bone marrow stromal cells (BMSC) or jaw periosteal cells (JPC) generated in Model III using Bio-Oss or Vitoss was quantified by computerized histomorphometry. Bone tissue amount is expressed as a percentage of the total implant area (% area) or of the total space available for tissue ingrowth (% tissue). The 2 percentages were equivalent when Vitoss was used because of the almost complete resorption of the material. *Statistically significant differences (P < 0.01) between BMSC and JPC constructs.
In Bio-Oss scaffolds, the area occupied by undegraded ceramic was about 40% of the total; therefore, only 60% of the total space was accessible for tissue and bone ingrowth. For this reason, the percentages of bone per total implant area (% area) and of bone per total space available for tissue ingrowth (% tissue) were distinct (Fig. 5). Instead, in Vitoss scaffolds, essentially all space was accessible for tissue and bone ingrowth; therefore, the amounts of bone expressed as % area or % tissue were equivalent. Consistent with the qualitative histologic assessment, for both BMSCs and JPCs, the amount of bone was similar using Bio-Oss or Vitoss scaffolds if calculated as % tissue, whereas it was higher using Vitoss scaffolds if calculated as % area.
In Vitro Gene Expression and In Vivo Bone Formation
BMSC or JPC primary cultures from different donors expressed markedly different mRNA levels of osteoblast-related genes in vitro and displayed differential bone forming capacity in vivo. We then tried to establish possible correlations between the expression levels of the different genes in vitro and to identify whether specific gene expression patterns in vitro could be predictive of bone formation in vivo. Significant positive correlations were found between the mRNA expression of BSP and OP in both Model I and Model II, and between the expression of BSP and osx or OP and osx, in Model II only (Table 1). The expression of cbfa-1 did not correlate with any of the other assessed genes. Figure 6 graphically displays the significant correlations and highlights that cell populations expressing high levels of the correlating osteoblast-related genes were able to form bone tissue in vivo. Statistically significant differences (P < 0.02) were found between bone-forming and non-bone-forming cultures in the mRNA expression of BSP and OP but not osx in Model I, and of BSP, OP, and osx in Model II. In particular, BMSCs or JPCs expressing in Model II mRNA levels of BSP, OP, and osx equal or greater than those measured in human osteoblasts were reproducibly forming bone tissue in Model III.
TABLE 1. Correlation Between the In Vitro mRNA Expression Levels of BSP, OP, and osx
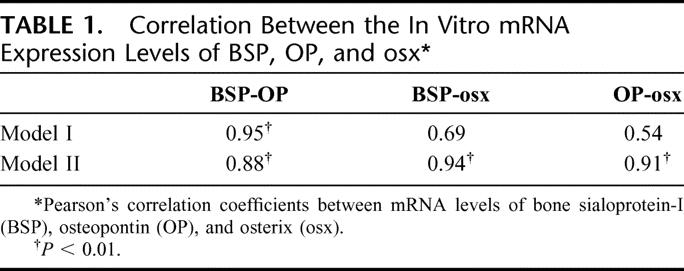

FIGURE 6. Correlation between the expression of osteoblast-related genes. Graphical representation of the statistically significant correlations (Table 1) between the mRNA expression levels of bone sialoprotein-I (BSP), osteopontin (OP), and osterix (osx) by bone marrow stromal cells or jaw periosteal cells from each donor. Correlation between BSP-OP in Model I (A) and Model II (B), or between BSP-osx (C) and OP-osx (D) in Model II. The graphs also indicate whether single cell cultures formed or not bone tissue in Model III.
DISCUSSION
In this study, using 3 different model systems, we compared the osteogenic potential of human BMSC and JPC from the same donors, in an attempt to determine whether JPCs could be an alternative cell source to BMSCs in cell-based approaches to bone repair. We demonstrated that, compared with BMSCs, JPCs exhibited a delayed and overall reduced capacity to differentiate toward the osteoblastic lineage in vitro, which is reflected by a lower frequency and amount of bone tissue formed ectopically following implantation in nude mice. Moreover, we found that cultures forming bone tissue in vivo expressed higher mRNA levels of BSP, OP, and osx in a 3D system than non-bone-forming cultures, indicating that specific in vitro assays may be used to predict osteoinductivity of cells from different donors or sites.
