

Abstract
Cellulose binding modules (CBMs) potentiate the action of cellulolytic enzymes on insoluble substrates. Numerous studies have established that three aromatic residues on a CBM surface are needed for binding onto cellulose crystals and that tryptophans contribute to higher binding affinity than tyrosines. However, studies addressing the nature of CBM–cellulose interactions have so far failed to establish the binding site on cellulose crystals targeted by CBMs. In this study, the binding sites of CBMs on Valonia cellulose crystals have been visualized by transmission electron microscopy. Fusion of the CBMs with a modified staphylococcal protein A (ZZ-domain) allowed direct immuno-gold labeling at close proximity of the actual CBM binding site. The transmission electron microscopy images provide unequivocal evidence that the fungal family 1 CBMs as well as the family 3 CBM from Clostridium thermocellum CipA have defined binding sites on two opposite corners of Valonia cellulose crystals. In most samples these corners are worn to display significant area of the hydrophobic (110) plane, which thus constitutes the binding site for these CBMs.
Typical cellulases contain distinct cellulose binding modules (CBMs), which have an important role in crystalline cellulose degradation (1). CBMs from different enzymes and different taxonomic origins have been classified into families with similar amino acid sequences and 3D structures (ref. 2 and http://afmb.cnrs-mrs.fr/∼pedro/CAZY/db.html). Fungal CBMs belong to family 1 (CBM1) characterized by a small wedge-shaped fold featuring a cellulose binding surface with three solvent-exposed aromatic residues (4, 5). Our earlier studies of CBMs of Trichoderma reesei cellobiohydrolases Cel6A and Cel7A and the endoglucanase Cel7B have shown these aromatic residues are critical for the binding of a CBM1 onto crystalline cellulose and that the presence of a Trp instead of a Tyr residue results in an increased affinity on crystalline cellulose (6–10). Finally, the mutagenesis of one of the three disulphide bridges essential for the structure of the Cel6A CBM1 resulted in higher off-rate on crystalline cellulose, implying that the molecular rigidity is also significant in the CBM1–cellulose interaction (10).
Structural studies indicate that the spacing of the three aromatic residues coincides with the spacing of every second glucose ring on a glucan chain (4). Therefore, it has been postulated that the aromatic amino acids of the CBMs form van der Waals interactions and aromatic ring polarization interactions with the pyranose rings exposed on the (110) crystalline face of cellulose (5, 6, 11). A few residues in some CBMs contribute hydrogen bonds but these are of less importance in the family 1 CBMs (7).
Valonia cellulose crystals occur predominantly as cellulose Iα with triclinic unit cells (12). Two of the four crystal faces, the (100) and (010) faces, present relatively large, hydrophilic surfaces rich in OH groups (Fig. 1). The other two faces, indexed as (110) and (1–10) faces, are sharp corners with essentially only one surface-exposed chain. Such an arrangement implies that, in perfect cellulose crystals, the surface area of the proposed binding site for the CBMs is very limited (6). For this reason, other possible binding sites for the CBMs have also been contemplated (13). However, previous observations made with electron microscopy (EM) indicate that the corners are often rounded because the chains with fewest interactions with the underlying body of the crystal are easily dissociated (14). Depending on their location, such “worn” corners present larger areas of the hydrophobic (110) face or hydrophilic (1–10) face (see Fig. 1), of which the hydrophobic face could provide binding sites for CBMs. This finding inspired us to attempt location-specific visualization of the CBM1s by transmission electron microscopy (TEM) using direct immuno-gold labeling of ZZ–CBM1 fusion proteins.
Figure 1.

Schematic presentation of the organization of the cellulose chains in the Iα allomorph of cellulose crystals and the shape of the complete crystal formed. As their unit cell parameters have been determined, Valonia crystals are known to be primarily in the Iα form (12) and the corresponding indexing is thus used throughout this article. The d-spacings characteristic to the different crystalline planes are indicated. The obtuse corner (circled), which exposes the (110) face in worn crystals, is the proposed binding site for the CBMs.
Materials and Methods
Bacterial Strains and Plasmids.
