

Abstract
Previous studies of the interactions of NO with human hemoglobin have implied the predominance of reaction channels that alternatively eliminate NO by converting it to nitrate, or tightly complex it on the α subunit ferrous hemes. Both channels could effectively quench NO bioactivity. More recent work has raised the idea that NO groups can efficiently transfer from the hemes to cysteine thiols within the β subunit (cysβ-93) to form bioactive nitrosothiols. The regulation of NO function, through its chemical position in the hemoglobin, is supported by response to oxygen and to redox agents that modulate the molecular and electronic structure of the protein. In this article, we focus on reactions in which Fe(III) hemes could provide the oxidative requirements of this NO-group transfer chemistry. We report a detailed investigation of the reductive nitrosylation of human met-Hb, in which we demonstrate the production of S-nitroso (SNO)-Hb through a heme-Fe(III)NO intermediate. The production of SNO-Hb is strongly favored (over nitrite) when NO is gradually introduced in limited total quantities; in this situation, moreover, heme nitrosylation occurs primarily within the β subunits of the hemoglobin tetramer. SNO-Hb can similarly be produced when Fe(II)NO hemes are subjected to mild oxidation. The reaction of deoxygenated hemoglobin with limited quantities of nitrite leads to the production of β subunit Fe(II)NO hemes, with SNO-Hb produced on subsequent oxygenation. The common theme of these reactions is the effective coupling of heme–iron and NO redox chemistries. Collectively, they establish a connectivity between hemes and thiols in Hb, through which NO is readily dislodged from storage on the heme to form bioactive SNO-Hb.
The transfer of NO groups within human hemoglobin from hemes to cys(β-93) thiols to form a bioactive nitrosothiol represents a novel intramolecular biochemistry that is both of fundamental interest and has considerable implications for understanding the physiological effects of NO in the regulation of vascular tension and blood flow. A requirement of this transfer, common to biological S-nitrosylation (1), is the redox activation of the NO group (2). In this article, we report the results of experiments that probe the idea that heme–iron valence change can support the oxidative requirements of NO-group transfer and thus efficiently lead to the production of S-nitroso (SNO)-Hb. As a model of the reaction between ferric hemes and NO, the reductive nitrosylation of human methemoglobin is examined in detail. Product distribution assays reveal that SNO-Hb is formed as a nitrosation product, which, moreover, is substantially favored over NO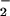 when NO is gradually introduced as a limiting reagent; furthermore, in this situation, heme nitrosylation occurs primarily within the β subunits of the Hb tetramer. A kinetic analysis unambiguously reveals the intermediacy of heme-Fe(III)NO in this reaction. To extend our observations to reactions that could mimic this chemistry but do not require an accumulation of the methemoglobin reactant, we additionally examined oxidation of Fe(II)NO in Hb as well as Hb reactions with nitrite. We report that SNO-Hb can similarly be produced when Fe(II)NO species are subjected to mild oxidation. Further, we show that on exposure to limited quantities of nitrite, oxy-Hb is unreactive, but deoxy-Hb leads to the production of β subunit Fe(II)NO hemes, with SNO-Hb produced on subsequent oxygenation. These reactions, which have the common characteristic of coupling of heme–iron valence change with NO chemistry, establish a connectivity between hemes and thiols in Hb, through which NO is readily dislodged from storage on the heme to form SNO-Hb, with selective processing in the β subunits.
when NO is gradually introduced as a limiting reagent; furthermore, in this situation, heme nitrosylation occurs primarily within the β subunits of the Hb tetramer. A kinetic analysis unambiguously reveals the intermediacy of heme-Fe(III)NO in this reaction. To extend our observations to reactions that could mimic this chemistry but do not require an accumulation of the methemoglobin reactant, we additionally examined oxidation of Fe(II)NO in Hb as well as Hb reactions with nitrite. We report that SNO-Hb can similarly be produced when Fe(II)NO species are subjected to mild oxidation. Further, we show that on exposure to limited quantities of nitrite, oxy-Hb is unreactive, but deoxy-Hb leads to the production of β subunit Fe(II)NO hemes, with SNO-Hb produced on subsequent oxygenation. These reactions, which have the common characteristic of coupling of heme–iron valence change with NO chemistry, establish a connectivity between hemes and thiols in Hb, through which NO is readily dislodged from storage on the heme to form SNO-Hb, with selective processing in the β subunits.
Materials and Methods
Solutions of human hemoglobin A0 (Apex Bioscience, Research Triangle Park, NC) were prepared in either 100 mM Hepes or PBS, pH 7.4, as described (3). Reductive nitrosylation reactions were conducted by slowly delivering aliquots, via gas-tight Hamilton syringes, to the methemoglobin solution at [NO]o/[heme] ratios varying nominally from 0.05 to 0.75 (where [NO]o is the initial concentration of the added NO). Solutions were mixed by vortexing immediately on the addition. In some experiments, a succession of additions was made, in the manner of a titration. The protein concentration was kept in excess of 75 μM in all experiments, to avoid substantial dissociation of Hb into dimers, and below 250 μM to maintain solution ideality. Reaction progress after each addition was monitored by UV/VIS spectroscopy. The spectra were analyzed by deconvolution of the contributions from met-, Fe(II)NO-, and Fe(III)NO-Hb, by methods similar to those described by us (3) and elaborated below. At selected time points, samples of the reacting mixture were taken for further product analysis: samples were either frozen by immersion in liquid nitrogen for characterization by EPR spectroscopy (4–7) or directly analyzed for nitrite and nitrosothiols by Greiss and Saville assays, following described methods (3). EPR spectra were recorded with a Varian E-109 X-band spectrometer that has been modified by the addition of a field-control unit obtained through the University of Denver (8). Samples were immersed in liquid nitrogen during the EPR measurements. The reactant/product analyses made before the completion of the reaction were used in the determination of the reaction kinetics. gepasi (9) and dynafit (10) were used in the kinetic analysis.
