

Abstract
For many diseases, mediation of pathogenesis by nitric oxide (NO) has been suggested. In this study, we explored NO-induced viral pathogenesis with a focus on nucleic acid damage as evidenced by 8-nitroguanosine formation in vivo. Wild-type mice and littermate mice deficient in inducible NO synthase (iNOS) were infected with influenza or Sendai virus. Formation of 8-nitroguanosine in virus-infected lungs was assessed immunohistochemically with an antibody specific for 8-nitroguanosine. Extensive nitration of RNA either treated with peroxynitrite or obtained from cultured RAW 264 cells expressing iNOS was readily detected by this antibody. Strong 8-nitroguanosine immunostaining was evident primarily in the cytosol of bronchial and bronchiolar epithelial cells of virus-infected wild-type mice but not iNOS-deficient mice. This staining colocalized with iNOS immunostaining in the lung. 8- Nitroguanosine staining disappeared after addition of exogenous authentic 8-nitroguanosine during the antibody reaction and after pretreatment of tissues with sodium hydrosulfite, which reduces 8-nitroguanosine to 8-aminoguanosine. NO was generated in excess in lungs of wild-type mice but was eliminated in iNOS-deficient mice after virus infection; this result also correlated well with formation of 8-nitroguanosine and 3-nitrotyrosine. One consequence of the lack of iNOS expression was marked improvement in histopathological changes in the lung and the lethality of the infection without effects on cytokine responses and viral clearance. It is intriguing that 8-nitroguanosine markedly stimulated superoxide generation from cytochrome P450 reductase and iNOS in vitro. The present data constitute a demonstration of 8-nitroguanosine formation in vivo and suggest a potential role for NO-induced nitrative stress in viral pathogenesis.
Nitric oxide (NO) has been suggested to be involved in the pathogenesis of a wide variety of diseases including various microbial infections, inflammatory and neurodegenerative diseases, and cancer (1–3). This NO is generated in excess by inducible NO synthase (iNOS). Although host defense and cytoprotective functions of NO have been demonstrated (4–8), accumulated evidence indicates that NO-derived reactive nitrogen-oxide species such as peroxynitrite (ONOO−) and nitrogen dioxide (NO2) also have a pathogenic potential in various diseases (3, 9–11).
The lethal effect of virus-induced pneumonia in mice is mediated in part by host responses rather than being a direct cytopathic result of viral replication in the lung (12). In fact, the pathogenesis of influenza virus can be attributed to the cytotoxic effect of oxygen radicals such as superoxide anion (O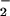 ) (13, 14). In addition, our previous study indicated that both NO and O
) (13, 14). In addition, our previous study indicated that both NO and O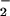 were produced in excess in an influenza model, in parallel with the development of pneumonia, and that pharmacological inhibition of NOS with Nω-monomethyl-L-arginine led to remarkable improvement in the pathological effects of the virus infection (15). More recently, we found that overproduction of NO by iNOS accelerated viral mutation in murine pneumonia caused by a recombinant Sendai virus (16). These data suggest that NO may play an important role in viral pathogenesis, but the molecular mechanism of NO toxicity occurring during virus infections is not fully understood.
were produced in excess in an influenza model, in parallel with the development of pneumonia, and that pharmacological inhibition of NOS with Nω-monomethyl-L-arginine led to remarkable improvement in the pathological effects of the virus infection (15). More recently, we found that overproduction of NO by iNOS accelerated viral mutation in murine pneumonia caused by a recombinant Sendai virus (16). These data suggest that NO may play an important role in viral pathogenesis, but the molecular mechanism of NO toxicity occurring during virus infections is not fully understood.
The chemical and biological activities of NO are affected greatly by concomitantly formed reactive oxygen species, particularly O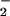 and hydrogen peroxide, via production of reactive nitrogen oxides, e.g., peroxynitrite and nitrogen dioxide (9, 10, 17). The pathophysiological action of reactive nitrogen oxides is quite important in the pathogenesis of virus infection, because these molecular species are known to be not only oxidants but also potent nitrating agents for proteins, nucleic acids, and unsaturated membrane lipids (9, 10, 17–21). In this context the genotoxicity of nitrogen oxides is currently of great interest, because reactive nitrogen oxides formed endogenously during infections are highly mutagenic for both invading viruses and hosts (12, 16), and cause cell and tissue injury in the host.
and hydrogen peroxide, via production of reactive nitrogen oxides, e.g., peroxynitrite and nitrogen dioxide (9, 10, 17). The pathophysiological action of reactive nitrogen oxides is quite important in the pathogenesis of virus infection, because these molecular species are known to be not only oxidants but also potent nitrating agents for proteins, nucleic acids, and unsaturated membrane lipids (9, 10, 17–21). In this context the genotoxicity of nitrogen oxides is currently of great interest, because reactive nitrogen oxides formed endogenously during infections are highly mutagenic for both invading viruses and hosts (12, 16), and cause cell and tissue injury in the host.