Escherichia coli strain XL1-blue (Stratagene) was used as host for plasmid constructions and protein production. The vector for secreted expression of the recombinant fusion protein proteins into the E. coli periplasmic space was the plasmid pEZZmp18 containing the Staphylococcus aureus protein A (SpA) promoter and the SpA signal sequence (15).
Recombinant DNA Techniques.
The DNA fragment coding for T. reesei cel6A CBM was amplified from plasmid pTI-p21 (16) by using the forward primer A and the reverse primer B (Table 1), which included cleavage sites for the restriction enzymes HindIII and EcoRI (MBI Fermentas, Newington, NH), respectively. DNA encoding T. reesei Cel7A CBM was prepared by PCR amplification of plasmid pTI-p21 using primers C and D.
Table 1.
Primers used for the DNA constructions
| A: | CGGGAATTCAGCGCGCGGCGGTGCGCAGGCTTGCTCAAGCGTCTGG |
| B: | CGGGAAGCTTGCTTAAAGACACTGGGAGTAATAGTCGTTGGA |
| C: | CGGGAATTCAGCGCGCAGCGGACCTACCCAGTCTCACTACGGC-3′ |
| D: | CGGGAAGCTTGCTTACAGGCACTGAGAGTAGTAAGGGTTC |
| E: | ACCCATTGGGGCCAGTGCGGTGGCATCGGCTAC-AGCGGTTGCAAAACCTGCACG |
| F: | GTAGTCGTTAGAATACGGCAGGTC GTGCCGGACGTGCAGGTTTTGCAACC |
| G: | CGGGAATTCAGCGCGCGGCGGTGCGAGCTGCACCCAGACCCATTGGGGCCAGTGC |
| H: | CGGGAAGCTTGCTTACAGGCACTGGCTATAGTAGTCGTTAGAATACTGGCAGGT |
| I: | CTTCGAATTCGTGTGGTGGTGCCTGGG |
| J: | GGGCTGCAGGCTGCGCGCAGGTCCTTGACATTGTGAATACCATTG |
| K: | CTTCGAATTCGAATTTGAAGGTTGAGTTTTAC |
| L: | GGG-CTGCAGGCTGCGCGCAGGTCCGGGTTCTTTACCCCATACAA |
| M: | GACGCG-AATTCAGCGCGCAGCGGACCTACCCAGTCTCACTGGGGCCA |
| N: | TGCCAAGCTTGCTTACAGGCACTGAGAGTACCAAGGGTTCA |
All primers are written from 5′ (left) to the 3′ (right) (see Materials and Methods).
The DNA fragment coding for T. reesei Cel7B CBM was constructed by annealing the overlapping oligonucleotides E and F by using a temperature gradient 95°C to 37°C. Klenow DNA polymerase (New England Biolabs) was added after annealing to assist complementary strand synthesis. After incubation at 37°C for 15 min, the polymerase was heat-inactivated at 72°C for 15 min. The complete Cel7B CBM DNA fragment was then amplified by using primers G and H (Table 1).
The sequence coding for the Neocallimastix patriciarum Cel6A CBM was amplified from plasmid pBSFCA (17) by using primers I and J including cleavage sites for EcoRI and PstI (MBI Fermentas) at the 5′ and 3′ ends, respectively. In the same way the fragment coding for the Clostridium thermocellum CipA CBM was amplified by using primers K and L. T. reesei Cel7A CBM mutants were generated by using oligonucleotides containing codons coding for tryptophan instead of tyrosine for the PCR. For the amplification of the Cel7A CBMY5W (numbering according to ref. 4), primers M and B were used. The double mutant CBMY5W:Y31W of T. reesei Cel7A CBM was amplified by PCR using sense primer I and antisense primer N. In all of the constructs, an arginine codon was introduced in the linker region between the sequence coding for the CBM and the ZZ-domains to enable preparation of free CBMs by tryptic digestion of fusion proteins. The cleaved PCR products were ligated with the digested plasmid pEZZmp18 at 15°C overnight with T4 ligase (MBI Fermentas). The nucleotide sequences of the final constructs were verified with sequencing performed on the MegaBACE sequencer (Amersham Pharmacia).
Protein Expression and Purification.