Other reactions and product analyses were carried out by analogous methods. Reactions with nitrite were conducted by mixing oxyhemoglobin solutions (≈4 mM in heme) with sodium nitrite at ≈100:1 heme/nitrite ratios. The solutions were allowed to stand for minutes to hours, then deoxygenated by sparging with argon. Samples were withdrawn just before or just after deoxygenation for product characterization. Hb(NO)4 oxidation was conducted in ≈2 mM solutions with an excess of potassium ferricyanide; the formation of products was followed by UV/VIS spectroscopy. Samples were cleansed of low molecular weight species by centrifugation through a G-25 column, then analyzed for products.
The procedure for deconvolution of UV/VIS spectra, described previously (3), was modified in the present study. We introduced, among the standards, the spectrum of authentic heme-Fe(III)NO Hb. To obtain this standard spectrum, the kinetic data obtained in reductive nitrosylation experiments were subject to principal component analysis (11), as described in detail elsewhere (12). The reference Fe(III)NO spectrum obtained by this procedure is similar to the spectrum reported by Alayash et al. (13) that is largely Hb[Fe(III)NO]4 (at pH 8) with apparent traces of unreacted met-hemes. The reference spectra used are exhibited in Fig. 1.
Figure 1.

Visible absorption spectra of species involved in the reductive nitrosylation reaction. Standard spectra, pH 7.4, of met-Hb (◊), Fe(II)NO-Hb (○), and Fe(III)NO-Hb (□). The methemoglobin and Fe(II) nitrosyl hemoglobin spectra are from authentic standards; the Fe(III) nitrosyl hemoglobin spectrum was determined by using factor analysis techniques and scaled as described in the text.
To improve the performance of the least-squares fitting routine used in the spectral analysis, all reference and experimental spectra were numerically differentiated; the least-squares fitting was done by using these derivative spectra. We also augmented the experimental and reference spectra to introduce constraints on the component concentrations. Application of constraints proved useful, for example, in proscribing nonphysical negative values for trace species.
EPR spectra of the samples generally included high- and low-spin methemoglobin components (14), as well as a feature assignable to Fe(II)NO. In conventional first-derivative EPR displays, the met-Hb signals have an effectively flat response in the region of the Fe(II)NO feature. The latter spectra were analyzed by using a deconvolution procedure (analogous to that used in the UV/VIS spectroscopy), involving two components, corresponding to NO occupying alternatively β or α subunits (with 6-fold coordination) (15–17). Hyperfine structure suggestive of penta-coordination of the α subunit Fe(II)NO hemes (15) was not observed in any EPR spectra.
Results
Reductive Nitrosylation Product Analysis.
Although the reductive nitrosylation of oxidized heme-proteins has been studied for >60 years (18–24), the reaction of NO with methemoglobin remains only partially understood. In the case of simple monomeric heme-proteins, reductive nitrosylation has been most simply summarized by the equation:
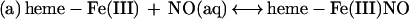 |
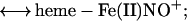 |
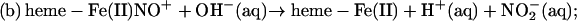 |
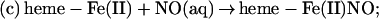 |
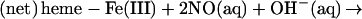 |
1 |
 |
It has been appreciated, however, that the situation with tetrameric hemoglobins, composed of distinct α and β subunits, is more complex (24, 25). Although subunit inequivalence has been reported in the uptake of NO (13, 24, 25), such effects in the latter steps of the reaction and their impact on the nature of distribution of the final reaction products have not been previously investigated. Moreover, this reaction has not been previously studied in situations where the NO is limiting, as in the biological situation.
By means of Saville assays, we determined that, in addition to the heme-Fe(II)NO and nitrite products of Eq. 1, a nitrosothiol derivative of Hb is among the species produced on exposure of methemoglobin to NO. Recent studies have clearly demonstrated that human Hb sustains S-nitrosylation at cys(β-93) of the β subunit (26–29). Collectively, these results implicate the formation of SNO-hemoglobin in the reductive nitrosylation of met-Hb. To verify that these three products account for all of the introduced and reacted NO, we compared the sum of their detected concentrations versus [NO]o (with all concentrations considered on a per-heme basis). These data are plotted in Fig. 2 Left. The linear variation with unit slope exhibited by the data demonstrate the material balance between the introduced NO and the Fe(II)NO, nitrite, plus nitrosothiol products.
Figure 2.

(Left) NO-group mass balance in the reductive nitrosylation of human met-Hb with NO. The total amount of the reaction products, heme-Fe(II)NO, nitrite, and S-nitrosothiol, is presented as a function of the amount of NO introduced. ○, Experimental determinations; solid line, the best linear fit to the experimental points (slope 1.06 ± 0.03; R = 0.990). (Right) Relative yields of ferrous heme nitrosyl products and the S-nitrosothiol plus nitrite products versus the total product yield. ○, Experimental determinations of ferrous heme nitrosyl; solid line, the best linear fit to these experimental points (slope 0.51 ± 0.01; R = 0.996). ●, Experimental determinations of S-nitrosothiol plus nitrite; dashed line, the best linear fit to these experimental points (slope 0.51 ± 0.02; R = 0.992). Amounts are tabulated relative to the amount of heme present in the reaction medium. Product analysis was carried out as described in the text.