In the present study we sought to elucidate the mechanism of the deleterious effect of NO in pneumotropic virus infections with particular attention on the strong nitrating potential of reactive nitrogen oxides, which may induce nitrative stress in virus-infected hosts. We analyzed in vivo guanosine nitration, i.e., 8-nitroguanosine formation, and the pathological consequences of NO production during virus infections by using iNOS-deficient and wild-type littermate mice infected with influenza or Sendai virus. We explored the biochemical function of 8-nitroguanosine in terms of its unique redox activity affecting NADPH-dependent reductases including NADPH-cytochrome P450 reductase (P450 reductase) and iNOS to produce O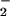 . Our results suggest that nitrative stress occurs during pneumotropic virus infections, as evidenced by 8-nitroguanosine formation and its potent O
. Our results suggest that nitrative stress occurs during pneumotropic virus infections, as evidenced by 8-nitroguanosine formation and its potent O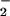 -generating activity, and is likely to contribute in a critical way to viral pathogenesis.
-generating activity, and is likely to contribute in a critical way to viral pathogenesis.
Materials and Methods
Animals and Production of Viral Pneumonia.
Heterozygous iNOS-deficient mice (iNOS+/−) were produced by mating homozygous iNOS−/− mice (The Jackson Laboratory) with their wild-type counterparts (iNOS+/+) in our laboratory. Littermates bred from the same iNOS+/− parents were used throughout the study. Influenza virus A/Kumamoto/Y5/67(H2N2) and Sendai virus Z strain were administered to 4-week-old male mice by inhalation of viral suspension at 2 × LD50, and virus yield in lungs was quantified via a plaque-forming assay (15, 16).
Synthesis of 8-Nitroguanosine.
8-Nitroguanosine was prepared from 8-bromoguanosine (Wako Pure Chemical, Osaka) by nucleophilic substitution with nitrite. 8-Bromoguanosine was reacted with sodium nitrite dissolved in anhydrous dimethyl sulfoxide followed by incubation at 70°C for 3 h. 8-Nitroguanosine thus produced was purified by reverse-phase high-performance liquid chromatography (HPLC). The purified 8-nitroguanosine was identified by its absorption spectrum as well as its molecular mass (327 Da). The yield of 8-nitroguanosine was 10–20% of the starting material (8-bromoguanosine).
Production of Anti-8-Nitroguanosine Antibody.
To obtain the conjugate used to raise the antibody, 8-nitroguanosine was conjugated to BSA (Sigma–Aldrich) via periodate oxidation according to the procedure of Erlanger and Beiser (22) with slight modifications. In brief, 8-nitroguanosine was treated with sodium periodate, resulting in its conjugation with BSA via the ribose ring that was split by periodate. 8-Nitroguanosine incorporated into the BSA conjugate was quantified, after acid hydrolysis (0.1 M HCl, 30 min, 100°C), by using its molar extinction coefficient (ɛ400, 9,144 M−1⋅cm−1) (23). The average number of 8-nitroguanosine nucleosides conjugated to BSA was 6.2 per 1 mol of BSA.
The polyclonal anti-8-nitroguanosine antibody was raised in rabbits by s.c. administration of the 8-nitroguanosine–BSA conjugate (20 μg) with Freund's complete adjuvant. A booster dose of the same antigen plus Freund's incomplete adjuvant was given four times every 2 weeks. The specific polyclonal IgG anti-8-nitroguanosine antibody was purified by use of a series of affinity chromatographic procedures including protein A- coupled Cellulofine (Seikagaku Kogyo, Tokyo) and 8- nitroguanosine-conjugated Cellulofine. Putative contamination with anti-BSA and antiguanosine antibodies was eliminated by means of BSA- and guanosine-coupled Cellulofine.
Characterization of Anti-8-Nitroguanosine Antibody.
The antibody was incubated in 96-well microtiter plates coated with the 8-nitroguanosine–BSA conjugate in the presence or absence of various nucleosides, and the antibody bound with the conjugate was detected by using peroxidase-labeled anti-rabbit IgG antibody with 1,2-phenylenediamine dihydrochloride as a substrate. For slot blot analysis, total RNA, obtained from CV-1 cells via an RNA-extraction kit (Purescript, Gentra Systems), was treated with bolus additions (three times) of peroxynitrite (2 mM). Total RNA was also extracted from cultured RAW 264 cells that had been stimulated or unstimulated with lipopolysaccharide (10 μg/ml) and a murine IFN-γ (100 units/ml) for 24 h as described (24). After extracted RNAs were denatured with 17.5% (vol/vol) formaldehyde and 50% formamide in 20 mM Mops buffer (pH 7.0) containing 5 mM sodium acetate and 1 mM EDTA, they were adsorbed onto Hybond-N+ membrane (Amersham Pharmacia) by using a slot blot apparatus. The RNA band that reacted immunologically with anti-8-nitroguanosine antibody (1 μg/ml) was detected by the ECL system (Amersham Pharmacia).
Immunohistochemistry, Histopathology, and Analysis of the Host Response.
8-Nitroguanosine formation was assessed with mouse lungs fixed in 2% periodate-lysine-paraformaldehyde (6-μm frozen sections) by the indirect immunoenzyme technique with anti-8-nitroguanosine antibody (10 μg/ml), alkaline phosphatase-conjugated secondary antibody, and the Vector red substrate kit I (Vector Laboratories) (15). Similarly, expression of 3-nitrotyrosine and iNOS in the lung was analyzed via the immunoperoxidase method as described (6, 15). Hematoxylin and eosin staining was used for the study of histopathological changes in the lung. Apoptotic changes in the lung were analyzed as reported (6). Induction of cytokines and xanthine oxidase in virus-infected mice was analyzed via bronchoalveolar lavage (BAL) fluid as described (15).