The transformed E. coli cells were grown for 16 h at 30°C in TSB [15 g/liter tryptic soy broth (Merck), 5 g/liter yeast extract] containing 100 μg/ml ampicillin. The cells were harvested by centrifugation, and the periplasmic fraction was separated by osmotic shock and subjected to affinity purification on an IgG-Sepharose column (Amersham Pharmacia). The column was washed with 10 bed vol of TST (25 mM Tris⋅HCl/150 mM NaCl/1.25 mM EDTA/0.05% Tween 20, pH 7.5) and 5 bed vol of 5 mM NH4Ac (pH 5.5). The elution of the fusion proteins with acetate buffer (0.2 M HAc, pH 3.3) was followed at 280 nm. The eluted proteins were purified further by using RESOURCE Mono-S cation exchanger on ÄKTA (both from Amersham Pharmacia). Proteins were eluted with a mild salt-pH gradient by using 50 mM acetate buffer (pH 4.0) as buffer A and 200 mM acetate buffer (pH 5.5) as buffer B. The peak fractions were collected and lyophilized. Correct mass and purity of the proteins were controlled with surface-enhanced laser desorption/ionization time-of-flight MS using H50 ProteinChip array (Ciphergen Biosystems, Fremont, CA), according to the manufacturer's instructions. In shake flask cultures 15–50 mg/liter of pure recombinant protein was routinely obtained.
Labeling of the Proteins.
Tritium labeling of the CBM fusion proteins was done by reductive alkylation as described (10, 18). Free label was separated from the labeled proteins by gel filtration on PD-10 columns (Amersham Pharmacia), including a buffer change into 50 mM sodium acetate, 50 mM NaCl, pH 5.0. The CBM-ZZ fusion proteins were concentrated by using Ultrafree centrifugal filters with a molecular cut-off of 5 kDa (Millipore), and the amount of free label in the filtrates was determined by using a liquid scintillation counter (Wallac). The amount of free label detected in the different protein batches was measured to vary between 0.3% and 1.7%.
Binding Studies.
The protein concentrations were determined spectrophotometrically at 280 nm by using molar absorption coefficients (ɛ) individually calculated for each fusion protein (T. reesei, CBMCel6A-ZZ 18140, ZZ-CBMCel7A 7920, ZZ-CBMY5W 12330, ZZ-CBMY5W:Y31W 16740, ZZ-CBMCel7B 13730; N. patriciarum, CBMCel6A-ZZ 15580; C. thermocellum, CBMCipA-ZZ 36840). Standard curves were determined for each protein, relating the protein amount to radioactivity, and the binding isotherms were determined on bacterial microcrystalline cellulose (BMCC) and Valonia cellulose in three independent experiments. BMCC and Valonia ventriculosa cellulose crystals were prepared as described (19, 20). The CBM fusion proteins were mixed with cellulose and incubated with agitation for 1 h at 4°C or 22°C in a buffer (50 mM acetate, pH 5.0/50 mM NaCl) containing 1% ovalbumin, Sigma) as a blocking agent. After the equilibrium was reached, the samples were filtered through 0.45-μm Millex-HV filters (Millipore), and the radioactivity of the filtrate was measured by using a liquid scintillation counter (Wallac).
The nonequilibrium desorption was studied by mixing a number of identical samples of the CBM-ZZ fusion proteins and BMCC (volume, 200 μl), allowing them to reach equilibrium (1 h). At different time points, the samples were diluted by adding 1 ml of buffer (50 mM sodium acetate/50 mM NaCl/1% ovalbumin) and filtrated followed by determination of the amount of free peptide in the filtrates. The diluted samples were then compared with reference samples, which had been allowed to reach equilibrium in protein concentration identical to the final dilution.
Sample Preparation for EM.
Valonia microcrystals were prepared as described (20) and overlaid on a carbon-coated EM grid. The cellulose and the supporting carbon film were blocked with 1% ovalbumin (Sigma) for 60 min. The CBM fusion proteins were dissolved in acetate buffer (50 mM sodium acetate/50 mM NaCl/1% ovalbumin, pH 5.5). A final concentration of 15 μM of the CBMs and the rabbit IgG gold conjugate (6 μg/ml) (GAF-013-5, EY Laboratories) were mixed and preincubated for 30 min. The grid overlaid by the Valonia cellulose crystals was incubated for 1 h on the drop of the preincubated protein solution at 4°C. Thereafter the samples were washed once with acetate buffer and three times with water. Uranyl acetate (1% in distilled water) was used for the negative staining of the sample.