The presence of a nitrosothiol product could be rationalized as reflecting a competition between the pathways involving formal NO+ transfer to, alternatively, hydroxide (reaction b) or protein thiol(ate) species:
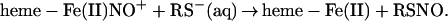 |
2 |
Either pathway would presumably lead to the concomitant production of an equivalent of heme-Fe(II), which is immediately converted to Fe(II)NO. To test this idea, we compared the total concentration of the products to: (i) the heme-Fe(II)NO concentration; and (ii) the sum of the nitrite and nitrosothiol concentrations, as shown in Fig. 2 Right. The correlations are essentially identical and demonstrate that protein nitrosothiol and nitrite are produced, in toto, on an equimolar basis with the nitrosyl ferrous-heme: Fe(II)NO and the sum of nitrite plus nitrosothiol each account for half of the reacted NO.
The factors that determine the distribution of products between nitrite and protein nitrosothiol remain to be more fully elucidated. We have observed that at low [NO]o/[heme] ratios (less than ≈0.1), protein nitrosothiol and ferrous nitrosyl heme account for essentially all of the products. The preference for nitrosothiol is a sharply decreasing function of the [NO]o/[heme] ratios: at ratios as small as 2, nitrite and protein nitrosothiol are produced in essentially equal amounts. Further analysis of the Fe(II)NO products by EPR spectroscopy reveal a striking and previously unrecognized preference for nitrosylation of β subunit hemes, as evidenced in the spectra shown in Fig. 3. We observed β vs. α subunit preferences ranging from ≈7:1 to ≈3:1 at [NO]o/[heme] ratios varying from 0.05 to 0.75, whereas at [NO]o/[heme] ratios ≥2, the (heme-Fe(II)NO)4 protein is obtained. The spectrum of this product, which of course exhibits no subunit preference, is also shown in Fig. 3. The fundamental trends in protein nitrosothiol vs. nitrite and β vs. α subunit preference are similar: strong preferences are exhibited only at low [NO]o/[heme] ratios; at ratios > ≈0.75, the relevant product ratios approach unity.
Figure 3.

EPR spectra of Fe(II)NO obtained by reductive nitrosylation of met-Hb with NO. EPR spectra were obtained at 9.29 GHz, with 10 mW incident power and field modulation of 10 G at 100 kHz. The external field was scanned over a range of 400 G in 2 min with a detection time constant of 0.128 sec. The experimental spectra shown were obtained with samples reacted with [NO]o/[heme] ratios of 0.2 (solid line) and 2 (dashed line). Reconstructions made as weighted combinations of α and β subunit spectra (dotted lines) are indistinguishable from the experimental spectra. The spectrum obtained with a [NO]o/[heme] ratio of 0.2 reflects a β vs. α subunit preference of 88 ± 8% (variance between methods was adapted from refs. 18–22). The spectrum obtained with a [NO]o/[heme] ratio of 2 reflects equal α and β subunit populations in the (Fe(II)NO)4 protein. EPR signals from residual met-Hb lie outside the field range included in the spectra presented here.
As a check on the possibility that the product distributions are influenced by “bolus” effects (30) associated with the addition of NO solutions, we conducted a series of experiments in which NO was derived from the decomposition of limited amounts of the nonoate DEA/NO (31), which was premixed into the Hb solution. In these experiments, reported in detail elsewhere (32), we also find a highly consistent preference for S-nitrosylation over nitrite formation and a β subunit preference of the Fe(II)NO product.
Collectively, our results suggest that the reductive nitrosylation of human Hb, with limiting amounts of NO, may most simply be considered to occur as a weighted sum of independent subunit reactions. We assume that the α subunits generate only nitrite, but that β subunits produce either nitrite or SNO. At progressively lower [NO]o/[heme] ratios, β subunit processing and SNO production are increasingly favored. At the very low [NO]o/[heme] ratios characteristic of the physiological situation, the chemistry would tend toward:
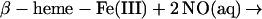 |
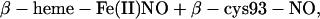 |
[3]
with nitrosothiol and β subunit ferrous heme nitrosyl as the exclusive products of this subunit selective reaction.
The dominance of reaction 3 at low NO levels implies a substantial subunit inequivalence in the binding of NO to form the ferric nitrosyl heme intermediate and/or the subsequent reaction of that intermediate to form the products. Previous studies of the NO binding to met-Hb (13, 24, 25) show that equilibrium favors the β subunit only by a factor of ≈1.5. Although the subunit dependence of Fe(III)NO reaction rates has not been examined in detail, it has been noted that the reductive nitrosylation occurs “considerably faster” with isolated β chains (β subunit tetrameric protein) than with α chains (25). In these previous studies, with NO present in excess, the reaction would presumably involve Hb (Fe(III)NO)4 intermediates. At very low [NO]o/[heme] ratios, however, hybrids such as α2(met)β(met)β(Fe(III)NO) would be the predominant initial reactive intermediates. It is tempting to suggest that the differential reactivity of the various intermediates mustered under different experimental conditions may play an important role in governing the substantial product/subunit selectivity and its dependence on [NO]o/[heme] ratios.
