

Abstract
Heterotrimeric G proteins, G12 and G13, have been shown to transduce signals from G protein-coupled receptors to activate Rho GTPase in cells. Recently, we identified p115RhoGEF, one of the guanine nucleotide exchange factors (GEFs) for Rho, as a direct link between Gα13 and Rho [Kozasa, T., et al. (1998) Science 280, 2109–2111; Hart, M. J., et al. (1998) Science 280, 2112–2114]. Activated Gα13 stimulated the RhoGEF activity of p115 through interaction with the N-terminal RGS domain. However, Gα12 could not activate Rho through p115, although it interacted with the RGS domain of p115. The biochemical mechanism from Gα12 to Rho activation remained unknown. In this study, we analyzed the interaction of leukemia-associated RhoGEF (LARG), which also contains RGS domain, with Gα12 and Gα13. RGS domain of LARG demonstrated Gα12- and Gα13-specific GAP activity. LARG synergistically stimulated SRF activation by Gα12 and Gα13 in HeLa cells, and the SRF activation by Gα12-LARG was further stimulated by coexpression of Tec tyrosine kinase. It was also found that LARG is phosphorylated on tyrosine by Tec. In reconstitution assays, the RhoGEF activity of nonphosphorylated LARG was stimulated by Gα13 but not Gα12. However, when LARG was phosphorylated by Tec, Gα12 effectively stimulated the RhoGEF activity of LARG. These results demonstrate the biochemical mechanism of Rho activation through Gα12 and that the regulation of RhoGEFs by heterotrimeric G proteins G12/13 is further modulated by tyrosine phosphorylation.
Members of the Rho family GTPases (Rho, Rac, and Cdc42) regulate a variety of cellular activities such as cell-cycle progression, chemotaxis, or axonal guidance by controlling actin cytoskeletal rearrangements or gene expression (1). Activation of Rho family GTPases is catalyzed by their guanine nucleotide exchange factors (GEFs). These GEFs share a Dbl homology domain and an adjacent pleckstrin homology domain (2). The Dbl homology domain is responsible for the capacity to stimulate GDP–GTP exchange of Rho family GTPases. Except for this common Dbl homology–pleckstrin homology structure, these GEFs contain various protein motifs that are implicated in signal transduction. However, the biochemical mechanism of regulation of these GEFs by upstream signals has been poorly understood.
Heterotrimeric G proteins G12 and G13 have been shown to mediate signals from G protein-coupled receptors to Rho GTPase activation (3–5). Recently, we identified p115RhoGEF, one of GEFs for Rho, as a direct link between heterotrimeric G13 and Rho (6, 7). Activated Gα13 stimulated the RhoGEF activity of p115 through the interaction with the N-terminal RGS (regulator of G protein signaling) domain. However, Gα12 did not activate Rho through p115 in reconstitution assays. Although the overexpression of a constitutively active mutant of Gα12 has demonstrated several evidences supporting Rho activation in cells (5, 8), the biochemical mechanism from Gα12 to Rho activation has not been understood.
Recently, several reports indicated the involvement of tyrosine phosphorylation in the regulation of GEF activity for Rho family GTPases. Tyrosine phosphorylation of Vav or Vav-2 was required for their GEF activity (9, 10). GEF activity of Dbl for Rho and Cdc42 was enhanced by tyrosine phosphorylation by ACK-1 (11). It was also demonstrated that several tyrosine kinase inhibitors blocked Gα12- or Gα13-mediated Rho activation in cells (12, 13). In addition, the involvement of Tec family tyrosine kinases in G12/13-mediated signaling pathway was demonstrated in cell-based assays as well as in in vitro experiments (14, 15). Tec kinases form a family of nonreceptor tyrosine kinases that share pleckstrin homology and Tec homology (TH) domains at the N-terminal region (16). These kinases are activated by various stimuli, including ligands for G protein-coupled receptors (17). However, their regulatory functions in cells remain unclear.
In this study, we investigated the possibility that RhoGEF other than p115 might be responsible for mediating signals from Gα12 to Rho. We found that leukemia-associated RhoGEF (LARG) could transduce Gα12-mediated Rho activation when it was phosphorylated by Tec tyrosine kinase.
Methods
Construction of Plasmids.