EM.
The samples were observed with a Jeol JEM-2000EXII microscope with a rotation-tilt goniometer and operated at an accelerating voltage of 100 kV. Under low-dose conditions, a Gatan Image Intensifier was used. A labeled microcrystal was first rotated to align its microfibril axis to the tilt axis of the microscope, and the crystal was then tilted around its microfibril axis. Images were taken at 0°, ±45° where after the image at 0° was recorded once more to ensure that the crystals were not deformed because of electron irradiation. Only the data from nondeformed crystals (identical 0° images before and after the tilting experiment) were used for further analysis. All images were recorded on Mitsubishi MEM EM films at a magnification of ×8,000. The labeled crystals were also subjected to microdiffraction analysis to determine their orientation on the supporting film as described (20, 21).
To deduce the 3D locations of the labeled points of the fiber their coordinates were collected from the three images with tilt 0° as well as ±45°. The coordinate system had the origin in the center of the bottom plane of the fiber, the y axis parallel with the axis of the fiber, and a z axis as the height of the fiber. From the picture of the untilted fiber the x and y coordinates were determined. The z coordinates were calculated from the tilt experiments without any assumptions about the fiber cross-section geometry. The coordinates were transformed to a polar coordinate system, as shown in Scheme S1.
Scheme 1.

The location of a labeled point in the fiber cross section is given by Cartesian coordinates x and z and polar coordinates ρ (distance from origin) and v (angle to z axis). The unknown z coordinate is given as z = ρ cosv. Upon rotation the distance ρ is constant whereas the x coordinate and the angle v to the z axis change. For the tilt experiments the following system of equations can be formulated:
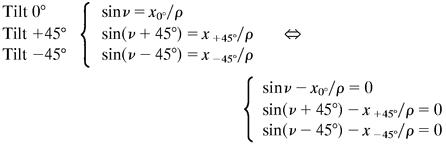 |
The overdetermined nonlinear system of equations was solved through the construction of a function f(ρ,v), such that:
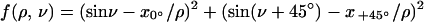 |
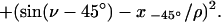 |
For data without any errors f(ρ,v) will be exactly 0, in all other cases >0. The function f(ρ,v) thus gives a measure of the quality of the data. The values of ρ and v needed for calculation of the z coordinate are given as the values that minimize f(ρ,v).
Results
Protein Design and Production.
Several WT and mutated family 1 and family 3 CBM fusion proteins were designed and produced to investigate their interactions with crystalline cellulose. Because a single Trp→Tyr mutation was known to decrease the affinity and increase the off-rate of Cel6A CBM1 on crystalline cellulose (8, 10), complementary mutations changing one or two of the tyrosines to tryptophans were introduced to the Cel7A CBM1 (ZZ-CBMY5W and ZZ-CBMY5W:Y31W). Two Z-domains, analogous to the domain B from S. aureus protein A (22), were genetically linked either to the N or the C termini of the CBMs, depending on the position of the CBM in the native enzyme. The IgG binding properties of the ZZ-domains were exploited in both the affinity purification of the CBM fusion proteins and the direct labeling with gold-conjugated antibodies for visualization with TEM. The linker peptide used resembles that previously used in a double CBM construction (16).
Binding Studies.
Binding isotherms for the T. reesei Cel6A and Cel7A WT and mutated CBMs were determined on BMCC and/or Valonia cellulose (Fig. 2). Consistent with earlier data on other CBM-fusion proteins (16), the presence of a ZZ-tag did not affect the binding isotherm of a CBM (CBMCel7A, Fig. 2C). The ZZ-domain alone did not bind to cellulose (Fig. 2A) nor does the tritium labeling interfere with the CBM adsorption (18). On both substrates, the mutated Cel7A CBMY5W and CBMY5W;Y31W showed increased binding with an affinity comparable to Cel6A WT CBM (Fig. 2B). The presence of two tryptophans in the double mutant CBMY5W;Y31W or in the N. patriciarum Cel6A CBM1 gave no further increase in binding. In line with earlier published data (23), the binding affinity of the CBMs decreased as a function of elevated temperature (+4°C to +22°C) but the relative order of the affinities between different CBMs was maintained in each temperature (data not shown). Nonequilibrium dilution experiments revealed fully reversible binding of the fusion proteins: at 4°C, 99–99.8% of each protein returned to the binding isotherm within 30 min of the time of dilution (data not shown).