The observed chemistry rationalizes a number of observations made in previous studies of this reaction. A fundamental issue is the lack of mass balance. Hoshino et al. (24) recently reported that the reductive nitrosylation of methemoglobin in excess NO yielded ≈0.9 heme-Fe(II)NO (detected as nitrate after oxidation in air) but only ≈0.4 NO per heme, even though hydroxide and water were considered as the only nucleophiles. [For whale myoglobin (24), the product yields closely followed Eq. 1.] These observations likely reflect S-nitrosylation of Hb in competition with (and independent of) hydroxide or other putative nitrosation targets. The product distribution reported by Hoshino et al. (24) in fact very closely follows the stoichiometry expected for a process that completes both α and β subunit reactions. The observation of Wade and Castro (23) that met-Hb is extremely ineffective, as compared with, for example, horse metmyoglobin, in promoting the apparent nitrosation of exogenous nucleophiles may, in part, reflect interference from the intramolecular nitrosothiol-forming reaction. Our results also clarify certain observations of Addison and Stephanos (22), which were interpreted as indicating that the protein product obtained by complete reductive nitrosylation of methemoglobin is modified in the course of the reaction: Addison and Stephanos, however, suggested that this modification entails nitrosation of a histidine or lysine rather than the thiol. Finally, the variation of the levels of heme-Fe(II)NO hemoglobin complexes that have been detected in blood by EPR and other methods (33–40) may be influenced by the levels of heme-Fe(III)NO and other EPR undetectable species that can interchange with the heme Fe(II)NO species under different methods of sample treatment.
per heme, even though hydroxide and water were considered as the only nucleophiles. [For whale myoglobin (24), the product yields closely followed Eq. 1.] These observations likely reflect S-nitrosylation of Hb in competition with (and independent of) hydroxide or other putative nitrosation targets. The product distribution reported by Hoshino et al. (24) in fact very closely follows the stoichiometry expected for a process that completes both α and β subunit reactions. The observation of Wade and Castro (23) that met-Hb is extremely ineffective, as compared with, for example, horse metmyoglobin, in promoting the apparent nitrosation of exogenous nucleophiles may, in part, reflect interference from the intramolecular nitrosothiol-forming reaction. Our results also clarify certain observations of Addison and Stephanos (22), which were interpreted as indicating that the protein product obtained by complete reductive nitrosylation of methemoglobin is modified in the course of the reaction: Addison and Stephanos, however, suggested that this modification entails nitrosation of a histidine or lysine rather than the thiol. Finally, the variation of the levels of heme-Fe(II)NO hemoglobin complexes that have been detected in blood by EPR and other methods (33–40) may be influenced by the levels of heme-Fe(III)NO and other EPR undetectable species that can interchange with the heme Fe(II)NO species under different methods of sample treatment.
Reductive Nitrosylation Kinetic Analysis.
As a check on the overall soundness of the deconvolution analysis and on the role of Fe(III)NO as an intermediate, we analyzed the UV/VIS spectra recorded before steady state was reached to obtain concentration versus time profiles, exhibited in Fig. 4 Left. The data are analyzed with a de minimis model composed of: (i) equilibrium uptake of NO by met-Hb (Eq. 1, reaction a); (ii) reduction of Fe(III)NO and formal loss of NO+ (Eq. 1, reaction b, or Eq. 2); and (iii) uptake of NO by the reduced heme (Eq. 1, reaction c).
Figure 4.

Kinetics of reductive nitrosylation in a deoxygenated aqueous solution prepared with met-Hb 1.2 mM in heme/0.7 mM NO/100 mM phosphate buffer, pH 7.4/150 mM NaCl. (Left) Time series of visible spectra of the reacting solution. Successive spectra were initiated at time intervals of 1 min. The first spectrum was recorded 22 sec after mixing the reagents. (Right) Concentration versus time profiles for methemoglobin (◊) and Fe(II) nitrosyl hemoglobin (○), and Fe(III) nitrosyl hemoglobin (□). The concentrations of the three species at each time point were determined by least-squares analysis of the corresponding visible absorption spectra (Left), as described in the text.
The solid lines in Fig. 4 Right show traces calculated on the basis of the kinetic model above, with parameter values obtained by least-squares adjustment. The quality of the fit is similar in all of the measurements. We followed the kinetic behavior from shortly after mixing to stationary phase in five samples at various buffer compositions (PBS, Hepes, and 100 mM phosphate) and initial heme/NO ratios (3:1–1:1). As detailed below, we obtain a precise determination of the rate constant for the reduction step ii, but only a rough estimate of the equilibrium constant for step i. The transfer of NO to reduced hemes (step iii) is likewise only found to be extremely fast relative to step ii.
In previous studies of met-heme interactions with NO in human Hb, Sharma et al. (25) and Alayash et al. (13) used stopped-flow methods to elucidate the detailed behavior of the initial binding step. Both groups reported equilibrium constants of ≈3 × 103 M−1, and a subunit inequivalence in the reaction, with the β subunit hemes exhibiting roughly 5-fold greater on- and off-rates than the α subunit hemes. Ford and coworkers (24) obtained an equilibrium constant of similar magnitude 13 × 103 M−1 by analyzing the effect of [NO] on the reduction rate. Within this range, our kinetic analysis was insensitive to the value of this equilibrium constant, and we opted to fix this parameter at 1.0 × 104 M−1 in the analysis. For the reduction step, the effective rate constant, first order in Fe(III)NO, was reported as 0.0018 sec−1 (pH 7, 25°C), by Addison and Stephanos (22), and 0.00063 sec−1 (pH 7, 20°C) by Alayash et al. (13) Ford and coworkers (24) measured the rate as a function of pH from which a value of 0.0019 sec−1 obtains at pH 7.4. The values we determine range from 0.0012 ± 0.0001 sec−1 (PBS) to 0.0027 ± 0.0001 sec−1 (Hepes, 100 mM phosphate), in good agreement with the previously determined values. The overall quality of the analysis and its consistency with prior observations provide strong support for the procedure used and thus identify the central role of the Fe(III)NO intermediate in this reaction.