BMSC and JPC osteogenic differentiation in vitro was assessed using a typical 2D culture system7,9,16 (Model I), as well as a 3D culture model including fibrin gel and ceramic-based porous scaffolds24 (Model II). Results from the 2 experimental systems consistently indicate that 1) BMSC differentiation was more efficient than that of JPC, as assessed by the higher AP activity, initial calcium accumulation, and mRNA expression of BSP, OP, and osx; and 2) the expression of these 3 genes was markedly higher if BMSCs or JPCs were cultured in Model II than in Model I, reaching the highest levels of expression already after 10 days. The enhanced and accelerated cell differentiation in Model II could be explained by the cell-cell interactions supported by a 3D environment,25 the presence of a ceramic substrate,26 the postulated osteoinductive property of fibrin,27 or combinations of these variables that remain to be further elucidated. The in vitro mRNA expression levels of BSP, OP, and osx by BMSCs and JPCs from different donors were highly variable, with differences covering up to 10 orders of magnitude. Strong positive correlations were identified between the expression of BSP and OP in Model I, and between BSP, OP, and osx in Model II. The fact that BSP and OP expression correlated with osx expression only in Model II may be explained by the overall negligible levels of osx expression detected in Model I. This reinforces the concept that a 3D culture system is required to induce osteogenic cells to fully display an osteogenic phenotype in vitro and therefore to model the physiology of early phases of bone tissue development.28 In contrast to osx, mRNA levels of cbfa-1, previously shown to be also required for osteoblastic differentiation,29,30 were similarly expressed by BMSCs or JPCs cultured in Model I or Model II. This finding is confirmatory of the thesis that cbfa-1 is a basic property of functionally different BMSCs31 and that osx should be considered as a more specific transcription factor for osteoblast differentiation, acting downstream of cbfa-1.23
BMSC and JPC bone formation capacity was assessed by ectopic implantation of cell-fibrin-scaffold constructs in nude mice9 (Model III). Despite the limitation of being representative of bone formation in unloaded conditions,32 the model addresses the intrinsic osteoinductive capacity of the graft, without interference from the process of osteoconduction in orthotopic models, and provides the unique opportunity to investigate the behavior of human osteogenic cells, biologically different from those of other animal species. Results from Model III demonstrated that BMSCs were overall capable of forming bone tissue at a higher frequency and total amount as compared with JPCs, consistent with data obtained using Model I and Model II. However, JPCs from the 2 youngest donors generated bone tissue qualitatively and quantitatively similar to that formed by BMSCs. This finding could be explained by a reduction of JPC osteogenic capacity with donor age, in contrast with previously published in vitro studies,16,33 or by an increased contamination of JPC cultures with fibroblasts starting from specimens of older individuals. Indeed, the thickness of jaw periosteum tends to be reduced with age and dissection of the fibrous layer, which consists mostly of fibroblasts, from the cambium layer, which is known to be the source of mesenchymal stem cells,34 may result less accurate.
BMSC and JPC osteogenic capacity was assessed in Model II and Model III using 2 types of ceramic-based scaffolds (Bio-Oss, Vitoss). Despite the several differences between the materials in terms of composition and architecture, the extent of cell differentiation in Model II and the frequency and percentage of bone formation within the available space (% tissue) in Model III were virtually identical. However, the relatively faster resorption rate of Vitoss was associated with an increased percentage of bone per total cross-sectional area (% area), with relevant amounts of immature bone growing in the spaces generated by the scaffold degradation. Taken together, these findings suggest that the different ceramic compositions and architectures do not overtly affect the biology of BMSCs or JPCs, whereas the different scaffold resorption rates regulate the pattern of bone tissue formation. These considerations may be useful in the selection of a scaffold for specific bone repair clinical applications: materials based on a certain amount of hydroxyapatite, like Bio-Oss, may be preferred where mechanical stability is required and the rate of new bone tissue formation is expected to be relatively slow; instead, pure tricalcium phosphate-based scaffolds, like Vitoss, may have preferential indications at sites where initial mechanical stability is not necessary and efficient bone tissue formation is expected.
The identification of in vitro predictive markers for in vivo bone formation is of particular clinical interest, since it may facilitate the selection of individuals being qualified for a cell-based bone repair approach, or indicate the need for an alternative strategy (eg, based on the supplementary use of bone growth factors). Kuznetsov et al35 reported that neither proliferation rate nor AP activity are positively correlated with the osteogenic potential of BMSCs, whereas Mendes et al36 concluded that procollagen I or OP expression in vitro, assessed by flow cytometry, is not of predictive value to the in vivo performance of BMSCs in nude mice. We found that high expression levels of specific genes (BSP and OP in Model I, or BSP, OP, and osx in Model II) were associated with cell osteoinductive capacity in Model III. In particular, cells expressing mRNA levels of BSP, OP, and osx equal or greater than osteoblasts in Model II were reproducibly forming bone tissue in vivo. These findings need to be confirmed using a larger numbers of donors but provide a proof of principle that osteoinductivity of cells from different donors or sites could be predicted using specific in vitro assays and that such prediction could be more consistent if a 3D model, as compared with a 2D environment, is used.