Figure 2.

Equilibrium adsorption isotherms of different CBM-ZZ fusion proteins: T. reesei CBMCel7A (⧫), T. reesei CBMCel6A (□), T. reesei CBMCel7B (●), N. patriciarum CBMCel6A (+), T. reesei CBMY5W (▵), T. reesei CBMY5W;Y31W (x), isolated T. reesei CBMCel7A (■), and ZZ (○). (A) Adsorption to BMCC (+4°). (B) Adsorption of Cel7A, Cel6A, and mutated CBM-fusion proteins to Valonia cellulose (+4°). (C) Adsorption of ZZ-CBMCel7A and isolated CBMCel7A to BMCC (+22°).
EM.
Despite extensive structure–function studies, the exact site of binding of the CBMs has not been conclusively determined on crystalline cellulose. Owing to the rectangular cross section of cellulose crystals, there are four possible sites available for the CBM binding (see Fig. 1). To investigate the exact binding site of the CBMs, TEM visualization of CBMs bound on Valonia cellulose crystals was carried out by using direct labeling of the ZZ-CBM1 fusion proteins with gold-labeled antibodies. Two different labeling approaches were tried: in the sequential approach, the CBM fusion proteins were first incubated with cellulose, where after the sample was washed briefly and incubated with the IgG-gold conjugate. In the direct labeling approach, the gold-conjugated IgG and the CBM fusion proteins were preincubated, allowing the CBM fusion proteins to form a complex with IgG-gold conjugate before adsorption to cellulose. The direct labeling minimized the number of washing steps required and revealed a lower level of background binding of the IgG–gold conjugate. Because no qualitative differences were observed between the two approaches, the direct labeling was used in the subsequent studies of the specificity of the CBM binding on the crystal surfaces.
To evaluate the binding sites of the CBMs on Valonia cellulose crystals, 10–20 independently prepared samples of each CBM were examined. Visual inspection of the different binding experiments suggested that the Cel7A and Cel6A CBMs bind along two sides of the Valonia crystals (Fig. 3). None of the images exhibited labeled proteins scattered around an isolated crystal. On the two preferred sites, binding of the CBMs was uniform with no apparent preference toward the chain ends or the mechanically damaged parts of the crystals (Fig. 3 A and B). Adsorption of the bacterial CBM3 from C. thermocellum CipA was similar to the fungal CBM1 binding. The only difference was a perhaps slightly increased tendency to bind close to the chain ends (Fig. 3C).
Figure 3.

Images obtained by TEM of the labeled CBMs binding to the Valonia cellulose crystals. (A) Cel7A CBM1. (B) Cel7B CBM1. (C) CipA CBM3. (D) Negative control (see text).
Identification of the two crystal surfaces, which constitute the observed preferential binding sites for CBMs, required determination of the orientation of the crystals on the underlying support. Owing to their cuboid shape, Valonia crystals can lie on the carbon support predominantly in four different orientations (see Fig. 4B), which can be distinguished by their characteristic microdiffraction patterns. The microdiffraction patterns were obtained as described (20) for a large number of individual, labeled crystals. The patterns obtained with labeled crystals revealed diffraction signals corresponding to d-spacings 0.61, 0.53, and 0.39 nm, which are characteristic to the interplanar distances of planes (100), (010), and (110), respectively (see Fig. 1). The dominant orientation for the labeled crystals was a (d100 = 0.61 nm), followed by c (d110 = 0.39 nm) and b (d010 = 0.53 nm) whereas the patterns characteristic to the orientation d were rarely observed. When nonlabeled crystals are observed, the dominant orientation is a followed by orientations b, c, and d at roughly equal frequencies. Fig. 4A shows a microdiffraction pattern characteristic to the orientation c.
Figure 4.