KIAA0382 was originally isolated as a partial cDNA lacking N-terminal PDZ and RGS domains (18). Full-length cDNA was obtained by 5′-RACE using KIAA0382 as a template and human brain cDNA library (CLONTECH). The full-length cDNA had an identical amino acid sequence with LARG. LARG (1–1543), ΔPDZ-LARG (307–1543), ΔN-LARG (617–1543), PDZ-RhoGEF, p115RhoGEF, Tec (1–629), and kinase domain-deleted Tec (Tec-KD) (1–358) were subcloned into pcDNA-myc vector with N-terminal myc-tag. cDNAs for Tec lacking TH domain (ΔTH-Tec) and the constitutively active form of Tec (mHTec), which has N-terminal myristoylation signal, were subcloned into pSRα mammalian expression vector (17, 19). cDNAs encoding the constitutively active Gα12 (Gα12Q229L) and Gα13 (Gα13Q226L) were subcloned into pCMV5 vector. SRE.L-luciferase reporter plasmid and an expression construct for GST-fused RhoA binding domain of Rhotekin (GST-RBD) were kindly provided by P. C. Sternweis (University of Texas Southwestern Medical Center) and G. Bokoch (The Scripps Research Institute, La Jolla, CA), respectively.
SRE-Luciferase Assay.
HeLa cells (6 × 104 cells per well) were plated onto 24-well plates 1 day before transfection. Cells were cotransfected with SRE.L-luciferase reporter plasmid (0.1 μg), pCMV-βgal (0.1 μg), and the indicated cDNAs. The cells were cultured in the presence of 10% FCS for 5 h and then serum-starved for 24 h. Luciferase activities in cell extracts were measured according to the manufacturer's instruction (Promega). Total amounts of transfected DNA were kept constant among wells by supplementing the empty vector DNA. β-Galactosidase activities of cell lysates were used to normalize for the transfection efficiency.
Expression and Purification of Proteins.
The constructs of LARG were subcloned into the pFastBacHT transfer vector with a six-histidine tag at the N terminus (Life Technologies, Grand Island, NY), and their recombinant baculoviruses were generated. Sf9 cells (1.8 × 106 cells per ml) were infected with corresponding recombinant baculovirus and harvested after 48 h. Cells were resuspended in lysis buffer (20 mM Hepes, pH 8.0/50 mM NaCl/0.1 mM EDTA/10 mM 2-mercaptoethanol and protease inhibitors) and lysed by nitrogen cavitation. The lysates were centrifuged at 100,000 × g and 4°C for 30 min. The supernatants were loaded onto Ni-NTA column equilibrated with buffer A (20 mM Hepes, pH 8.0/100 mM NaCl/10 mM 2-mercaptoethanol). The column was washed with 10 column volumes of buffer B (buffer A containing 400 mM NaCl and 10 mM imidazole). Recombinant LARG was eluted by 10 column volumes of buffer C (buffer A containing 150 mM imidazole). The elution fractions were concentrated and the buffer was exchanged with buffer D (buffer A containing 10% glycerol).
p115RhoGEF and RhoA were prepared as described (6, 7). Gα12 and Gα13 were purified using the Sf9-baculovirus expression system as described (20), with the following modification for Gα13 purification. Instead of 1% octylglucoside, 0.2% n-dodecyl-β-D-maltoside and 10% glycerol were included in the elution buffer of Gα13 from Ni-NTA column.
RhoGEF Assay.
RhoA loaded with [3H]GDP (100 nM, 2,000 cpm/pmol) was incubated with the indicated proteins at 20°C in GEF assay buffer (50 mM Tris⋅HCl, pH 7.5/50 mM NaCl/1 mM EDTA/1 mM DTT/10 mM MgCl2/5 μM GTPγS/0.1% C12E10) in a final volume of 20 μl. G protein α subunits were preincubated in the presence of AMF (30 μM AlCl3/5 mM MgCl2/10 mM NaF) and added to the GEF reaction mixture. The reactions were stopped by the addition of 2 ml of washing buffer (20 mM Tris⋅HCl, pH 7.5/40 mM MgSO4/100 mM NaCl), followed by filtration through BA-85 filters (Schleicher & Schuell). The amount of [3H]GDP that remained on the filter was determined by a liquid scintillation counter.