Electron Spin Resonance (ESR) Analysis.
NO production in virus-infected lung was assessed by ESR spectroscopy. The N-dithiocarboxy(sarcosine)-Fe2+ complex served as a spin trap for NO and its related compounds including nitrosothiols during X-band ESR spectroscopy at 110 K (15, 16, 25).
3-Nitrotyrosine Quantification.
The amount of 3-nitrotyrosine formed in the lung was quantified with BAL protein obtained from virus-infected mice as described (15). BAL protein, enzymatically hydrolyzed as reported (26), was processed in an HPLC-based electrochemical-detection system (Eicom, Kyoto) equipped with a reduction cell (−900 mV) and a detection cell (+300 mV).
Analysis for O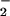 Generation from P450 Reductase and iNOS.
Generation from P450 Reductase and iNOS.
We examined the effect of 8-nitroguanosine on O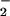 generation from P450 reductase and iNOS by means of ESR spin trapping with 5,5-dimethyl-1-pyrroline-N-oxide (DMPO). P450 reductase and human recombinant iNOS were prepared and purified according to the literature (27, 28). Each enzyme preparation was confirmed as fully active on the basis of NADPH oxidation and L-arginine oxidation to form L-citrulline (15). X-band ESR spectrometry (double integration of the ESR spectrum) was used to quantify the DMPO adduct (DMPO-OOH) as described (28).
generation from P450 reductase and iNOS by means of ESR spin trapping with 5,5-dimethyl-1-pyrroline-N-oxide (DMPO). P450 reductase and human recombinant iNOS were prepared and purified according to the literature (27, 28). Each enzyme preparation was confirmed as fully active on the basis of NADPH oxidation and L-arginine oxidation to form L-citrulline (15). X-band ESR spectrometry (double integration of the ESR spectrum) was used to quantify the DMPO adduct (DMPO-OOH) as described (28).
Results
Guanosine Nitration and Specificity of Anti-8-Nitroguanosine Antibody.
Guanosine in RNA was nitrated more effectively compared with nitration of deoxyguanosine and DNA after treatment of the nucleic acids and nucleosides with peroxynitrite (Fig. 1A). The higher efficacy of 8-nitroguanosine formation from guanosine and in RNA may be due to better stability of its nitrated guanine moiety than that in deoxyguanosine and in DNA (29). The competitive enzyme immunoassay showed that only 8-nitoguanosine and 8-nitroguanine, not the other nucleotides and nucleosides, completely inhibited binding between the antibody and the 8-nitroguanosine–BSA conjugate (Fig. 1B).
Figure 1.

Efficacy of 8-nitroguanosine formation by peroxynitrite in nucleic acids and nucleosides and specificity of anti-8-nitroguanosine antibody. (A) Guanosine (Guo, 0.1 mg/ml), deoxyguanosine (dGuo, 0.1 mg/ml), DNA (calf thymus, 0.5 mg/ml), and RNA (yeast tRNA, 0.5 mg/ml) were reacted with peroxynitrite in 100 mM sodium phosphate buffer (pH 7.4) followed by 10 min of incubation at room temperature. 8-Nitroguanosine (8-NitroGuo) thus formed was quantified by HPLC/electrochemical-detection analysis as described by Yermilov et al. (29). (B) Competitive enzyme immunoassay for the reaction of anti-8-nitroguanosine antibody with 8-nitroguanosine–BSA conjugate. Gua, guanine; 3-NitroTyr, 3-nitrotyrosine; 8-OxoGua, 8-oxoguanine. (Inset) Binding of anti-8-nitroguanosine antibody to BSA or 8-nitroguanosine–BSA conjugate fixed on microtiter plates. (C) Endogenous formation of 8- nitroguanosine in cells. Total RNA from RAW 264 cells stimulated or unstimulated with IFN-γ and lipopolysaccharide were analyzed by slot blotting coupled with the immunoperoxidase method for visualization of binding of anti-8-nitroguanosine antibody. Peroxynitrite-treated total RNA from CV-1 cells served as a positive control. (Lower) Immunoreactivity of total RNA from RAW 264 cells and peroxynitrite-treated RNA that were treated with 0.5 M Na2S2O4 in 0.1 M Tris⋅HCl buffer (pH 9.0) for 5 min at room temperature before adsorption onto the membrane. RNA was stained with ethidium bromide and Sybr green II (Molecular Probes).
It is notable that not only peroxynitrite-treated RNA from CV-1 cells (control) but also total RNA from RAW 264 cells expressing iNOS showed strong immunoreactivity with the antibody (Fig. 1C). Low-level 8-nitroguanosine immunoreactivity was observed with RNA from RAW 264 cells without stimulation for iNOS induction. The intensity of the band was greater 24 h after stimulation with lipopolysaccharide and IFN-γ, but this intensity was reduced appreciably by treatment with Nω-monomethyl-L-arginine. Immunoreactivity was eliminated almost totally by sodium hydrosulfite (Na2S2O4) treatment, which reduces nitroguanosine to aminoguanosine, before slot blot analysis (Fig. 1C).
These results lead us to two important conclusions. First, the antibody that we developed is truly specific for 8-nitroguanosine and recognizes its epitope in RNA. Second, an appreciable amount of 8-nitroguanosine is endogenously formed in the cells producing NO.
Formation of 8-Nitroguanosine in Virus-Infected Lungs.