(A) Microdiffraction pattern corresponding to the d-spacing of 0.39 nm, which is characteristic of the (110) plane. This plane is parallel to the electron beam in crystal orientation c. (B) Schematic representation of four alternative orientations of the cellulose crystals relative to the underlying carbon support as discussed in the text.
Second, the coordinates of the individual CBMs bound on isolated Valonia crystals were determined by 3D mapping of the labels (Fig. 5). Each individual crystal was rotated at exact angles of 0° and ±45° (20), and the spatial positions of the labeled points on the microfibrils were recorded for each orientation (Fig. 5 A and B). As seen in Fig. 5A, rotation of the crystal −45° brought the labels in the middle of the crystal image whereas rotation of +45° led them further away form the edge of the crystal image. This finding is consistent with labeling at the (110) edge/plane when observing crystals with an initial orientation a (see Figs. 4B and 5B).
Figure 5.

(A) Electron micrographs showing a typical distribution of gold-labeled CBM1Cel7A on an individual Valonia cellulose crystal rotated from the initial 0° position to ±45° along its microfibril axis. The white arrows in A indicate the labels examined and B shows schematically the outcome of the rotation experiment. The black circles represent the labeled CBMs as seen on the cellulose crystals rotated in the three different angles. (C) An example of the 3D mapping of the distribution of the gold particles (filled symbols) recorded in the rotating experiment. Each symbol corresponds to one bound CBM1Cel7A on a single Valonia cellulose crystal. The symbol at the x axis indicates the location of the origin of the coordinate system used (see Materials and Methods for an explanation). (Magnifications: ×250.)
The coordinates of the labeled points were then calculated as described in Materials and Methods. Several independent fiber rotation experiments confirmed that the labeled CBMs were concentrated into two distinct groups on two edges of the crystals, rather than two broad surfaces or evenly around the crystal (Fig. 5C). The distance between the two groups of particles across the microfibril is in the range of 20–25 nm, which is in good agreement with the width of the Valonia microfibril (18 nm) (24). Because a somewhat broader (lower-left) scattering of the label was observed on one side of the crystal and somewhat tighter (upper-right) scattering on the other (Fig. 5C), it is most likely that the crystals analyzed laid in the orientations a or b than the orientation c (see Fig. 4B).
Discussion
Consistent with earlier investigations (7, 12, 25, 26), our current data confirm that tryptophan residues contribute higher energy of binding than tyrosines on a CBM binding surface. However, the effect is limited to one Trp residue; addition of two Trp residues led to no further improvement in binding.
All of the ZZ-CBM fusion proteins studied showed fully reversible binding at 4°C and retained a dynamic equilibrium when diluted. This finding is in contrast to earlier data showing that isolated Cel6A CBM1 from T. reesei and some bacterial CBMs show irreversible binding at 4°C (10, 18, 26, 27). However, when bound to crystalline cellulose, the CBM2a from Cellulomonas fimi is apparently able to diffuse laterally on the cellulose surface (28) and the tryptophans on its binding face are accessible to oxidation with N-bromosuccinimide (26). Even in the case of the Cel6A CBM1, the irreversible binding behavior seems to be borderline behavior, which can be reversed by small changes in the CBM1 structure, the binding conditions, or substrate (10). Because the intact Cel6A enzyme also exhibits partially reversible binding (29), it is possible that the presence of the ZZ-fusion partner alone is sufficient to reverse the apparently irreversible binding of the CBM1Cel6A. The role of the CBMs as a part of the often processively acting cellulases necessitates a dynamic binding behavior. Especially in the case of T. reesei cellulases naturally operating in tropical temperatures, the irreversible binding observed at 4°C may not have any biological significance at all.
CBMs specific to crystalline cellulose are found in enzymes with different roles in lignocellulose degradation. Nevertheless, with one exception (23), no specificity reflecting the activity of the catalytic module has been observed. On the contrary, EM and molecular modeling studies of intact T. reesei Cel7A (CBHI) suggested binding to the edges rather than, for example, to the ends of the crystals (30, 31).
