

Abstract
Previous studies in Creutzfeldt–Jakob disease (CJD) have shown that myeloid cells in the periphery as well as derivative microglial cells in the brain are infectious. Microglia can show an activated phenotype before prion protein (PrP) pathology is detectable in brain, and isolated infectious microglia contain very little PrP. To find whether a set of inflammatory genes are significantly induced or suppressed with infection, we analyzed RNA from isolated microglia with relevant cDNA arrays, and identified ≈30 transcripts not previously examined in any transmissible spongiform encephalopathy. This CJD expression profile contrasted with that of uninfected microglia exposed to prototypic inflammatory stimuli such as lipopolysaccharide and IFN-γ, as well as PrP amyloid. These findings underscore inflammatory pathways evoked by the infectious agent in brain. Transcript profiles unique for CJD microglia and other myeloid cells provide opportunities for more sensitive preclinical diagnoses of infectious and noninfectious neurodegenerative diseases.
Most studies in Creutzfeldt–Jakob disease (CJD) and scrapie have emphasized neurons as the principal site of agent replication, based in part on high neuronal expression of PrP, a host protein required for infection and pathogenesis (1). During the course of infection, PrP adopts pathological properties, often accumulating in the brain as amyloid aggregates. Nonneuronal brain cells including microglia are assumed to play only a reactive role in disease, releasing neurotoxic substances in response to extracellular PrP fibrils (2). Alternatively, microglia may be a carrier or a productive factory for at least some strains of these infectious agents (3). As in other viral infections of the brain, infectivity can be recovered from various immune cell types outside the nervous system, including lymphocytes, macrophages, and dendritic cells (4–6). Recently, we have shown that microglia, the myeloid cells that can migrate to the brain from the periphery, contain substantial levels of CJD infectivity. Agent titers in these purified microglia are surprisingly close to starting brain homogenates replete with neuronal components (7). We decided that further studies of microglia in CJD could reveal hidden aspects of the agent life cycle and illuminate new targets for diagnosis and therapy.
The CJD agent induces an activated phenotype in microglia, including alterations in morphology and gene expression (3, 7). This activation may result from (i) direct interaction of microglia with the agent itself, (ii) a secondary response to soluble proinflammatory molecules, or (iii) a direct effect of accumulating pathologic PrP. To develop a molecular signature of the infectious state within microglia, we compared mRNA expression profiles of microglia isolated from CJD and uninfected mouse brains and evaluated normal microglia activated by relevant stimuli. These studies identified patterns of gene expression that were unique to CJD microglia and not readily attributable to nonspecific inflammatory activation or responses to PrP amyloid.
Methods
Microglia and Astrocyte Cultures.
Microglia (≈95% CD11b+ cells) were isolated as described (7) from the brains of normal mice or mice infected with the Fukuoka (FU) strain of CJD (5). For stimulation experiments, 100 ng/ml bacterial lipopolysaccharide (LPS) (serotype 055:B5, Sigma), 100 ng/ml IFNγ (100 units/ng, BioSource International, Camarillo, CA), or PrP (see below) was included in the medium during the 16- to 18-h incubation. Neonatal astrocytes (>98% pure) were also prepared as described (7).
cDNA Expression Array Analysis.
Total RNA was isolated from cultures or whole brain by using Trizol (Invitrogen). First-strand cDNA was labeled with 32P and hybridized to Mouse 1.2 II expression arrays according to the manufacturer's instructions (CLONTECH), and exposed at −70°C to Biomax MS films and intensifying screens (Kodak). Films in the linear range of exposure were quantitated with NIH IMAGE without manipulation. For quantitative comparisons, signals were normalized for differences in original RNA quantity based upon the average intensity of GAPDH, which was located in multiple spots on the array.
Semiquantitative RT-PCR Analysis.
RT-PCR from 500 ng of total RNA was performed as described (7). Cycle numbers for detection within the exponential phase, determined empirically for each transcript, are listed along with annealing temperatures and primer sequences in Table 1, which is published as supporting information on the PNAS web site, www.pnas.org. Biotinylated PCR products were separated by agarose gel electrophoresis and visualized by chemiluminescence, as described (7).