Very little immunostaining for 8-nitroguanosine was observed in normal lung and in lungs obtained early after the initiation of influenza virus infection [i.e., 3 days postinfection (dpi)] (Fig. 2 Aa and Ab). The immunostaining became obvious at 6 dpi (Fig. 2Ac), reached a maximum at 8 dpi (Fig. 2Ad), and declined thereafter at 10 dpi (Fig. 2Ae). 8-Nitroguanosine in bronchial and bronchiolar epithelial cells showed intense staining; in inflammatory cells such as macrophages infiltrating the tissues staining was lighter. The time profile of 8-nitrogunosine production correlated with that of NO production and 3-nitrotyrosine generation after influenza virus infection, as mentioned below. Tissue from Sendai virus-infected lungs showed a similar pattern of immunostaining: strong staining in bronchiolar epithelial cells together with relatively extensive staining in alveolar epithelial cells and macrophages that had infiltrated parenchymal tissues (Fig. 2Af). Confocal laser scanning microscopy demonstrated localization of 8-nitroguanosine mainly in the cytosol of bronchiolar epithelial cells (Fig. 2B).
Figure 2.

8-Nitroguanosine immunohistochemistry in virus-infected lungs. (A a–e) Immunostaining in lungs obtained 0 (control noninfection), 3, 6, 8, and 10 days after influenza virus infection, respectively. (Af) Immunostaining for Sendai virus-infected lung (8 dpi). (B Upper) Tissue section (8 dpi, influenza) stained with anti-8-nitroguanosine antibody. (B Lower) The same section viewed by a confocal laser scanning microscope (Fluoroview FV300, Olympus, Nagano, Japan), in which strong fluorescence due to emission of Vector red is evident in the cytosol.
Incubation of tissue sections with the antibody and with authentic 8-nitroguanosine (1 mM) totally nullified the 8- nitroguanosine immunostaining (Fig. 3A). 3-Nitrotyrosine and other guanosine derivatives including 8-oxoguanosine and 8- bromoguanosine had no effect on the immunostaining (data not shown). When tissue sections were treated with 0.5 M Na2S2O4, as for RNA (Fig. 1C), the immunostaining was prevented almost completely (Fig. 3B). Moreover, 8-nitroguanosine staining colocalized with iNOS immunostaining, particularly in bronchial cells (Fig. 3C). 8-Nitroguanosine immunostaining was absent in airways of homozygous iNOS-deficient (iNOS−/−) mice infected with influenza virus (Fig. 3D).
Figure 3.

Specificity of immunostaining with anti-8-nitroguanosine antibody and colocalization of this staining with iNOS immunostaining in virus-infected lung. 8-Nitroguanosine immunostaining in lungs infected with influenza virus was nullified by authentic 8-nitroguanosine (8-NitroGuo, 1 mM) (A) and by pretreatment of tissue sections with Na2S2O4 (B). (C) Localization of 8- nitroguanosine immunostaining compared with immunostaining of iNOS. Serial adjacent sections were used for each panel (A–C). (D) 8-Nitroguanosine immunostaining in the lung of an iNOS−/− mouse infected with influenza virus (8 dpi).
Tyrosine Nitration in Lungs.
In wild-type mice, 3-nitrotyrosine formed in BAL fluid became detectable at 6 dpi, reached a peak at 8 dpi, and remained at an appreciable level until 10 dpi (Fig. 4A). 3-Nitrotyrosine was not detected in BAL fluid from iNOS−/− mice throughout the infection. A very low level of 3-nitrotyrosine was identified in concentrated BAL fluid collected from 20 mice; the average 3-nitrotyrosine value per mouse was 7.6 ± 0.5 fmol/ml. Immunohistochemical analysis revealed extensive 3-nitrotyrosine formation in the lung of iNOS+/+ mice at 7 dpi (Fig. 4B). Immunostaining was noted mainly in infiltrated inflammatory cells such as macrophages, in epithelial cells, and in alveolar exudates. Nitrotyrosine formation was reduced significantly in both iNOS-deficient littermates. Similar results were obtained for mice with Sendai virus infection (data not shown). Generation of both 3-nitrotyrosine and 8-nitroguanosine correlated well with NO production as assessed by ESR analysis with wild-type and iNOS-deficient mice (Fig. 4C).
Figure 4.

3-Nitrotyrosine and NO formation in influenza virus-infected lungs from wild-type and iNOS-deficient mice. (A) HPLC/electrochemical-detection analysis of protein in BAL fluid was used to determine tyrosine nitration. Concentrations of protein-bound 3-nitrotyrosine in BAL fluid (means ± SE, n = 3) are plotted versus time. (B) Immunohistochemistry for 3-nitrotyrosine formation in lung tissues. (C) Typical ESR signals of the NO-N-dithiocarboxy(sarcosine)-Fe2+ adduct for each corresponding group in B. The amounts of NO-N-dithiocarboxy(sarcosine)-Fe2+ as assessed by double integration of ESR spectra (15) are shown (means ± SE, n = 3) (P < 0.01 by t test for the value for iNOS+/+ vs. values for iNOS+/− and iNOS−/−).
Reduced Mortality and Improved Pathological Changes in iNOS-Deficient Mice.