To prepare Tec for GEF assays, COS1 cells were transfected with myc-tagged Tec. After 24 h, cells were lysed in the lysis buffer (50 mM Tris⋅HCl/150 mM NaCl/1% Nonidet P-40/1 mM EDTA/1 mM DTT/10 mM β-glycerophosphate/10 mM Na3VO4 and protease inhibitors) and centrifuged at 200,000 × g for 20 min. The supernatants were incubated with anti-myc antibody 9E10 (Covance). Tec was immunoprecipitated using protein G-agarose (Santa Cruz Biotechnology) and resuspended in GEF buffer.
To prepare phosphorylated LARG, Tec immunoprecipitated from COS1 cells was mixed with LARG and incubated at 20°C for 40 min in GEF buffer with 100 μM ATP. Then, [3H]GDP-loaded RhoA (100 nM) and AlF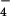 -activated Gα were added to the GEF reaction mixture. The mixture was further incubated at 20°C for the indicated time. The dissociation of GDP from RhoA was measured as described above.
-activated Gα were added to the GEF reaction mixture. The mixture was further incubated at 20°C for the indicated time. The dissociation of GDP from RhoA was measured as described above.
To measure RhoGEF activity in cells, endogenous GTP-bound RhoA in cell lysate were detected by their association with GST–RBD as described by Ren and Schwartz (21).
Phosphorylation Assay.
Tec or Tec-KD was overexpressed in COS1 cells, prepared as described above, and was resuspended in the kinase buffer (20 mM Tris⋅HCl, pH 7.4/50 mM NaCl/10 mM MgCl2/2 mM MnSO4/100 μM ATP). RhoGEF with or without Gα12/13 was incubated with Tec in the kinase buffer at 30°C for 20 min. The reactions were terminated by adding SDS/PAGE sample buffer, and the samples were separated by SDS/PAGE, followed by immunoblotting using anti-Tec antibody (17) or antiphosphotyrosine antibody PY20 (Zymed).
For the assessment of phosphorylation in vivo, HEK293 cells were cotransfected with myc-tagged ΔPDZ-LARG, ΔN-LARG, or p115 and the constitutively active Tec (mHTec). After 24 h, cells were lysed and LARG was immunoprecipitated by anti-myc antibody. The immunoprecipitates were subjected to SDS/PAGE and analyzed by immunoblotting with PY20 antibody.
Miscellaneous Procedures.
Immunoblotting was performed using the chemiluminescent detection system (Pierce). GTPase assays for Gα subunits were performed as described (6).
Results
In addition to p115RhoGEF, two mammalian RhoGEFs, PDZ-RhoGEF (KIAA0380) and LARG, were identified to have an RGS domain in their N-terminal region (refs. 18 and 22; Fig. 1A). It was shown that PDZ-RhoGEF and LARG interacted with constitutively active mutants of Gα12 and Gα13 through their RGS domains (23, 24). However, the biochemical mechanism to regulate the RhoGEF activity of PDZ-RhoGEF or LARG by Gα12/13 has not been elucidated. To examine whether PDZ-RhoGEF or LARG can mediate the signal from Gα12 or Gα13 to Rho activation, we first performed SRE-luciferase reporter assays. It has already been shown that Gα12/13-mediated Rho activation could be monitored in cells by SRF activation (25). As shown in Fig. 1B, overexpression of a constitutively active mutant of Gα12 (Gα12Q229L) or Gα13 (Gα13Q226L) modestly stimulated SRF activity, whereas coexpression of these mutants with LARG or PDZ-RhoGEF synergistically potentiated SRF activation. In particular, SRF activation by PDZ-RhoGEF or LARG and Gα12 was almost similar to the level with these RhoGEFs and Gα13. We could not detect similar synergistic SRF activation by using p115RhoGEF in the assay. The results suggest that PDZ-RhoGEF or LARG may transduce the signal from both Gα12 and Gα13 to Rho activation. In this study, we focused on the function of LARG in G12/13-mediated signaling.
Figure 1.

Involvement of LARG and PDZ-RhoGEF in Gα12/13-mediated SRF activation. (A) Domain structure of RhoGEFs. Domains of p115RhoGEF, LARG, and PDZ-RhoGEF are schematically represented. PDZ, PDZ domain; RGS, RGS domain; DH, Dbl homology domain; PH, pleckstrin homology domain. The constructs of LARG that were used in this study are shown at the top. (B) SRF activation by Gα12/13-RhoGEF. HeLa cells were cotransfected with 0.1 μg of SRE.L-luciferase reporter plasmid and the indicated constructs: 0.01 μg of Gα12QL, 0.01 μg of Gα13QL, 0.1 μg of PDZ-RhoGEF, 0.1 μg of LARG, or 0.02 μg of p115RhoGEF. SRF activities of cell lysates were measured 24 h after transfection as described in Methods. The expression of RhoGEFs in lysates was detected by immunoblotting using anti-myc antibody as shown (Lower).