PrP Preparations.
Digestion of CJD brain homogenates (10 mg equivalent) with proteinase K and confirmation of PrP-res by Western blotting was performed as described (8). Homogenates from uninoculated mice were processed in parallel as a control. PrP fibrils or control material from normal brains were diluted into culture medium to ≈2 ng/ml, based on estimates of 10 μg of PrP fibrils per gram of brain homogenate. Freshly isolated microglia were treated for 16–18 h before RNA collection.
Results
RNA samples for array analysis were derived from purified microglia allowed to recover for 16 h in vitro, precluding any spurious changes associated with either acute stress or prolonged artificial culture conditions. We chose cDNA expression arrays that, although modest in size (1,174 sequences), included named genes with well characterized functions related to myeloid cell inflammatory responses. These membranes also offered increased sensitivity of radioactive vs. fluorescent detection, and allowed us to use the small amounts of RNA derived from limited numbers of microglial cells, while avoiding amplification techniques that might skew representation of transcripts in a heterogeneous RNA population.
Initial comparison of RNA from normal brain microglia, whole brain, and normal astrocyte cultures revealed mRNA expression profiles that were obviously different and largely nonoverlapping (Fig. 1A). To visualize differences in expression easily, all autoradiograms are pseudocolored such that normal unstimulated microglia were represented as a red reference signal, and experimental samples for comparison were displayed as green signals. With superimposition, equivalent signals thus appear yellow. Without cell purification, the distinctive microglial expression patterns are obscured by the complexity of the many other cell types in brain. As expected, comparison of normal microglia under basal conditions, and after exposure to activating stimuli, showed fewer differences than between normal microglia and astrocytes (compare Fig. 1 A vs. B). Nevertheless, robust differences were clearly apparent between stimulated and unstimulated microglia (for example, arrowheads in Fig. 1B Upper). Likewise, microglia isolated from end-stage CJD brain exhibited profound differences relative to normal brain microglia, as seen by simple superimposition of autoradiograms in Fig. 1B (Upper Left, N-Mg/CJ-Mg). An example of a completely CJD-specific transcript is also obvious as a green signal seen in only one of the six panels in Fig. 1 (white arrow).
Figure 1.

Specific hybridization patterns on expression arrays. Representative patterns of normal isolated microglia (red reference signal) with other experimental samples (green) superimposed. (A) Comparisons of normal microglia (N-Mg) to normal total brain (N-Br) or normal astrocytes (N-Ast) reveal little or no coincident expression signals (yellow). (B) Microglia from CJD brain (CJD-Mg) and normal microglia are shown as a composite (Upper Left). For comparison, normal microglia treated with LPS (LPS-Mg) or IFNγ (IFNγ-Mg) are shown in other panels as labeled. Comparisons and patterns were all reproducible in independent experiments. (Lower Right) Comparisons of normal microglia treated with PrP amyloid (N-Mg + PrP-res; green) to microglia treated with digested normal brain with no PrP-res (N-Mg control; red). Arrows show a CJD-specific transcript (a green signal seen in only one of the six panels). Arrowheads indicate a microglial-enriched transcript that is slightly up-regulated in CJD microglia but down-regulated with LPS.
Fig. 2 summarizes the quantitative analysis of differentially expressed transcripts in microglia from CJD brain, using the same pseudocoloring scheme. Each data point is representative of multiple experiments typically performed in triplicate; we found the array hybridization patterns were highly reproducible in more than eight experiments with normal untreated microglia. Various mRNAs involved in inflammatory functions were substantially increased (5- to 20-fold) in CJD microglia. These transcripts included IL-1, enzymes responsible for the macrophage respiratory burst (gp91phox and p22phox), and several other leukocytic cell-surface molecules. The complement cascade components C1q, properdin, and factor H were also significantly up-regulated in CJD microglia, as was the complement receptor subunit CD18. Complement components have previously been linked to scrapie pathogenesis in the periphery (9), but the current results additionally indicate that alternative complement pathways are activated in the nervous system. This finding may help explain why a deficiency in some complement components has no effect on scrapie pathogenesis after intracerebral inoculation (9), and perturbation of more complement pathways may also result in little demonstrable effect in a complex disorder like CJD.