The lethal effects of influenza and Sendai viruses were markedly blunted in both iNOS+/− and iNOS−/− mice, whereas iNOS+/+ mice became moribund at ≈6–7 dpi (Fig. 5 A Left and B Left). This reduced mortality is thought to result from improvement in the pathological characteristics of virus-infected lungs caused by a lack of iNOS and NO production. In both influenza and Sendai virus infections, iNOS+/+ mice had extensive inflammatory cell infiltration and alveolar exudates as well as destruction of pulmonary architecture (Fig. 5 A Right and B Right). Such damage was reduced significantly in iNOS-deficient mice. Similarly, apoptotic changes were reduced significantly in lungs of iNOS−/− mice at 7 days after influenza and Sendai virus infections compared with lung tissues from virus-infected wild-type mice (data not shown). No significant differences were found, however, in virus yields and host immune responses, as assessed by the enzyme immunoassay specific for various cytokines (IFN-γ, IL-4, and tumor necrosis factor-α) and induction of xanthine oxidase, among the three genotypes throughout the course of infection (data not shown).
Figure 5.

Survival and pathological pulmonary changes in wild-type and iNOS-deficient mice after influenza (A) and Sendai (B) virus infections. The time profile of survival of virus-infected mice (Left) and histopathological changes at 8 dpi (hematoxylin and eosin staining) (Right) are shown. The statistical difference in survival rates (wild-type vs. iNOS-deficient mice) was analyzed by Fisher's exact probability test.
Stimulation of O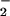 Generation from P450 Reductase and iNOS by 8-Nitroguanosine.
Generation from P450 Reductase and iNOS by 8-Nitroguanosine.
8-Nitroguanosine is classified as a nitroarene on the basis of its chemical structure. Some nitroarenes possess redox-active properties with certain reductases such as P450 reductase (30). This finding motivated us to hypothesize that 8-nitroguanosine may have redox activity that modulates biological reactions involving reductases. Our ESR analysis revealed that 8-nitroguanosine markedly stimulated production of O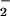 from the NADPH–P450 reductase system as evidenced by DMPO-OOH generation that was sensitive to superoxide dismutase (Fig. 6A). More importantly, iNOS generated an appreciable amount of O
from the NADPH–P450 reductase system as evidenced by DMPO-OOH generation that was sensitive to superoxide dismutase (Fig. 6A). More importantly, iNOS generated an appreciable amount of O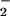 in the presence of NADPH and 8- nitroguanosine (Fig. 6B), which was not affected significantly by L-arginine. Such O
in the presence of NADPH and 8- nitroguanosine (Fig. 6B), which was not affected significantly by L-arginine. Such O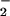 production was evident with even low micromolar concentrations of 8-nitroguanosine as shown by increased formation of DMPO-OOH after the addition of 8-nitroguanosine (Fig. 6C) (P < 0.05 and P < 0.01 by t test, n = 3), no nitroguanosine vs. 0.5 μM, and 1.0 μM nitroguanosine (ESR spectra not shown), respectively. This result suggests that 8-nitroguanosine stimulates the electron uncoupling of iNOS to form O
production was evident with even low micromolar concentrations of 8-nitroguanosine as shown by increased formation of DMPO-OOH after the addition of 8-nitroguanosine (Fig. 6C) (P < 0.05 and P < 0.01 by t test, n = 3), no nitroguanosine vs. 0.5 μM, and 1.0 μM nitroguanosine (ESR spectra not shown), respectively. This result suggests that 8-nitroguanosine stimulates the electron uncoupling of iNOS to form O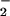 through a mechanism similar to the redox cycling that operates in the P450 reductase system.
through a mechanism similar to the redox cycling that operates in the P450 reductase system.
Figure 6.

8-Nitroguanosine-stimulated O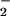 generation from P450 reductase (A) and iNOS (B). (A) The complete reaction system contained 8-nitroguanosine (8-NitroGuo) (10 μM), P450 reductase (0.2 μM), NADPH (0.1 mM), diethylenetriamine pentaacetic acid (0.1 mM), and DMPO (45 mM) in sodium phosphate buffer (25 mM, pH 7.4) and was incubated for 1 min at room temperature. SOD, superoxide dismutase (310 units/ml). The ESR signal of DMPO-OOH adduct (aN = 1.43 mT, a
generation from P450 reductase (A) and iNOS (B). (A) The complete reaction system contained 8-nitroguanosine (8-NitroGuo) (10 μM), P450 reductase (0.2 μM), NADPH (0.1 mM), diethylenetriamine pentaacetic acid (0.1 mM), and DMPO (45 mM) in sodium phosphate buffer (25 mM, pH 7.4) and was incubated for 1 min at room temperature. SOD, superoxide dismutase (310 units/ml). The ESR signal of DMPO-OOH adduct (aN = 1.43 mT, a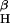 = 1.15 mT, and a
= 1.15 mT, and a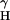 = 0.13 mT) was observed in the complete reaction system (28). (B) iNOS (0.2 μM) was incubated with NADPH (0.1 mM) in the absence or presence of 10 μM 8-nitroguanosine, and ESR analysis was performed in the same manner as described for A. L-Arg, L-arginine (1 mM). (C) Increase in DMPO-OOH formation with the iNOS/NADPH system after addition of various concentrations of 8-nitroguanosine (means ± SD, n = 3).
= 0.13 mT) was observed in the complete reaction system (28). (B) iNOS (0.2 μM) was incubated with NADPH (0.1 mM) in the absence or presence of 10 μM 8-nitroguanosine, and ESR analysis was performed in the same manner as described for A. L-Arg, L-arginine (1 mM). (C) Increase in DMPO-OOH formation with the iNOS/NADPH system after addition of various concentrations of 8-nitroguanosine (means ± SD, n = 3).