CONCLUSION
Our data indicate that under our experimental conditions JPCs would not be as reliable as BMSCs for cell-based bone tissue engineering approaches, and suggest that in vitro tests may help to determine case-by-case whether the expanded cell populations would be osteoinductive in vivo.
ACKNOWLEDGMENTS
The authors thank Dr. D. Wendt and Dr. U. Gueller for helpful advice with the statistical analyses, Dr. A. Goessl (Baxter, Austria) for the generous supply of fibrin gel and critical revision of the manuscript, Dr. J. F. Clemence and Dr. C. Goerlach (Geistlich, Switzerland) for the kind provision of Bio-Oss, and Verena Winkelmann (University of Bern) for the histologic processing of the specimens.
Discussions
Dr. Friess: I must congratulate Dr. Jacquiery for his excellent presentation and the interesting study he and his coworkers have carried out. In this study, you have compared different sources of mesenchymal progenitor cells for their use in bone tissue engineering. Apart from the crucial in vivo bone-forming potential of the cells, you have also identified some predictive markers for in vivo bone formation. This study will most certainly stimulate further research based on the clinical relevance of your findings.
I take this opportunity to ask you some questions that arose in my mind during your presentation and while reading your manuscript.
The first is the clinical applications you and your coworkers envisage: Realistically, how far away do you think we are from the clinical use that potentially arises from your findings? The second question relates to the cell numbers generated for your experiments. How were you able to generate so many cells out of a small periosteal flap to do all of your experiments in all the samples for all those different models described? My next question: why have you chosen, for example, BSP, as a marker for osteoblastic differentiation, since it has been shown in the literature that there are much better markers such as osteocalcin? Why have you not measured this parameter and instead measured the parameters you depicted today?
Dr. Jaquiery: Thank you for asking these relevant questions. With regard to the first question, this study identifies that mesenchymal cells from bone marrow (BMSC) have a superior intrinsic osteogenic capacity than those from jaw periosteum (JPC). In order to introduce BMSC-based engineered tissues in the routine clinical praxis, several challenges have to be addressed, especially related to the prediction and possibly reduction of donor-to-donor variability, and to the efficient vascularization of the implanted grafts. In addition, generation of constructs has to be streamlined, possibly reducing manufacturing costs and improving logistics and standardization. We are currently working on the use of perfusion bioreactor systems to simplify the generation of osteoinductive grafts: starting from a few ml of bone marrow aspirate, osteoprogenitor cells are seeded and expanded in porous scaffolds within a closed bioreactor system; after about 3 weeks of automated culture, the osteoinductive grafts would be ready to be transferred to the patient, bypassing repeated passaging and maintaining a higher osteogenic capacity of the cells (see Braccini et al., Stem Cells 2005).
The initial number of BMSC and JPC available from each donor was in the range of a few thousand cells. During culture for 14 to 17 days in FGF-2 and Dexamethasone, which have been shown to commit the cells to the osteogenic lineage while increasing proliferation rate, cells were expanded for a total number of 12 doublings. This made it possible to obtain at least 10 million cells from each cell preparation, sufficient for use in the 3 different Models.
Regarding osteogenic markers, I agree that osteocalcin is important for osteoblastic differentiation. However, as previously demonstrated (see Frank et al., J Cell Biochem 2002), mRNA levels of bone sialoprotein and osteopontin appear more specifically associated to osteogenic cell differentiation than osteocalcin. That is the reason why we have chosen bone sialoprotein as characteristic marker for osteogenic differentiation. Indeed in our study, mRNA expression levels of osteocalcin by different cultures were highly variable and were not significantly related to in vivo bone formation as shown for bone sialoprotein. In addition to these markers, we have also identified that a transcription factor named osterix could be of relevance to monitor in vitro osteogenic differentiation and predict in vivo bone formation.
Dr. Thorlacius: My comment also relates to the selection of markers. When it comes to bone formation in the normal growth and in fracture healing, cartilage requires vascularization for turning into bone. Vascularization is also necessary in this ectopic bone formation model. The potential of the cells to induce bone will be directly related to its proangiogeneic potential, and there are data showing, for example, that in the endochondral bone formation of collagen 10 may be important for vascularization. I think it would be useful to look for proangiogeneic factors as markers for bone formation in this model.