Figure 2.

Quantitative microglial profiles, where red represents the control microglia. Densitometry results from original (nonpseudocolored) autoradiograms are expressed in terms of fold induction (green) or fold suppression (red), according to the scale (Bottom Right). Each data point is representative of at least two completely independent experiments, using RNAs derived from separate microglial preparations, hybridized to arrays from different lot numbers. Genes are grouped according to function and sorted in order from most induced to most suppressed in CJD microglia.
The arrays highlighted alterations in lipid and cholesterol metabolism that have been linked to membrane function and acute inflammatory reactions. Cholesterol-rich lipid rafts support the life cycle of several viruses (10). In CJD, most infectivity associates with synaptosomal membranes (11), and membrane cholesterol content has been reported to influence PrP changes (12). CJD microglia also exhibited increased transcript levels for several mediators of lipid uptake and transport (Fig. 2), which could have eventual consequences for membrane physiology. Lipoprotein lipase, CD36, and CD68 promote cholesterol uptake in the form of low-density lipoproteins (LDL). In contrast, the 12-fold increase in apolipoprotein C-I (apo C-I) would be expected to inhibit both LDL uptake and lipoprotein lipase activity, and apo C-I has been linked to other neurodegenerative disorders such as Alzheimer's disease (13). Serum amyloid A3 (SAA3), which was also increased 12-fold in CJD microglia, becomes a major constituent of high-density lipoprotein during inflammatory reactions, and SAA isoforms can affect cholesterol storage and release (14). These mRNA changes in CJD microglia may reflect homeostatic mechanisms to cope with membrane pathology. On the other hand, lipid processing may prove to be important in early agent interactions.
To determine whether the activation of CJD microglia resembled a standard immune response, we examined the expression profile of normal microglia that had been exposed to LPS, a prototypical inflammatory stimulus. Although LPS treatment increased mRNAs for IL-1 and several other proinflammatory genes, most of the transcripts altered in CJD microglia were unaffected by LPS (Fig. 1B and Fig. 2). Indeed, levels of C1q, the colony stimulating factor-1 receptor, and the growth-promoting factor granulin were regulated in opposite directions in LPS and CJD microglia.
CJD infection of microglia also induced expression of a range of molecules linked to IFN signaling. Because a few other microglial transcripts up-regulated in scrapie brain are also known to be IFNγ-inducible (15), we asked whether the expression profile of CJD microglia could be mimicked by IFNγ treatment of normal cells. All of the IFN-related transcripts up-regulated in CJD microglia were also increased in normal microglia exposed to IFNγ (Fig. 2). However, Ia-associated invariant chain and the costimulatory molecule CD86, two molecules important for MHC class II-dependent antigen presentation, were strongly induced in microglia treated with IFNγ, but not in CJD microglia. Therefore, other factors in CJD must be activating a program that is different from that elicited by IFNγ. Because the effects of α- and β-interferons overlap partially with those of IFNγ, we further examined transcript levels for these molecules with semiquantitative RT-PCR. IFNγ, IFNβ, and various IFNα family members were undetectable at the RNA level on our arrays or by RT-PCR (Fig. 3A and data not shown). Other factors, perhaps even the CJD agent itself, may be activating similar transcriptional programs via an IFNβ-independent mechanism.
Figure 3.

Confirmation of specific microglial patterns by semiquantitative RT-PCR. Representative blots are shown for interferons (A), transcripts specifically regulated in CJD microglia (B), unchanged transcripts (C), transcripts showing the same pattern in both CJD and LPS-treated microglia (D), and transcripts similarly regulated in CJD and IFNγ-treated microglia (E).