Additional SNO-Hb-Producing Reactions.
The salient feature of the reductive nitrosylation reaction is the coupling of met-heme reduction with formal NO oxidation to trigger nitrosation. The signature of this reaction in Hb, when the NO is limiting, is the SNO formation in the nitrosation reaction and the β subunit preference in the heme nitrosylation. This interplay of met-hemes with NO could be approached by a number of different starting conditions, which might nevertheless lead to the same set of products. In a physiological context, reactions that do not require the presence of a preexisting reservoir of met-Hb for reaction with incident NO are of particular interest.
The activity of NO as an oxidant of hemoglobin in both oxy and deoxy forms is well documented (3, 41, 42). Accordingly, a sequence composed of an oxidative reaction that forms met-Hb, followed by a reductive nitrosylation leading to SNO-Hb and restoration of a reduced-heme, is in principle possible. In this scheme, heme-redox acts in a cyclic fashion, with no met-Hb reservoir required. In a reaction with oxygenated Hb, this sequence could lead to (i) SNO production and (ii) the formation of a Fe(II)NO in an amount that exceeds the number of originally available vacant Fe(II) centers, thus an apparent cooperativity in NO addition (3, 42), which stems from changes in vacant heme concentrations during NO uptake rather than favorable changes in rate or equilibrium constants. This process could assume special significance for hemoglobins repeatedly exposed to NO, for example, membrane-bound hemoglobins (43, 51) exposed to a slow flux of NO passing into a cell. The loss (to nitrate or nitroxyl) of one equivalent of NO in this scheme would be circumvented if other oxidants were involved. Accumulation of met-Hb would still not be required provided that the oxidation could follow NO addition, that is, if the reaction entails the oxidation of Fe(II)NO.
To explore the feasibility of the latter process, we have initiated studies to assay the production of SNO-Hb accompanying the oxidation of heme-Fe(II)NO species. In Fig. 5, we present results of experiments that show that Hb(NO)4 reacts in the presence of excess ferricyanide to convert Fe(II)NO hemes to met-hemes, with approximately one SNO-Hb produced for every two Fe(II)NO lost. Overall, this product distribution suggests the formal oxidation of Fe(II)NO to a Fe(III)NO species that then reacts to form S-nitrosothiol (or nitrite products), liberating a vacant heme-Fe(II), which, being more easily oxidized than heme-Fe(II)NO [because the association equilibrium constant for NO and heme-Fe(II) is much greater than for heme-Fe(III) and NO], readily accedes to oxidation under these conditions.
Figure 5.

Conversion of heme-Fe(II)NO centers to heme-Fe(III) and SNO-Hb on exposure of Hb(NO)4 to ferricyanide. Samples of the reaction medium are withdrawn at various points in the course of the reaction for analysis of the amounts of heme-Fe(II)NO, heme-Fe(III), and nitrosothiol, relative to the total heme in the medium. ○, Heme-Fe(III) production compared to heme-Fe(II)NO depletion; solid line, the best linear fit to these experimental points (slope −1.15 ± 0.01; intercept = 1.14 ± 0.01; R = 0.9998) indicative of a one-to-one conversion of heme-Fe(II)NO to heme-Fe(III). ●, Experimental S-nitrosothiol production compared to heme-Fe(II)NO depletion; dashed line, the best linear fit to these experimental points (slope −0.50 ± 0.11; intercept 0.46 ± 0.07; R = 0.96). Product analysis was carried out as described in the text.
The reaction of nitrite with hemoglobin provides a further route to the simultaneous presentation of heme Fe(III) and NO (41, 44–46). The possible significance in the context of nitric oxide biology has been thoughtfully considered by Reutov and Sorokina (47), who posited that because of the well known nitrite reductase activity of hemoglobin, nitrite could be considered an integral component of NO biochemistry, rather than a metabolic dead end. To examine this idea and to determine whether SNO-Hb is produced in this interaction, we first incubated oxyhemoglobin with ≈0.01 equivalents (on a per-heme basis) of nitrite. No discernible changes were present in the EPR spectrum over a time span of as much as 15 min. The only feature in the spectrum derived from the small fraction (≈1%) of met-heme in the oxy-Hb solution, which was not discernibly changed by the nitrite addition. Deoxygenation of these mixtures after various incubation times led to an immediate change in the EPR spectrum with roughly equal increases in met-heme and β heme-Fe(II)NO signals, as illustrated in Fig. 6 Upper and Lower, respectively). These results are consistent with the simple understanding of the nitrite reaction with deoxy-Hb according to the following scheme (41, 44):
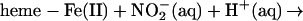 |
 |
[4]
Figure 6.