Dr. Jaquiery: I agree that vascularization is a relevant problem in bone tissue engineering, in particular if large constructs have to be generated. Looking at the size of our scaffolds, which is in the range of about 3 to 5 mm, vascularization was not a critical issue. Your suggestion to investigate expression of proangiogenic factors is an interesting approach to possibly extend our results to a larger graft scale. Other options currently investigated include the co-culture of endothelial cells, the controlled release of proangiogenic factors, or the initial prefabrication of flaps around the graft, prior to orthotopic transplantation.
Footnotes
Reprints: Claude Jaquiéry, MD, DMD, Clinic of Reconstructive Surgery, University Hospital Basel, Spitalstrasse 21, 4031 Basel, Switzerland. E-mail: cjaquiery@uhbs.ch.
REFERENCES
- 1.Schnurer SM, Gopp U, Kuhn KD, et al. Bone substitutes. Orthopade. 2003;32:2–10. [DOI] [PubMed] [Google Scholar]
- 2.Jaquiery C, Rohner D, Kunz C, et al. Reconstruction of maxillary and mandibular defects using prefabricated microvascular fibular grafts and osseointegrated dental implants: a prospective study. Clin Oral Implants Res. 2004;15:598–606. [DOI] [PubMed] [Google Scholar]
- 3.Hartman EH, Spauwen PH, Jansen JA. Donor-site complications in vascularized bone flap surgery. J Invest Surg. 2002;15:185–197. [DOI] [PubMed] [Google Scholar]
- 4.Bianco P, Gehron RP. Marrow stromal stem cells. J Clin Invest. 2000;105:1663–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Owen M. Marrow stromal stem cells. J Cell Sci Suppl. 1988;10:63–76. [DOI] [PubMed] [Google Scholar]
- 6.Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. [DOI] [PubMed] [Google Scholar]
- 7.Frank O, Heim M, Jakob M, et al. Real-time quantitative RT-PCR analysis of human bone marrow stromal cells during osteogenic differentiation in vitro. J Cell Biochem. 2002;85:737–746. [DOI] [PubMed] [Google Scholar]
- 8.Jaiswal N, Haynesworth SE, Caplan AI, et al. Osteogenic differentiation of purified, culture-expanded human mesenchymal stem cells in vitro. J Cell Biochem. 1997;64:295–312. [PubMed] [Google Scholar]
- 9.Haynesworth SE, Goshima J, Goldberg VM, et al. Characterization of cells with osteogenic potential from human marrow. Bone. 1992;13:81–88. [DOI] [PubMed] [Google Scholar]
- 10.Quarto R, Mastrogiacomo M, Cancedda R, et al. Repair of large bone defects with the use of autologous bone marrow stromal cells. N Engl J Med. 2001;344:385–386. [DOI] [PubMed] [Google Scholar]
- 11.Banfi A, Muraglia A, Dozin B, et al. Proliferation kinetics and differentiation potential of ex vivo expanded human bone marrow stromal cells: Implications for their use in cell therapy. Exp Hematol. 2000;28:707–715. [DOI] [PubMed] [Google Scholar]
- 12.Derubeis AR, Cancedda R. Bone marrow stromal cells (BMSCs) in bone engineering: limitations and recent advances. Ann Biomed Eng. 2004;32:160–165. [DOI] [PubMed] [Google Scholar]
- 13.Phinney DG, Kopen G, Righter W, et al. Donor variation in the growth properties and osteogenic potential of human marrow stromal cells. J Cell Biochem. 1999;75:424–436. [PubMed] [Google Scholar]
- 14.Mendes SC, Tibbe JM, Veenhof M, et al. Bone tissue-engineered implants using human bone marrow stromal cells: effect of culture conditions and donor age. Tissue Eng. 2002;8:911–920. [DOI] [PubMed] [Google Scholar]
- 15.Muschler GF, Boehm C, Easley K. Aspiration to obtain osteoblast progenitor cells from human bone marrow: the influence of aspiration volume. J Bone Joint Surg Am. 1997;79:1699–1709. [DOI] [PubMed] [Google Scholar]
- 16.Koshihara Y, Hirano M, Kawamura M, et al. Mineralization ability of cultured human osteoblast-like periosteal cells does not decline with aging. J Gerontol. 1991;46:B201–B206. [DOI] [PubMed] [Google Scholar]