The most potently induced transcript in CJD microglia was that of lysozyme M, an important bacteriolytic enzyme expressed in activated macrophages. Lysozyme M has previously been identified as an up-regulated gene in CJD brain (16) and Sindbis virus infections of the nervous system (17). In CJD infection, this up-regulation likely reflects a general increase in lysosomal proteolytic activity, because it was accompanied by the induction of several lysosomal proteases, with concomitant down-regulation of protease inhibitors (Fig. 2). These other transcripts may reflect a response to pathologic PrP in the brain or a reaction to less specific neurodegenerative changes. Additionally, pathologic PrP accumulates in lysosomes in scrapie-infected neuroblastoma cells (18) and can be detected in subsets of microglia and other myeloid cells (3, 19). Therefore, it was pertinent to test the direct effects of pathologic PrP on microglia.
High concentrations of synthetic fibrillary or amyloid peptides, including those related to pathologic PrP and Alzheimer's disease, have been shown to activate inflammatory signaling pathways in microglia (20, 21). To determine whether a subset of the changes in CJD microglia was caused by pathologic PrP, we stimulated microglia with preparations enriched in PrP that were resistant to proteolysis (PrP-res; Fig. 4). Electron microscopy of such PrP-res preparations has revealed aggregates of typical PrP fibrils (22). Remarkably, the vast majority of altered mRNAs in CJD microglia were unaffected or oppositely regulated in microglia treated with PrP-res at high but reasonably physiological concentrations of ≈2 ng/ml. Nevertheless, this dose of PrP-res was sufficient to alter the expression patterns of ≈20 genes by more than twofold (data not shown). In contrast, other studies have used synthetic PrP peptides at ≈150 μg/ml (80 μM) to elicit neuronal and microglial changes. This concentration is at least 1,000-fold greater than the amyloid in our experiments, and we have some concern about the meaning of the effects caused by such high peptide doses. Only three transcripts (CD48, CD84, and a 47-kDa IFN-responsive protein) were increased in both CJD microglia and microglia exposed to PrP-res (Fig. 2). Furthermore, the CJD expression profile was distinct from that of adult microglia treated with Alzheimer's disease β-amyloid peptides (21). For example, β-amyloid treatment of microglia increased fos-related antigen-1 and decreased CD86 mRNA levels, the opposite of their patterns in microglia from CJD brain. Together, all these results suggest that neither pathologic PrP itself nor an amyloid protein structure was the principal cause of the many changes we found in CJD microglia.