Discussion
We have demonstrated here that in murine viral pneumonia an effective nitration reaction is taking place, not only of tyrosine moieties of proteins but also of the nucleotide (nucleoside) base guanosine via iNOS-dependent NO overproduction. Our results show generation of 8-nitroguanosine via NO formed endogenously in vivo. It is now well documented that reactive nitrogen oxides effectively nitrate guanosine at least in cell-free chemical reaction systems (Fig. 1A) (19, 20). Also, Masuda et al. (21) recently demonstrated that 8-nitroguanosine was formed in total RNA of human lung carcinoma cells in culture after treatment with exogenous peroxynitrite; no evidence was presented, however, for endogenous, NO-dependent 8-nitroguanosine production in cells.
Effects of this 8-nitroguanosine formation during viral infection seem to be closely related to pathological consequences caused by NO and its oxidized reactive derivatives. In fact, we unequivocally verified that iNOS and excessive NO production are significantly involved in the pathogenesis of influenza and Sendai virus pneumonia. Results supporting this statement include the striking alteration in the pattern of tissue damage to virus-infected lungs and the higher survivability, with no effect on antiviral host defense, in iNOS-deficient mice. We thus suggest that NO-induced guanosine nitration may be deleterious to hosts infected with pneumotropic viruses.
In our earlier reports, we described remarkable improvements in pathological conditions in the lung and in the survival rate of virus-infected mice after treatment with Nω-monomethyl-L-arginine or O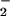 antidotes (superoxide dismutase and the xanthine oxidase inhibitor allopurinol) (13, 14). The beneficial effects of these pharmacological interventions indicate that peroxynitrite could be an important molecular species in the pathogenesis of influenza virus-induced pneumonia in mice. Similar mechanisms involving NO, O
antidotes (superoxide dismutase and the xanthine oxidase inhibitor allopurinol) (13, 14). The beneficial effects of these pharmacological interventions indicate that peroxynitrite could be an important molecular species in the pathogenesis of influenza virus-induced pneumonia in mice. Similar mechanisms involving NO, O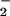 , and possibly peroxynitrite in the pathogenesis of virus-induced lung tissue injuries have been reported by other groups (12, 31–33).
, and possibly peroxynitrite in the pathogenesis of virus-induced lung tissue injuries have been reported by other groups (12, 31–33).
It is particularly important that staining for 8-nitroguanosine was most extensive in bronchiolar epithelial cells, which are the primary site of pneumotropic virus infections. We recently clarified that NO accelerates viral mutation during in vivo infection (16); mutation of a recombinant Sendai virus, i.e., GFP-constructed Sendai virus, was accelerated markedly by NO overproduction in virus-infected lungs. Because Sendai virus propagates mainly in airway epithelial cells, where 8-nitroguanosine is formed (as mentioned earlier), guanosine nitration may contribute to this NO-dependent viral mutagenesis. In fact, we found in a separate experiment that authentic 8-nitroguanosine significantly increased the mutation frequency of GFP-constructed Sendai virus when it was given exogenously to virus-infected cells in culture (unpublished observation).
NO per se is not a truly cytotoxic molecule, and many of its pathological effects thus are thought to be mediated by reactive nitrogen oxides, such as peroxynitrite produced through a coupling reaction of NO and O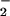 , and nitrogen dioxide generated from a nitrite plus a hydrogen peroxide-dependent and peroxidase-catalyzed reaction (3, 9, 10, 17). However, how such reactive nitrogen intermediates effect the cytotoxicity is still obscure. Eiserich et al. (34) showed that 3-nitrotyrosine is incorporated into α-tubulin, which in turn distorts microtubule structures, leading to altered cellular morphologies and functions, e.g., epithelial barrier functions. 3-Nitrotyrosine formation therefore may contribute in part to cytotoxicity induced by reactive nitrogen oxides. However, a causal role for tyrosine nitration in inflammatory tissue injury is not confirmed yet (35). Alternatively, there may be a mechanism for NO-induced cytotoxicity independent of tyrosine nitration; e.g., nucleic acid damage caused by reactive nitrogen oxides may account for the NO-induced cytotoxicity.
, and nitrogen dioxide generated from a nitrite plus a hydrogen peroxide-dependent and peroxidase-catalyzed reaction (3, 9, 10, 17). However, how such reactive nitrogen intermediates effect the cytotoxicity is still obscure. Eiserich et al. (34) showed that 3-nitrotyrosine is incorporated into α-tubulin, which in turn distorts microtubule structures, leading to altered cellular morphologies and functions, e.g., epithelial barrier functions. 3-Nitrotyrosine formation therefore may contribute in part to cytotoxicity induced by reactive nitrogen oxides. However, a causal role for tyrosine nitration in inflammatory tissue injury is not confirmed yet (35). Alternatively, there may be a mechanism for NO-induced cytotoxicity independent of tyrosine nitration; e.g., nucleic acid damage caused by reactive nitrogen oxides may account for the NO-induced cytotoxicity.