Valonia cellulose crystals are large in size and can be treated as single crystals, which allowed detailed microscopic and crystallographic investigations of the binding sites of the CBMs. Visual inspection of the microscopic data together with the microdiffraction analysis and 3D mapping of the CBMs provide direct evidence of their preferred binding on the hydrophobic (110) plane of Valonia crystals instead of the adjacent (100) and (010) faces. Even the binding pattern earlier obtained by immuno-gold labeling and TEM on a C. fimi CBM2a (32) seems very similar to ours. Moreover, prelabeling the crystals with the antibody–ZZ-CBM complex was found to influence the orientations in which the crystals lie on the carbon support. Labeling the crystals with the antibody–ZZ-CBM complex clearly increased the incidence of the orientation c, which was not abundant in nonlabeled crystals, whereas orientation d, common with nonlabeled crystals, was only rarely observed with the labeled crystals. Because loading the (110) obtuse corner with the labeled antibody–CBM complexes could be expected to disfavor orientation d, it seems very likely that the (110) plane is the primary binding site for the CBMs.
However, because evidence of two different binding sites for some CBMs has been obtained (27), it cannot be ruled out that the initial, perhaps lower affinity, binding occurs in the (100) and (010) faces followed by tighter binding at the (110) plane as the hydrolysis proceeds. As predicted (3), this process would result in more enzyme binding per unit of cellulose on prolonged cellulose degradation, which has indeed been recently observed for T. reesei Cel6A and Cel7A (29).
Acknowledgments
We thank Pierre Beguin for the kind gift of the plasmid encoding the C. thermocellum CipA CBM3 and David Wilson for helpful comments on the manuscript. Financial support from the Knut and Alice Wallenberg Foundation, Sweden, is gratefully acknowledged.
Abbreviations
- CBM
cellulose binding module
- EM
electron microscopy
- TEM
transmission EM
- BMCC
bacterial microcrystalline cellulose
References
- 1.Linder M, Teeri T T. J Biotechnol. 1997;57:15–28. doi: 10.1016/s0168-1656(97)00088-6. [DOI] [PubMed] [Google Scholar]
- 2.Tomme P, Warren R A J, Miller R C, Kilburn D G, Gilkes N R. ACS Symp Ser. 1995;618:143–163. [Google Scholar]
- 3.Reinikainen T, Teleman O, Teeri T T. Proteins. 1995;22:392–403. doi: 10.1002/prot.340220409. [DOI] [PubMed] [Google Scholar]
- 4.Kraulis J, Clore G M, Nilges M, Jones T A, Pettersson G, Knowles J, Gronenborn A M. Biochemistry. 1989;28:7241–7257. doi: 10.1021/bi00444a016. [DOI] [PubMed] [Google Scholar]
- 5.Mattinen M L, Linder M, Teleman A, Annila A. FEBS Lett. 1997;407:291–296. doi: 10.1016/s0014-5793(97)00356-6. [DOI] [PubMed] [Google Scholar]
- 6.Reinikainen T, Ruohonen L, Nevanen T, Laaksonen L, Kraulis P, Jones T A, Knowles J K, Teeri T T. Proteins. 1992;14:475–482. doi: 10.1002/prot.340140408. [DOI] [PubMed] [Google Scholar]
- 7.Linder M, Mattinen M L, Kontteli M, Lindeberg G, Ståhlberg J, Drakenberg T, Reinikainen T, Pettersson G, Annila A. Protein Sci. 1995;4:1056–1064. doi: 10.1002/pro.5560040604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Linder M, Lindeberg G, Reinikainen T, Teeri T T, Pettersson G. FEBS Lett. 1995;372:96–98. doi: 10.1016/0014-5793(95)00961-8. [DOI] [PubMed] [Google Scholar]
- 9.Srisodsuk M, Lehtio J, Linder M, Margolles-Clark E, Reinikainen T, Teeri T T. J Biotechnol. 1997;57:49–57. doi: 10.1016/s0168-1656(97)00088-6. [DOI] [PubMed] [Google Scholar]