Figure 4.
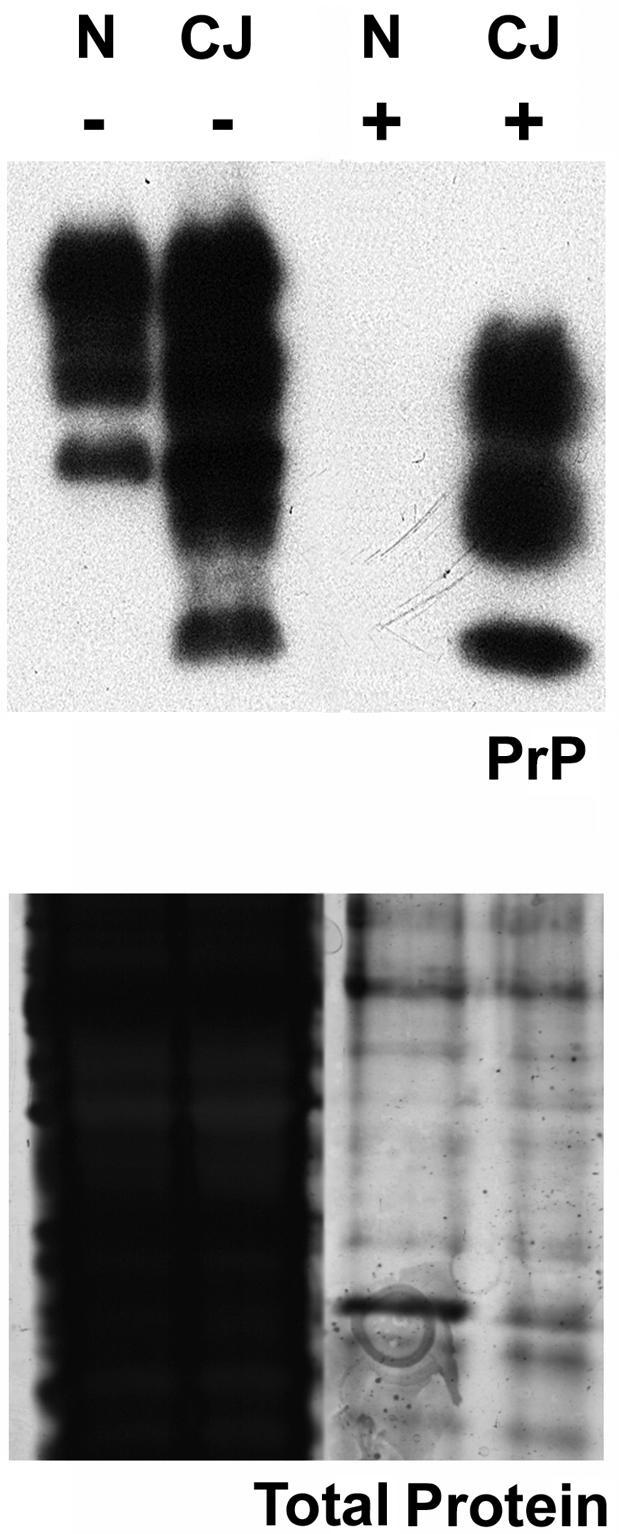
Control and PrP-res preparations for treatment of microglia. Homogenates from normal brain (N) or end-stage CJD brain (CJ) were examined on polyacrylamide gels either before (−) or after (+) treatment with proteinase K and precipitation of PrP-res. Samples were analyzed for PrP by Western blotting (Upper) or total protein content by silver staining of polyacrylamide gels (Lower). Equivalent amounts of starting brain homogenates were loaded in each lane. The control preparations made from normal brain contain residual protein fragments similar to those from CJD brain, but show no PrP-res. These parallel preparations made with the same solutions were used to discriminate effects of PrP-res on microglia in Figs. 2 and 3.
Because we identified a set of transcripts that were highly CJD-specific, we used semiquantitative RT-PCR to confirm further the array patterns. Lysozyme M and properdin were specifically induced in CJD microglia, as was the CD72 antigen (Fig. 3B). Because CD72 influences the inflammatory signaling properties of macrophages and dendritic cells (23), we also confirmed the up-regulation in CJD microglia of three closely related molecules (CD48, CD84, and CD229) that influence communication between inflammatory cells (24). The regulation of these transcripts could indicate cell interactions in the nervous system representing a nascent or abortive immune response. Because CD48 and CD84 were also up-regulated to a similar extent in cells exposed to PrP-res, these changes could be part of an initial recognition or ligand binding of pathologic PrP by microglia.
CJD-infected microglia also showed specific suppression of six transcripts that were unaltered by LPS, IFNγ, or PrP-res treatment (Fig. 2). RT-PCR of the two mRNAs with the greatest down-regulation confirmed specific inhibition in CJD microglia (Fig. 3B), whereas other transcripts were unaltered in all conditions (Fig. 3C) or were similarly changed in CJD and LPS-treated microglia (Fig. 3D). The 10-fold decrease of plasminogen activator inhibitor type 2 (PAI-2) would be expected to increase fibrinolysis, which could in turn facilitate microglial migration through extracellular matrix. Previously identified increases in the chemotactic ligands and receptors IP-10, BLC/CXCL13, and CCR5 in CJD microglia (7, 25) could also support the enhanced trafficking of CJD-infected microglia and related myeloid cells (5).