- 17.Arnold U, Lindenhayn K, Perka C. In vitro-cultivation of human periosteum derived cells in bioresorbable polymer-TCP-composites. Biomaterials. 2002;23:2303–2310. [DOI] [PubMed] [Google Scholar]
- 18.Schantz JT, Hutmacher DW, Chim H, et al. Induction of ectopic bone formation by using human periosteal cells in combination with a novel scaffold technology. Cell Transplant. 2002;11:125–138. [PubMed] [Google Scholar]
- 19.Schimming R, Schmelzeisen R. Tissue-engineered bone for maxillary sinus augmentation. J Oral Maxillofac Surg. 2004;62:724–729. [DOI] [PubMed] [Google Scholar]
- 20.Martin I, Muraglia A, Campanile G, et al. Fibroblast growth factor-2 supports ex vivo expansion and maintenance of osteogenic precursors from human bone marrow. Endocrinology. 1997;138:4456–4462. [DOI] [PubMed] [Google Scholar]
- 21.Maniatopoulos C, Sodek J, Melcher AH. Bone formation in vitro by stromal cells obtained from bone marrow of young adult rats. Cell Tissue Res. 1988;254:317–330. [DOI] [PubMed] [Google Scholar]
- 22.Martin I, Mastrogiacomo M, De Leo G, et al. Fluorescence microscopy imaging of bone for automated histomorphometry. Tissue Eng. 2002;8:847–852. [DOI] [PubMed] [Google Scholar]
- 23.Nakashima K, Zhou X, Kunkel G, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. [DOI] [PubMed] [Google Scholar]
- 24.Le Guehennec L, Layrolle P, Daculsi G. A review of bioceramics and fibrin sealant. Eur Cell Mater. 2004;8:1–11. [DOI] [PubMed] [Google Scholar]
- 25.Kale S, Biermann S, Edwards C, et al. Three-dimensional cellular development is essential for ex vivo formation of human bone. Nat Biotechnol. 2000;18:954–958. [DOI] [PubMed] [Google Scholar]
- 26.Heng BC, Cao T, Stanton LW, et al. Strategies for directing the differentiation of stem cells into the osteogenic lineage in vitro. J Bone Miner Res. 2004;19:1379–1394. [DOI] [PubMed] [Google Scholar]
- 27.Abiraman S, Varma HK, Umashankar PR, et al. Fibrin glue as an osteoinductive protein in a mouse model. Biomaterials. 2002;23:3023–3031. [DOI] [PubMed] [Google Scholar]
- 28.Huang W, Carlsen B, Wulur I, et al. BMP-2 exerts differential effects on differentiation of rabbit bone marrow stromal cells grown in two-dimensional and three-dimensional systems and is required for in vitro bone formation in a PLGA scaffold. Exp Cell Res. 2004;299:325–334. [DOI] [PubMed] [Google Scholar]
- 29.Komori T, Yagi H, Nomura S, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. [DOI] [PubMed] [Google Scholar]
- 30.Ueta C, Iwamoto M, Kanatani N, et al. Skeletal malformations caused by overexpression of Cbfa1 or its dominant negative form in chondrocytes. J Cell Biol. 2001;153:87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Satomura K, Krebsbach P, Bianco P, et al. Osteogenic imprinting upstream of marrow stromal cell differentiation. J Cell Biochem. 2000;78:391–403. [PubMed] [Google Scholar]
- 32.Muraglia A, Martin I, Cancedda R, et al. A nude mouse model for human bone formation in unloaded conditions. Bone. 1998;22(suppl):131–134. [DOI] [PubMed] [Google Scholar]
- 33.Koshihara Y, Honda Y. Age-related increase in collagen production in cultured human osteoblast-like periosteal cells. Mech Ageing Dev. 1994;74:89–101. [DOI] [PubMed] [Google Scholar]
- 34.Marini RP, Stevens MM, Langer R, et al. Hydraulic elevation of the periosteum: a novel technique for periosteal harvest. J Invest Surg. 2004;17:229–233. [DOI] [PubMed] [Google Scholar]
- 35.Kuznetsov SA, Krebsbach PH, Satomura K, et al. Single-colony derived strains of human marrow stromal fibroblasts form bone after transplantation in vivo. J Bone Miner Res. 1997;12:1335–1347. [DOI] [PubMed] [Google Scholar]
- 36.Mendes SC, Tibbe JM, Veenhof M, et al. Relation between in vitro and in vivo osteogenic potential of cultured human bone marrow stromal cells. J Mater Sci Mater Med. 2004;15:1123–1128. [DOI] [PubMed] [Google Scholar]