- 10.Carrard G, Linder M. Eur J Biochem. 1999;262:637–643. doi: 10.1046/j.1432-1327.1999.00455.x. [DOI] [PubMed] [Google Scholar]
- 11.Tormo J, Lamed R, Chirino A J, Morag E, Bayer E A, Shoham Y, Steitz T A. EMBO J. 1996;15:5739–5751. [PMC free article] [PubMed] [Google Scholar]
- 12.Sugiyama J, Vuong R, Chanzy H. Macromolecules. 1991;24:4168–4175. [Google Scholar]
- 13.McLean B W, Bray M R, Boraston A B, Gilkes N R, Haynes C A, Kilburn D G. Protein Eng. 2000;13:801–809. doi: 10.1093/protein/13.11.801. [DOI] [PubMed] [Google Scholar]
- 14.Sugiyama J, Harada H, Fujiyoshi Y, Uyeda N. Planta. 1985;166:161–168. doi: 10.1007/BF00397343. [DOI] [PubMed] [Google Scholar]
- 15.Ståhl S, Sjölander A, Nygren P A, Berzins K, Perlmann P, Uhlén M. J Immunol Methods. 1989;124:43–52. doi: 10.1016/0022-1759(89)90184-1. [DOI] [PubMed] [Google Scholar]
- 16.Linder M, Salovuori I, Ruohonen L, Teeri T T. J Biol Chem. 1996;271:21268–21272. doi: 10.1074/jbc.271.35.21268. [DOI] [PubMed] [Google Scholar]
- 17.Denman S, Xue G P, Patel B. Appl Environ Microbiol. 1996;62:1889–1896. doi: 10.1128/aem.62.6.1889-1896.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Linder M, Teeri T T. Proc Natl Acad Sci USA. 1996;93:12251–12255. doi: 10.1073/pnas.93.22.12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilkes N R, Jervis E, Henrissat B, Tekant B, Miller R C, Jr, Warren R A, Kilburn D G. J Biol Chem. 1992;267:6743–6749. [PubMed] [Google Scholar]
- 20.Imai T, Boisset C, Samejima M, Igarashi K, Sugiyama J. FEBS Lett. 1998;432:113–116. doi: 10.1016/s0014-5793(98)00845-x. [DOI] [PubMed] [Google Scholar]
- 21.Koyama M, Helbert W, Imai T, Sugiyama J, Henrissat B. Proc Natl Acad Sci USA. 1997;94:9091–9095. doi: 10.1073/pnas.94.17.9091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nilsson B, Moks T, Jansson B, Abrahmsen L, Elmblad A, Holmgren E, Henrichson C, Jones T A, Uhlén M. Protein Eng. 1987;1:107–113. doi: 10.1093/protein/1.2.107. [DOI] [PubMed] [Google Scholar]
- 23.Carrard G, Koivula A, Soderlund H, Beguin P. Proc Natl Acad Sci USA. 2000;97:10342–10347. doi: 10.1073/pnas.160216697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Revol J. Carbohydr Polym. 1982;2:123–134. [Google Scholar]
- 25.Din N, Forsythe I J, Burtnick L D, Gilkes N R, Miller R C, Jr, Warren R A, Kilburn D G. Mol Microbiol. 1994;11:747–755. doi: 10.1111/j.1365-2958.1994.tb00352.x. [DOI] [PubMed] [Google Scholar]
- 26.Bray M R, Johnson P E, Gilkes N R, McIntosh L P, Kilburn D G, Warren R A. Protein Sci. 1996;5:2311–2318. doi: 10.1002/pro.5560051117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Creagh A L, Ong E, Jervis E, Kilburn D G, Haynes C A. Proc Natl Acad Sci USA. 1996;93:12229–12234. doi: 10.1073/pnas.93.22.12229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jervis E J, Haynes C A, Kilburn D G. J Biol Chem. 1997;272:24016–24023. doi: 10.1074/jbc.272.38.24016. [DOI] [PubMed] [Google Scholar]
- 29.Palonen H, Tenkanen M, Linder M. Appl Environ Microbiol. 1999;65:5229–5233. doi: 10.1128/aem.65.12.5229-5233.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chanzy H, Henrissat B, Vuong R. FEBS Lett. 1984;172:193–197. [Google Scholar]
- 31.Henrissat B, Vigny B, Buleon A, Perez S. FEBS Lett. 1988;231:177–182. [Google Scholar]
- 32.Gilkes N R, Kilburn D G, Miller R C, Jr, Warren R A, Sugiyama J, Chanzy H, Henrissat B. Int J Biol Macromol. 1993;15:347–351. doi: 10.1016/0141-8130(93)90052-n. [DOI] [PubMed] [Google Scholar]