The reproducible down-regulation of the 65-kDa FK-506 binding protein (FKBP65) may be linked to accumulation of PrP-res. FKBP65 is thought to regulate proper protein folding in the endoplasmic reticulum (ER) via cis-trans proline isomerization (26), and loss of this function could contribute to ER stress and PrP aggregation. As noted above, PrP-res collects in some migrating microglia in CJD brain (3). The protease inhibitor cystatin F may also contribute to amyloid formation (27), including that of PrP. Notably, cystatin F was the transcript most potently induced in both CJD microglia and IFNγ-treated microglia (Fig. 3E). However, cystatin family members are well known to inhibit viral replication in a variety of systems (28, 29). We suspect that cystatin F is part of an underappreciated host defense mechanism in CJD.
We were surprised to find reduced mRNA levels for nerve growth factor (NGF) in CJD microglia and in IFNγ-treated cells (Fig. 3E). This response contrasts with models of nervous system injury, in which microglial cells often increase NGF expression (30). Thus, neurodegeneration itself should have enhanced NGF in CJD microglia. Stimulation of microglia with LPS also increased NGF levels here, as in earlier studies (31). Interestingly, treatment of microglia with PrP fibrils, but not normal parallel brain preparations, similarly increased NGF mRNA. The microglial expression profiles after LPS or PrP-res were very different, indicating that contamination of the PrP-res material with exogenous LPS was highly unlikely. Furthermore, untreated microglia showed few differences from cells treated with normal brain, again indicating undetectable LPS contamination of solutions. Nevertheless, one intriguing possibility is that the PrP-res preparations from CJD brains contain some endogenous LPS-like component. Regardless, the divergent profiles of CJD microglia and normal microglia exposed to pathologic PrP again underscore the differences between these two conditions. Because NGF suppresses MHC class II expression in microglia (32), the NGF down-regulation in CJD microglia could help relieve the inhibition of inflammatory responses to the agent.
Discussion
The current results reveal a sophisticated pattern of responses in isolated microglia with high levels of the CJD agent despite low PrP expression (7). They also underscore the central role of microglia in CJD, with changes that cannot be ascribed to either pathologic PrP or neurodegeneration. Hence, some of these alterations may occur very early in CJD infection. The PrP-res and LPS comparisons here further demonstrate a distinctive CJD profile that obviously differ from more simplified and stereotypic secondary responses. The mRNAs identified in our experiments, moreover, do not rely on PrP-res, which is often undetectable early in the course of CJD infection. These studies also emphasize several pathways that have been associated with host IFN responses to neurotrophic viruses and double-stranded RNA (17, 33). Although previous experiments have failed to find active IFN in scrapie (34), up-regulation of IFN-related genes in CJD might involve other factors. This latter type of IFN-independent transcriptional induction has already been noted in monocytes infected with HIV (35); it can also be caused by direct interactions between viral proteins and host transcriptional machinery (36). Conventional viral elements could conceivably be involved in the transcriptional program observed in CJD microglia, given the continued failure of PrP-res to elicit all of the changes associated with infection. Many studies have shown little correlation between infectious titers and PrP-res during fractionation of brain as well as in many different in vivo models (8, 37). Moreover, despite numerous efforts, no form of PrP itself has ever reproduced infection in any system, including transgenic mice and PrP-res amplified >100-fold in vitro.
Our studies further implicate microglia as a nexus for IFN-related host inflammatory responses, spread of CJD infectivity, and PrP amyloid formation. In CJD and scrapie, approaches based on PrP alone have not been sufficient to accurately assess infectivity, and PrP pathology may be a consequence of infection rather than the agent itself. The current experiments identified ≈30 significantly and repeatedly altered (>4-fold) transcripts that have not been previously examined in CJD or scrapie. Six of these mRNAs were specifically regulated in CJD microglia, and represent new markers for myeloid cells that could serve as diagnostic and therapeutic targets both peripherally as well as in the brain. We are currently examining these myeloid cell transcript profiles in early asymptomatic disease, when diagnosis has the greatest potential for protecting human and animal health. Additionally, regulation of these genes may also provide robust comparative assays in Alzheimer's disease and other neurodegenerative disorders.