We examined the biochemical interaction of Gα12/13 with LARG in vitro by using purified components. Gα subunits and RhoGEFs were expressed in and purified from Sf9 cells (Fig. 2A). As shown in Fig. 2B, the constructs of LARG that contain the RGS domain demonstrated GAP activity for Gα12 or Gα13 similar to p115RhoGEF. However, a construct of LARG lacking the RGS domain did not show any GAP activity. The RGS domain of LARG did not have GAP activity for Gαs, Gαi, Gαo, and Gαq (data not shown). Thus, the RGS domain of LARG serves as a specific GAP for Gα12 or Gα13 similar to that of p115RhoGEF.
Figure 2.

GAP activity of LARG for Gα12/13 and the regulation of RhoGEF activity of LARG by Gα12/13. (A) Coomassie brilliant blue staining of Gα12/13 and RhoGEFs. Purified Gα12, Gα13, LARG, and p115RhoGEF (50 pmol each) were separated by SDS/PAGE and stained by Coomassie brilliant blue. (B) Stimulation of GTPase activity of Gα12 and Gα13 by LARG. Hydrolysis of GTP bound to Gα12 or Gα13 was measured at 15°C without (□) or with (■) 25 nM LARG, 25 nM ΔPDZ-LARG (●), 25 nM ΔN-LARG (▴), or 25 nM p115RhoGEF (◊). (C) Stimulation of the RhoGEF activity of LARG. Dissociation of GDP from RhoA was measured at 20°C: ○, control; □, 25 nM ΔPDZ-LARG; ▵, 25 nM ΔPDZ-LARG + 80 nM AlF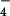 -activated Gα12; and ▿, 25 nM ΔPDZ-LARG + 80 nM AlF
-activated Gα12; and ▿, 25 nM ΔPDZ-LARG + 80 nM AlF -activated Gα13.
-activated Gα13.
We also examined the regulation of RhoGEF activity of LARG by Gα12/13. In the case of p115RhoGEF, Gα13, but not Gα12, stimulated its RhoGEF activity (7). As shown in Fig. 2C, AlF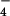 -activated Gα13 stimulated the RhoGEF activity of LARG. However, AlF
-activated Gα13 stimulated the RhoGEF activity of LARG. However, AlF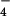 -activated Gα12 did not demonstrate RhoGEF activation. Thus, although SRF assays suggested that Gα12-LARG mediated Rho activation in HeLa cells, we could not reconstitute that pathway in vitro. The results suggest that additional factors or some modification on Gα12 or LARG will be necessary for activation of Rho through the Gα12-LARG pathway.
-activated Gα12 did not demonstrate RhoGEF activation. Thus, although SRF assays suggested that Gα12-LARG mediated Rho activation in HeLa cells, we could not reconstitute that pathway in vitro. The results suggest that additional factors or some modification on Gα12 or LARG will be necessary for activation of Rho through the Gα12-LARG pathway.
Because the involvement of Tec kinase has been reported in the Gα12-mediated pathway, we tested the possibility that Tec tyrosine kinase might be involved in Rho activation through Gα12/13-LARG. As shown in Fig. 3A, coexpression of Tec kinase in HeLa cells potently stimulated both Gα12- and Gα13-LARG-mediated SRF activation. However, we did not observe a similar effect of Tec when Gα12/13 or LARG was expressed alone. Coexpression of Tec did not stimulate SRF activation mediated by Gα12/13-p115RhoGEF (data not shown). In addition, a kinase-deficient mutant of Tec (Tec-KD) failed to stimulate the Gα12/13-LARG-mediated SRF activation. GTP-bound Rho pull-down assay also demonstrated that Rho activation by Gα12-LARG in HeLa cells was further stimulated by Tec (Fig. 3B). These results suggest that Tec tyrosine kinase regulates Gα12/13-LARG-mediated Rho activation by phosphorylating some component of the pathway.
Figure 3.