In fact, we discovered a unique biochemical feature of 8- nitroguanosine (Fig. 6). Some redox-active compounds, e.g., quinone-containing cytotoxic and mutagenic agents such as doxorubicin, have been shown to be reduced by P450 reductase and NOSs to generate O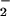 from molecular oxygen (30, 36). Electron uncoupling occurring at the reductase domain of NOSs seems to be involved in this redox-cycling reaction of NOSs (37). In this study, we found that 8-nitroguanosine greatly stimulated O
from molecular oxygen (30, 36). Electron uncoupling occurring at the reductase domain of NOSs seems to be involved in this redox-cycling reaction of NOSs (37). In this study, we found that 8-nitroguanosine greatly stimulated O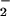 generation from P450 reductase in the presence of the electron donor NADPH by promoting an electron-transfer reaction from the enzyme to molecular oxygen. Also, 8-nitroguanosine may uncouple iNOS to induce production of a significant amount of O
generation from P450 reductase in the presence of the electron donor NADPH by promoting an electron-transfer reaction from the enzyme to molecular oxygen. Also, 8-nitroguanosine may uncouple iNOS to induce production of a significant amount of O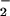 . Expression of P450 reductase in the lung has been described (38), and our present immunohistochemical analysis showed colocalization of 8-nitroguanosine with iNOS in virus-infected lung. 8-Nitroguanosine thus formed in the cells may have pathophysiological consequences via enhancing oxidative and nitrative damage of cells rather than serving as an inert footprint of biological nitration. In summary, guanosine nitration may be responsible for NO-induced inflammatory lung injury in murine viral pneumonia, and it could be an important mediator of nitrative stress in the pathogenesis of various diseases.
. Expression of P450 reductase in the lung has been described (38), and our present immunohistochemical analysis showed colocalization of 8-nitroguanosine with iNOS in virus-infected lung. 8-Nitroguanosine thus formed in the cells may have pathophysiological consequences via enhancing oxidative and nitrative damage of cells rather than serving as an inert footprint of biological nitration. In summary, guanosine nitration may be responsible for NO-induced inflammatory lung injury in murine viral pneumonia, and it could be an important mediator of nitrative stress in the pathogenesis of various diseases.
Taken together, the present data unambiguously confirm our earlier interpretation that NO and its reactive nitrogen-oxide derivatives are involved in a critical way in the pathogenesis of murine pneumotropic virus infections. Viral pathogenesis that depends on NO may be mediated by nitration of nucleic acids rather than nitration of proteins and lipids. The present verification of in vivo 8-nitroguanosine formation may shed light on the possible impact of nitrative stress imposed by the potential toxicity of NO and its byproducts formed during infection and inflammation and even in neurodegenerative diseases. Identification of 8-nitroguanosine formed in various diseases, the physiological and pathological significance of this nitrated nucleoside, and the fate of nitrated nucleotides remain challenges for further research.
Acknowledgments
We thank Ms. Judith B. Gandy for editing the manuscript and Dr. Dennis J. Stuehr for his generous supply of recombinant iNOS. Thanks are also due to Drs. Hiroshi Ohshima, Albert van der Vliet, and Tetsuhiko Yoshimura for helpful discussion on biochemical analysis for 8-nitroguanosine. This work is supported by a Grant-in-Aid from the Ministry of Education, Science, Sports and Culture of Japan and a grant from the Ministry of Health and Welfare of Japan.
Abbreviations
- iNOS
inducible NO synthase
- BAL
bronchoalveolar lavage
- ESR
electron spin resonance
- DMPO
5,5-dimethyl-1-pyrroline-N-oxide
- dpi
days postinfection
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Akaike T, Maeda H. In: Nitric Oxide: Biology and Pathobiology. Ignarro L J, editor. San Diego: Academic; 2000. pp. 733–745. [Google Scholar]
- 2.Nathan C. J Clin Invest. 1997;100:2417–2423. doi: 10.1172/JCI119782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Estévez A G, Crow J P, Sampson J B, Reiter C, Zhuang Y, Richardson G J, Tarpey M M, Barbeito L, Beckman J S. Science. 1999;286:2498–2500. doi: 10.1126/science.286.5449.2498. [DOI] [PubMed] [Google Scholar]