Supplementary Material
Acknowledgments
These studies were supported by National Institutes of Health Grants NS034569 and NS012674. The authors have no competing conflicts of interest.
Abbreviations
- CJD
Creutzfeldt–Jakob disease
- LPS
bacterial lipopolysaccharide
- NGF
nerve growth factor
References
- 1.Büeler H, Aguzzi A, Sailer A, Greiner R-A, Autenried P, Auget M, Weissmann C. Cell. 1993;73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 2.Brown D R, Schmidt B, Kretzschmar H A. Nature. 1996;380:345–347. doi: 10.1038/380345a0. [DOI] [PubMed] [Google Scholar]
- 3.Maneulidis L, Fritch W, Xi Y G. Science. 1997;277:94–98. doi: 10.1126/science.277.5322.94. [DOI] [PubMed] [Google Scholar]
- 4.Raeber A J, Klein M A, Frigg R, Flechsig E, Aguzzi A, Weissmann C. EMBO J. 1999;18:2702–2706. doi: 10.1093/emboj/18.10.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manuelidis L, Zaitsev I, Koni P, Lu Z Y, Flavell R A, Fritch W. J Virol. 2000;74:8614–8622. doi: 10.1128/jvi.74.18.8614-8622.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aucouturier P, Geissmann F, Darnotte D, Saborio G P, Meeker H C, Kascsak R, Carp R I, Wisniewski T. J Clin Invest. 2001;108:703–708. doi: 10.1172/JCI13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker C A, Martin D, Manuelidis L. J Virol. 2002;76:10905–10913. doi: 10.1128/JVI.76.21.10905-10913.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manuelidis L, Fritch W. Virology. 1996;216:46–59. doi: 10.1006/viro.1996.0033. [DOI] [PubMed] [Google Scholar]
- 9.Mabbot N A, Bruce M E, Botto M, Walport M J, Pepys M B. Nat Med. 2001;7:485–487. doi: 10.1038/86562. [DOI] [PubMed] [Google Scholar]
- 10.Campbell S M, Crowe S M, Mak J. J Clin Virol. 2001;22:217–227. doi: 10.1016/s1386-6532(01)00193-7. [DOI] [PubMed] [Google Scholar]
- 11.Manuelidis L, Manuelidis E E. Banbury Rep. 1983;15:399–412. [Google Scholar]
- 12.Taraboulous A, Scott M, Semenov A, Avrahami D, Lazlo L, Prusiner S B, Avrahami D. J Cell Biol. 1995;129:121–132. doi: 10.1083/jcb.129.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petit-Turcotte C, Stohl S M, Beffert U, Cohn J S, Aumont N, Tremblay M, Dea D, Yang L, Poirier J, Shachter N S. Neurobiol Dis. 2001;8:953–963. doi: 10.1006/nbdi.2001.0441. [DOI] [PubMed] [Google Scholar]
- 14.Ely S, Bonatesta R, Ancsin J B, Kindy M, Kisilevsky R. Amyloid. 2001;8:169–181. doi: 10.3109/13506120109007360. [DOI] [PubMed] [Google Scholar]
- 15.Riemer C, Queck I, Simon D, Kurth R, Baier M. J Virol. 2000;74:10245–10248. doi: 10.1128/jvi.74.21.10245-10248.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kopacek J, Sakaguchi S, Shigematsu K, Nishida N, Atarashi R, Nakaoke R, Moriuchi R, Niwa M, Katamine S. J Virol. 2000;74:411–417. doi: 10.1128/jvi.74.1.411-417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnston C, Jiang W, Chu T, Levine B. J Virol. 2001;75:10431–10445. doi: 10.1128/JVI.75.21.10431-10445.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKinley M P, Taraboulous A, Kenaga L, Serban D, Stieber A, DeArmond S J, Prusiner S B, Gonatas N. Lab Invest. 1991;65:622–630. [PubMed] [Google Scholar]