EPR spectra from Fe(II)NO and Fe(III) hemes obtained by exposure of hemoglobin to nitrite. EPR spectra were obtained at 9.28 GHz, with 10 mW incident power and field modulation of 20 G at 100 kHz. The external field was scanned for 4 min with a detection time constant of 0.128 sec over to range: 800−1,300 G to detect high-spin heme-Fe(III) (Upper) or 3,050−3,450 G to detect heme-Fe(II)NO (Lower). A spectrum of a sample of a neat 3.8 mM oxyhemoglobin solution, frozen in liquid nitrogen, is displayed (▴, pink) in both panels; the trace (Lower) was used as a base-line for subtraction from subsequent scans. Spectra were subsequently obtained from samples withdrawn from the solution after incubation with 37 μM nitrite for periods of 5 (□, blue), 12 (⧫, purple), and 24 (○, teal) min, followed by deoxygenation and freezing in liquid nitrogen. SNO-Hb is observed on reoxygenation of such samples. A spectrum obtained without deoxygenation after an incubation time of 10 min (◊, green) is also shown (Upper).
NO will ultimately lodge at available heme vacancies to form Fe(II)NO. Interestingly, as we have reported elsewhere (43), oxygenation leads to elimination of the Fe(II)NO and the formation of SNO-Hb, in effect the reverse of reaction 4, with nitrosothiol formation (Eq. 2), rather than nitrite regeneration. Overall, this chemistry reveals a catalytic role for Hb, in conjunction with its oxygenation/deoxygenation cycle, in converting nitrite to the SNO-Hb.
Discussion
We have shown that NO can be transported from the heme-binding site (distal pocket) to the thiol of β-cys93, which lies on the opposite face of the heme (next to the proximal histidine residue). The production of SNO-Hb in this transfer can be supported by heme-iron redox chemistry. When NO is limiting, SNO-Hb is the major product formed in the reductive nitrosylation reaction, with preference for the β subunits of hemoglobin. We have also shown that SNO-Hb production rather generally accompanies the formal interaction of NO and heme-Fe(III), regardless of the sequence of NO addition and heme oxidation, sequences including NO reaction with heme-Fe(III), oxidation of heme-Fe(II)NO, or reaction of nitrite with deoxy-Hb in an oxygenation/deoxygenation cycle.
The chemistry reported here has several implications of potential importance to NO hemoglobin interactions in blood. Inasmuch as the NO group is present in limited concentrations, any reductive nitrosylation reaction would tend to process the NO group exclusively within the β subunit. This subunit selective processing could provide a means by which NO could avoid being trapped by the α hemes to yield the five-coordinate α heme-Fe(II)NO complexes that could quench NO bioactivity (48). Moreover, our results reveal a means by which the NO group can be dislodged from heme-Fe(II)NO complexes to make the nitrosothiol hemoglobin derivative: the perceived unfavorability of NO dissociation from nitrosylated ferrous hemes (49) can be circumvented by oxidation to the ferric heme-nitrosyl species, thus facilitating NO-group transfer to protein thiol. In conjunction with this oxidation, reaction 3 depicts NO-group transfer from a β subunit heme species that holsters NO bioactivity [heme-Fe(II)NO] to one that arms it (protein–SNO) for potential release via, for example, transfer to other thiols (50, 51). Inasmuch as NO is a potent oxidant of both oxy- and deoxy-Hb, the methemoglobin requirement may be potentiated through NO itself, in a cycle of heme oxidation/reductive nitrosylation, which does not require or involve any met-Hb accumulation. The oxidative and NO-group requirements can also be supported by interaction of heme vacancies with nitrite as part of a greater NO-group biological chemistry.
The results presented here clarify some issues relevant to SNO-Hb function raised in several recent publications. The ferricyanide oxidation experiments raise questions about the analytical methods and interpretations of Gladwin et al. (40). Apparent cooperativity in the formation of heme-Fe(II)NO by exposure of oxygenated hemoglobin to NO has been observed under certain conditions in vitro by us and in red blood cell preparations by Huang et al. (52) and Han et al. (53). Moreover, although Huang et al. (52) suggest that their in vitro experiments do not show this apparent cooperativity, the systematic differences between the experimental data and the theoretical predictions indicate that, whereas such effects are attenuated under their conditions, an excess of heme-Fe(II)NO is produced at higher oxygen saturations. Preference for the β subunits is evident in the Fe(II)NO EPR spectra presented in the work of Huang et al. (52). The latter authors also measure SNO production and obtain nearly equivalent yields of Fe(II)NO and SNO-Hb under a variety of conditions. In related experiments, Joshi et al. (30) fail to detect Fe(II)NO at very high oxygenation and nonphysiological NO levels. Notably, they do observe the production of SNO-Hb in yields that are similar to those reported by Gow et al. (3). At odds with their conclusions, the production is independent of the manner of NO addition. The observations reported in all of these studies share features that are common to the reductive nitrosylation chemistry reported here.
Han et al. (53) suggest, on the basis of the cyanide blocking experiments, that reductive nitrosylation is not significant to the production of SNO-Hb. Their results, however, only compel the conclusion that in the presence of high cyanide levels, some met-heme independent SNO-Hb production occurs. Our mechanistic conclusions are supported by direct observations of all of the relevant chemical species, detailed material balance, and global kinetic analysis; there is no ambiguity regarding the centrality of the heme-Fe(III)NO intermediate.