Effect of Tec for Rho activation by Gα12/13-LARG. (A) Activation of SRF activity by Tec. HeLa cells were cotransfected with 0.1 μg of SRE.L-luciferase reporter plasmid with the indicated constructs: 0.01 μg of Gα12QL, 0.01 μg of Gα13QL, 0.1 μg of ΔPDZ-LARG, 0.1 μg of Tec, and 0.1 μg of Tec-KD. SRF activities of cell lysates were measured after 24 h. (B) RhoA activation by Gα12-LARG and Tec in HeLa cells. HeLa cells were transiently transfected with the plasmids encoding ΔPDZ-LARG, Gα12QL, or Tec. GTP-bound RhoA in cell lysates was detected using GST-RBD pull-down assay. The result shown is a representative of three separate experiments with similar results. (C) Direct interaction of Tec with Gα12. COS1 cells were transfected with myc-tagged Tec with or without Gα12QL. Tec was immunoprecipitated from the lysate, and the immunoprecipitates were analyzed by immunoblotting using Gα12 antibody. (D) Requirement of the TH domain of Tec in Gα12/13-LARG signaling pathway. HeLa cells were cotransfected with 0.1 μg of SRE.L-luciferase reporter plasmid with indicated plasmids as described for A or with 0.1 μg of Tec-ΔTH. SRF activities of cell lysates were measured after 24 h.
The interaction of Gα12 with Btk, another member of the Tec family, through its pleckstrin homology–TH domain was recently demonstrated (15). As shown in Fig. 3C, we could also observe the interaction between constitutively active Gα12QL and Tec by coimmunoprecipitation. Furthermore, a Tec construct lacking TH domain did not show the stimulatory effect on Gα12-LARG-mediated SRF activation, indicating that the TH domain of Tec is required for its effect on the Gα12/13-LARG pathway (Fig. 3D).
We next examined whether Tec can directly phosphorylate Gα12/13 or LARG. Myc-tagged Tec was overexpressed in COS1 cells, immunoprecipitated by anti-myc antibody, and used for in vitro phosphorylation assays. As shown in Fig. 4A, ΔPDZ-LARG was phosphorylated on tyrosine by Tec. However, p115RhoGEF, Gα12, or Gα13 did not serve as a substrate for Tec. Moreover, the activated Gα12 or Gα13 did not affect the phosphorylation of LARG by Tec. We also examined tyrosine phosphorylation of LARG in cells. A Tec construct with an N-terminal myristoylation signal (mHTec) was targeted to the plasma membrane and exhibited constitutive activity (19). As shown in Fig. 4B, ΔPDZ-LARG, but not p115, was tyrosine phosphorylated in HEK293 cells when coexpressed with mHTec. However, we could not detect tyrosine phosphorylation of ΔN-LARG under the same condition. These results suggest that Tec phosphorylates LARG in vivo as well as in vitro. Furthermore, the phosphorylation site on LARG is likely in the region including its RGS domain (amino acid residues 307–617 of LARG).
Figure 4.

Tyrosine phosphorylation of LARG by Tec. (A) Tyrosine phosphorylation of LARG by Tec in vitro. Myc-tagged Tec or Tec-KD was overexpressed in COS1 cells, immunoprecipitated by anti-myc antibody, and used for kinase assays. Recombinant ΔPDZ-LARG or p115RhoGEF (600 nM each) with or without 1 μM AlF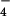 -activated Gα12 or Gα13 were incubated with immunoprecipitated Tec in the presence of ATP for 20 min at 30°C. Proteins were separated by SDS/PAGE, followed by immunoblotting with antiphosphotyrosine antibody. (B) Tyrosine phosphorylation of LARG by Tec in vivo. HEK293 cells were cotransfected with myc-tagged ΔPDZ-LARG, ΔN-LARG, or p115 with or without mHTec. RhoGEFs were immunoprecipitated by anti-myc antibody from the cell lysates, and the immunoprecipitates were separated by SDS/PAGE, followed by immunoblotting using antiphosphotyrosine antibody or anti-myc antibody. (C) Stimulation of GDP dissociation from RhoA by Gα12, LARG, and Tec. GDP dissociation from RhoA by ΔPDZ-LARG was assayed for 20 min at 20°C in the presence of the indicated proteins (as described in Methods): 10 nM ΔPDZ-LARG, 200 nM AlF