- 19.Radebold K, Chernyak M, Martin D, Manuelidis L. BMC Infect Dis. 2001;1:20. doi: 10.1186/1471-2334-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peyrin J M, Lasmezas C I, Haik S, Tagliavini F, Salmona M, Williams A, Richie D, Deslys J P, Dormont D. NeuroReport. 1999;10:723–729. doi: 10.1097/00001756-199903170-00012. [DOI] [PubMed] [Google Scholar]
- 21.Walker D G, Lue L F, Beach T G. Neurobiol Aging. 2001;22:957–966. doi: 10.1016/s0197-4580(01)00306-2. [DOI] [PubMed] [Google Scholar]
- 22.Merz P A, Somerville R A, Wisniewski H M, Manuelidis L, Manuelidis E E. Nature. 1983;306:474–476. doi: 10.1038/306474a0. [DOI] [PubMed] [Google Scholar]
- 23.Kumanogoh A, Kikutani H. Trends Immunol. 2001;22:670–676. doi: 10.1016/s1471-4906(01)02087-7. [DOI] [PubMed] [Google Scholar]
- 24.Sayos J, Martin M, Chen A, Simarro M, Howie D, Morra M, Engel P, Terhorst C. Blood. 2001;97:3867–3874. doi: 10.1182/blood.v97.12.3867. [DOI] [PubMed] [Google Scholar]
- 25.Baker C A, Lu Z Y, Zaitsev I, Manuelidis L. J Virol. 1999;73:5089–5097. doi: 10.1128/jvi.73.6.5089-5097.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davis E C, Broekelmann T J, Ozawa Y, Mecham R P. J Cell Biol. 1998;140:295–303. doi: 10.1083/jcb.140.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Staniforth R A, Giannini S, Higgins L D, Conroy M J, Hounslow A M, Jerala R, Craven C J, Waltho J P. EMBO J. 2001;20:4774–4781. doi: 10.1093/emboj/20.17.4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collins A R, Grubb A. Oral Microbiol Immunol. 1998;13:59–61. doi: 10.1111/j.1399-302X.1998.tb00753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bjorck L, Grubb A, Kjellen L. J Virol. 1990;64:941–943. doi: 10.1128/jvi.64.2.941-943.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krenz N R, Weaver L C. J Neurochem. 2000;74:730–739. doi: 10.1046/j.1471-4159.2000.740730.x. [DOI] [PubMed] [Google Scholar]
- 31.Heese K, Fiebich B L, Bauer J, Otten U. Glia. 1998;22:401–407. doi: 10.1002/(sici)1098-1136(199804)22:4<401::aid-glia9>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 32.Neumann H, Misgeld T, Matsumuro K, Wekerle H. Proc Natl Acad Sci USA. 1998;95:5779–5784. doi: 10.1073/pnas.95.10.5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taniguchi T, Takaoka A. Curr Opin Immunol. 2002;14:111–116. doi: 10.1016/s0952-7915(01)00305-3. [DOI] [PubMed] [Google Scholar]
- 34.Gresser I, Maury C, Chandler R L. J Gen Virol. 1983;64:1387–1389. doi: 10.1099/0022-1317-64-6-1387. [DOI] [PubMed] [Google Scholar]
- 35.Baca L M, Genis P, Kalvakolanu D, Sen G, Meltzer M S, Zhou A, Silverman R, Gendelman H E. J Leukoc Biol. 1994;55:299–309. doi: 10.1002/jlb.55.3.299. [DOI] [PubMed] [Google Scholar]
- 36.tenOever B R, Servant M J, Grandvaux N, Lin R, Hiscott J. J Virol. 2002;76:3659–3669. doi: 10.1128/JVI.76.8.3659-3669.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manuelidis L, Sklaviadis T, Akowitz A, Fritch W. Proc Natl Acad Sci USA. 1995;92:5124–5128. doi: 10.1073/pnas.92.11.5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.