- 4.Granger D L, Hibbs J B, Jr, Perfect J R, Durack D T. J Clin Invest. 1988;81:1129–1136. doi: 10.1172/JCI113427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.James S L. Microbiol Rev. 1995;59:533–547. doi: 10.1128/mr.59.4.533-547.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alam M S, Akaike T, Miyamoto Y, Okamoto S, Kubota T, Tamura F, Maeda H. Infect Immun. 2002;70:3130–3142. doi: 10.1128/IAI.70.6.3130-3142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nathan C, Shiloh M U. Proc Natl Acad Sci USA. 2000;97:8841–8848. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mannick J B, Asano K, Izumi K, Kieff E, Stamler J S. Cell. 1994;79:1137–1146. doi: 10.1016/0092-8674(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 9.Beckman J, Koppenol W H. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 10.Rubbo H, Darley-Usmar V, Freeman B A. Chem Res Toxicol. 1996;9:809–820. doi: 10.1021/tx960037q. [DOI] [PubMed] [Google Scholar]
- 11.Stamler J, Singel D, Loscalzo J. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- 12.Akaike T. Rev Med Virol. 2001;11:87–101. doi: 10.1002/rmv.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oda T, Akaike T, Hamamoto T, Suzuki F, Hirano T, Maeda H. Science. 1989;244:974–976. doi: 10.1126/science.2543070. [DOI] [PubMed] [Google Scholar]
- 14.Akaike T, Ando M, Oda T, Doi T, Ijiri S, Araki S, Maeda H. J Clin Invest. 1990;85:739–745. doi: 10.1172/JCI114499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akaike T, Noguchi Y, Ijiri S, Setoguchi K, Suga M, Zheng Y M, Dietzschold B, Maeda H. Proc Natl Acad Sci USA. 1996;93:2448–2453. doi: 10.1073/pnas.93.6.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akaike T, Fujii S, Kato A, Yoshitake J, Miyamoto Y, Sawa T, Okamoto S, Suga M, Asakawa M, Nagai Y, et al. FASEB J. 2000;14:1447–1454. doi: 10.1096/fj.14.10.1447. [DOI] [PubMed] [Google Scholar]
- 17.Eiserich J P, Hristova M, Cross C E, Jones A D, Freeman B A, Halliwell B, van der Vliet A. Nature. 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- 18.Sawa T, Akaike T, Maeda H. J Biol Chem. 2000;275:32467–32474. doi: 10.1074/jbc.M910169199. [DOI] [PubMed] [Google Scholar]
- 19.Sodum R S, Fiala E S. Chem Res Toxicol. 2001;14:438–450. doi: 10.1021/tx000189s. [DOI] [PubMed] [Google Scholar]
- 20.Niles J C, Wishnok J S, Tannenbaum S R. J Am Chem Soc. 2001;123:12147–12151. doi: 10.1021/ja004296k. [DOI] [PubMed] [Google Scholar]
- 21.Masuda M, Nishino H, Ohshima H. Chem Biol Interact. 2002;139:187–197. doi: 10.1016/s0009-2797(01)00299-x. [DOI] [PubMed] [Google Scholar]
- 22.Erlanger B F, Beiser S M. Proc Natl Acad Sci USA. 1964;52:68–74. doi: 10.1073/pnas.52.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen H J, Chen Y M, Wang T F, Wang K S, Shiea J. Chem Res Toxicol. 2001;14:536–546. doi: 10.1021/tx0002334. [DOI] [PubMed] [Google Scholar]
- 24.Akaike T, Inoue K, Okamoto T, Nishino H, Otagiri M, Fujii S, Maeda H. J Biochem. 1997;122:459–466. doi: 10.1093/oxfordjournals.jbchem.a021774. [DOI] [PubMed] [Google Scholar]
- 25.Tsuchiya K, Kirima K, Yoshizumi M, Houchi H, Tamaki T, Mason R P. Biochem J. 2002;367:771–779. doi: 10.1042/BJ20020310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crow J P. Methods Enzymol. 1999;301:151–160. doi: 10.1016/s0076-6879(99)01078-2. [DOI] [PubMed] [Google Scholar]
- 27.Ichimori K, Stuehr D J, Atkinson R N, King S B. J Med Chem. 1999;42:1842–1848. doi: 10.1021/jm980232x. [DOI] [PubMed] [Google Scholar]
- 28.Sato K, Akaike T, Kojima Y, Ando M, Nagao M, Maeda H. Jpn J Cancer Res. 1992;83:1204–1209. doi: 10.1111/j.1349-7006.1992.tb02746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yermilov V, Rubio J, Ohshima H. FEBS Lett. 1995;376:207–210. doi: 10.1016/0014-5793(95)01281-6. [DOI] [PubMed] [Google Scholar]
- 30.Mason R P. In: Free Radicals in Biology. Pryor W A, editor. Vol. 5. New York: Academic; 1982. pp. 196–212. [Google Scholar]
- 31.Karupiah G, Chen J H, Mahalingam S, Nathan C F, MacMicking J D. J Exp Med. 1998;188:1541–1546. doi: 10.1084/jem.188.8.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adler H, Beland J L, Del-Pan N C, Kobzik L, Brewer J P, Martin T R, Rimm I J. J Exp Med. 1997;185:1533–1540. doi: 10.1084/jem.185.9.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suliman H B, Ryan L K, Bishop L, Folz R J. Am J Physiol. 2001;280:L69–L78. doi: 10.1152/ajplung.2001.280.1.L69. [DOI] [PubMed] [Google Scholar]
- 34.Eiserich J P, Estévez A G, Bamberg T V, Ye Y Z, Chumley P H, Beckman J S, Freeman B A. Proc Natl Acad Sci USA. 1999;96:6365–6370. doi: 10.1073/pnas.96.11.6365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baldus S, Castro L, Eiserich J P, Freeman B A. Am J Respir Crit Care Med. 2001;163:308–310. doi: 10.1164/ajrccm.163.2.ed2000c. [DOI] [PubMed] [Google Scholar]
- 36.Vasquez-Vivar J, Martasek P, Hogg N, Masters B S, Pritchard K A, Jr, Kalyanaraman B. Biochemistry. 1997;36:11293–11297. doi: 10.1021/bi971475e. [DOI] [PubMed] [Google Scholar]
- 37.Stuehr D, Pou S, Rosen G M. J Biol Chem. 2001;276:14533–14536. doi: 10.1074/jbc.R100011200. [DOI] [PubMed] [Google Scholar]
- 38.Hall P M, Stupans I, Burgess W, Birkett D J, McManus M E. Carcinogenesis. 1989;10:521–530. doi: 10.1093/carcin/10.3.521. [DOI] [PubMed] [Google Scholar]