Finally, it is worth pointing out that in the experiments of Han et al. (53) and Huang et al. (52), very high protein concentrations are used. Although this choice reflects the concentrations within red blood cells, it can obscure the fundamental chemistry of hemoglobin at the molecular level. Analysis of the chemical behavior of concentrated protein solutions that is significantly depleted in the activity of water may require more elaborate models for bolus and diffusion effects and attention to chemical activities. Ultimately, with a clearer picture of the relevant molecule scale behavior of hemoglobins, an integrated model of cardiovascular NO function, encompassing molecular, cellular, and system wide variables, may be developed.
Acknowledgments
We gratefully acknowledge thoughtful discussions with Joseph Bonaventura. This work was supported by the National Institutes of Health (HL52529, HL59130, and HL66179-02), the ALS Association, MONT (National Science Foundation Experimental Program to Stimulate Competitive Research), and the National Science Foundation (MCB 00981228).
Abbreviation
- SNO
S-nitroso
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Stamler J S. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 2.Stamler J S, Singel D J, Loscalzo J. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- 3.Gow A J, Luchsinger B P, Pawloski J R, Singel D J, Stamler J S. Proc Natl Acad Sci USA. 1999;96:9027–9032. doi: 10.1073/pnas.96.16.9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hille R, Olson J S, Palmer G. J Biol Chem. 1979;254:12110–12220. [PubMed] [Google Scholar]
- 5.Hille R, Palmer G, Olson J S. J Biol Chem. 1977;252:403–405. [PubMed] [Google Scholar]
- 6.Reisberg P, Olson J S, Palmer G. J Biol Chem. 1976;251:4379–4383. [PubMed] [Google Scholar]
- 7.Taketa F, Antholine W E, Chen J Y. J Biol Chem. 1978;253:5448–5451. [PubMed] [Google Scholar]
- 8.Quine R W, Eaton G R, Eaton S S, Koska A, Bowman M K. J Magn Reson. 1986;69:371–374. [Google Scholar]
- 9.Mendes P. Trends Biochem Sci. 1997;22:361–363. doi: 10.1016/s0968-0004(97)01103-1. [DOI] [PubMed] [Google Scholar]
- 10.Kuzmic P. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 11.Malinowski E R. Factor Analysis in Chemistry. New York: Wiley; 1991. [Google Scholar]
- 12.Luchsinger B P. Ph.D. thesis. Bozeman: Montana State University; 2003. [Google Scholar]
- 13.Alayash A I, Fratantoni J C, Bonaventura C, Bonaventura J, Cashon R E. Arch Biochem Biophys. 1993;303:332–338. doi: 10.1006/abbi.1993.1292. [DOI] [PubMed] [Google Scholar]
- 14.Henry Y, Banerjee R. J Mol Biol. 1970;50:99–110. doi: 10.1016/0022-2836(70)90107-5. [DOI] [PubMed] [Google Scholar]
- 15.Kon H. J Biol Chem. 1968;243:4350–4357. [PubMed] [Google Scholar]
- 16.Shiga T, Hwang K J, Tyuma I. Biochemistry. 1969;8:378–383. doi: 10.1021/bi00829a052. [DOI] [PubMed] [Google Scholar]
- 17.Henry Y, Banerjee R. J Mol Biol. 1973;73:469–482. doi: 10.1016/0022-2836(73)90094-6. [DOI] [PubMed] [Google Scholar]
- 18.Hartree E F, Keilin D. Nature. 1937;139:548. [Google Scholar]
- 19.Chien J C W. J Am Chem Soc. 1969;91:2166–2168. doi: 10.1021/ja01036a085. [DOI] [PubMed] [Google Scholar]
- 20.Ehrenberg A, Szczepkowski T W. Acta Chem Scand. 1960;14:1684–1692. [Google Scholar]
- 21.Wayland B B, Olson L W. J Am Chem Soc. 1974;96:6037–6041. doi: 10.1021/ja00826a013. [DOI] [PubMed] [Google Scholar]
- 22.Addison A W, Stephanos J J. Biochemistry. 1986;25:4104–4113. doi: 10.1021/bi00362a018. [DOI] [PubMed] [Google Scholar]
- 23.Wade R S, Castro C E. Chem Res Toxicol. 1990;3:289–291. doi: 10.1021/tx00016a002. [DOI] [PubMed] [Google Scholar]
- 24.Hoshino M, Maeda M, Konishi R, Seki H, Ford P C. J Am Chem Soc. 1996;118:5702–5707. [Google Scholar]
- 25.Sharma V S, Traylor T G, Gardiner R, Mizukami H. Biochemistry. 1987;26:3837–3843. doi: 10.1021/bi00387a015. [DOI] [PubMed] [Google Scholar]
- 26.Jia L, Bonaventura C, Bonaventura J, Stamler J S. Nature. 1996;380:221–226. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 27.Stamler J S, Jia L, Eu J P, McMahon T J, Demchenko I T, Bonaventura J, Gernert K, Piantadosi C A. Science. 1997;276:2034–2037. doi: 10.1126/science.276.5321.2034. [DOI] [PubMed] [Google Scholar]
- 28.Chan N L, Rogers P H, Arnone A. Biochemistry. 1998;37:16459–16464. doi: 10.1021/bi9816711. [DOI] [PubMed] [Google Scholar]
- 29.Ferranti P, Malorni A, Mamone G, Sannolo N, Marino G. FEBS Lett. 1997;400:19–24. doi: 10.1016/s0014-5793(96)01258-6. [DOI] [PubMed] [Google Scholar]
- 30.Joshi M S, Ferguson T B, Jr, Han T H, Hyduke D R, Liao J C, Rassaf T, Bryan N, Feelisch M, Lancaster J R., Jr Proc Natl Acad Sci USA. 2002;99:10341–10346. doi: 10.1073/pnas.152149699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keefer L K, Nims R W, Davies K M, Wink D A. Methods Enzymol. 1996;268:281–293. doi: 10.1016/s0076-6879(96)68030-6. [DOI] [PubMed] [Google Scholar]
- 32. Williams, E. M. (2001) B.S. Honors thesis (Middlebury College, Middlebury, VT).
- 33.Kosaka H, Watanabe M, Yoshihara H, Harada N, Shiga T. Biochem Biophys Res Commun. 1992;184:1119–1124. doi: 10.1016/0006-291x(92)90708-s. [DOI] [PubMed] [Google Scholar]
- 34.Kosaka H, Tanaka S, Yoshii T, Kumura E, Seiyama A, Shiga T. Biochem Biophys Res Commun. 1994;204:1055–1060. doi: 10.1006/bbrc.1994.2569. [DOI] [PubMed] [Google Scholar]
- 35.Jacob T D, Nakayama D K, Seki I, Exler R, Lancaster J R, Jr, Sweetland M A, Yousem S, Simmons R L, Billiar T R, Peitzman A B, et al. J Appl Physiol. 1994;76:1794–1801. doi: 10.1152/jappl.1994.76.4.1794. [DOI] [PubMed] [Google Scholar]
- 36.Romand J A, Pinsky M R, Firestone L, Zar H A, Lancaster J R., Jr J Appl Physiol. 1994;76:1356–1362. doi: 10.1152/jappl.1994.76.3.1356. [DOI] [PubMed] [Google Scholar]
- 37.Roccatello D, Mengozzi G, Alfieri V, Pignone E, Menegatti E, Cavalli G, Cesano G, Rossi D, Formica M, Inconis T, et al. Nephrol Dial Transplant. 1997;12:292–297. doi: 10.1093/ndt/12.2.292. [DOI] [PubMed] [Google Scholar]
- 38.Weinberg J B, Gilkeson G S, Mason R P, Chamulitrat W. Free Radical Biol Med. 1998;24:191–196. doi: 10.1016/s0891-5849(97)00217-7. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi Y, Kobayashi H, Tanaka N, Sato T, Takizawa N, Tomita T. Am J Physiol. 1998;274:H349–H357. doi: 10.1152/ajpheart.1998.274.1.H349. [DOI] [PubMed] [Google Scholar]
- 40.Gladwin M T, Ognibene F P, Pannell L K, Nichols J S, Pease-Fye M E, Shelhamer J H, Schechter A N. Proc Natl Acad Sci USA. 2000;97:9943–9948. doi: 10.1073/pnas.180155397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doyle M P, Pickering R A, DeWeert T M, Hoekstra J W, Pater D. J Biol Chem. 1981;256:12393–12398. [PubMed] [Google Scholar]
- 42.Gow A J, Stamler J S. Nature. 1998;391:169–173. doi: 10.1038/34402. [DOI] [PubMed] [Google Scholar]
- 43.McMahon T J, Moon R E, Luchsinger B P, Carraway M S, Stone A E, Stolp B W, Gow A J, Pawloski J R, Watke P, Singel D J, et al. Nat Med. 2002;8:711–717. doi: 10.1038/nm718. [DOI] [PubMed] [Google Scholar]
- 44.Stepuro I I, Chaikovskaya N A, Solodunov A A, Artsukevich A N. Biochemistry (Moscow) 1997;62:960–966. [PubMed] [Google Scholar]
- 45.Kosaka H, Tyuma I, Imaizumi K. Biomed Biochim Acta. 1983;42:S144–S148. [PubMed] [Google Scholar]
- 46.Tomoda A, Tsuji A, Yoneyama Y. Biochem J. 1981;193:169–179. doi: 10.1042/bj1930169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reutov V P, Sorokina E G. Biochemistry (Moscow) 1998;63:874–884. [PubMed] [Google Scholar]
- 48.Yonetani T, Tsuneshige A, Zhou Y, Chen X. J Biol Chem. 1998;273:20323–20333. doi: 10.1074/jbc.273.32.20323. [DOI] [PubMed] [Google Scholar]
- 49.Gibson Q H, Roughton F J W. J Physiol. 1957;136:507–511. doi: 10.1113/jphysiol.1957.sp005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McMahon T J, Exton Stone A, Bonaventura J, Singel D J, Stamler J S. J Biol Chem. 2000;275:16738–16745. doi: 10.1074/jbc.M000532200. [DOI] [PubMed] [Google Scholar]
- 51.Pawloski J R, Hess D T, Stamler J S. Nature. 2001;409:622–626. doi: 10.1038/35054560. [DOI] [PubMed] [Google Scholar]
- 52.Huang Z, Louderback J G, Goyal M, Azizi F, King S B, Kim-Shapiro D B. Biochim Biophys Acta. 2001;1568:252–260. doi: 10.1016/s0304-4165(01)00227-6. [DOI] [PubMed] [Google Scholar]
- 53.Han T H, Hyduke D R, Vaughn M W, Fukuto J M, Liao J C. Proc Natl Acad Sci USA. 2002;99:7763–7768. doi: 10.1073/pnas.122118299. [DOI] [PMC free article] [PubMed] [Google Scholar]