-activated Gα12 or Gα13 were incubated with immunoprecipitated Tec in the presence of ATP for 20 min at 30°C. Proteins were separated by SDS/PAGE, followed by immunoblotting with antiphosphotyrosine antibody. (B) Tyrosine phosphorylation of LARG by Tec in vivo. HEK293 cells were cotransfected with myc-tagged ΔPDZ-LARG, ΔN-LARG, or p115 with or without mHTec. RhoGEFs were immunoprecipitated by anti-myc antibody from the cell lysates, and the immunoprecipitates were separated by SDS/PAGE, followed by immunoblotting using antiphosphotyrosine antibody or anti-myc antibody. (C) Stimulation of GDP dissociation from RhoA by Gα12, LARG, and Tec. GDP dissociation from RhoA by ΔPDZ-LARG was assayed for 20 min at 20°C in the presence of the indicated proteins (as described in Methods): 10 nM ΔPDZ-LARG, 200 nM AlF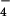 -activated Gα12, and 200 nM AlF
-activated Gα12, and 200 nM AlF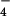 -activated Gα13. *, P < 0.01, significant difference from the data with Gα12 + LARG + Tec. The results shown are from a representative experiment of three such experiments with similar results. (D) Time course of GDP dissociation from RhoA. GDP dissociation from RhoA was measured with the indicated proteins: ○, control; □, 10 nM ΔPDZ-LARG; ◊, 10 nM ΔPDZ-LARG + Tec; ▵, 10 nM ΔPDZ-LARG + 200 nM Gα12; ▿, 10 nM ΔPDZ-LARG + 200 nM Gα13; ▴, 10 nM ΔPDZ-LARG + 200 nM Gα12 + Tec; or ▾, 10 nM ΔPDZ-LARG + 200 nM Gα13 + Tec.
-activated Gα13. *, P < 0.01, significant difference from the data with Gα12 + LARG + Tec. The results shown are from a representative experiment of three such experiments with similar results. (D) Time course of GDP dissociation from RhoA. GDP dissociation from RhoA was measured with the indicated proteins: ○, control; □, 10 nM ΔPDZ-LARG; ◊, 10 nM ΔPDZ-LARG + Tec; ▵, 10 nM ΔPDZ-LARG + 200 nM Gα12; ▿, 10 nM ΔPDZ-LARG + 200 nM Gα13; ▴, 10 nM ΔPDZ-LARG + 200 nM Gα12 + Tec; or ▾, 10 nM ΔPDZ-LARG + 200 nM Gα13 + Tec.
Finally, we examined the effect of phosphorylation of LARG on its RhoGEF activity (Fig. 4 C and D). LARG was first phosphorylated by preincubation with Tec, and then RhoGEF assays were started by adding RhoA and Gα subunits. The preincubation with Tec did not affect the basal RhoGEF activity of LARG. However, Gα12 stimulated the RhoGEF activity of LARG in a phosphorylation-dependent manner. This stimulation was almost similar to the level by Gα13 without Tec. The stimulatory effect by Gα12 was also dependent on the presence of ATP during preincubation (data not shown), confirming that the phosphorylation of LARG is required for Gα12 to activate Rho. These results indicate that Gα12 can activate Rho through phosphorylated LARG. Tyrosine phosphorylation of LARG also modestly stimulated RhoGEF activity mediated by Gα13 (Fig. 4D).
Discussion
The results of this study demonstrated the biochemical mechanism of Rho activation through Gα12. Although Gα12 and Gα13 are similar in amino acid sequences and biochemical properties and both are involved in Rho activation, several studies indicated the functional differences between these two Gα subunits. The most striking difference was demonstrated in Gα13 gene knockout mice, which showed embryonic lethality due to the defect of vascular system formation (26). Gα12 could not rescue the function of Gα13 in these mice. In reconstitution experiments, Gα13 stimulated the GEF activity of p115RhoGEF. However, Gα12 could not stimulate the GEF activity of p115 and competitively inhibited the stimulatory effect of Gα13 (7). In this report, we demonstrated that the tyrosine phosphorylation of LARG is required for Gα12 to activate Rho. Although tyrosine phosphorylation of LARG could further stimulate the effect of Gα13, it was not required for Rho activation by Gα13. These differences in the regulatory mechanisms of Gα12- and Gα13-mediated pathways may be responsible for the different cellular effects induced by Gα12 or Gα13.
Because the TH domain was required for Tec to stimulate Gα12-LARG-mediated SRF activation, this domain may be involved in the interaction with Gα12 similar to the case of Btk. However, we could not detect the activation of Tec kinase by Gα12 in vitro (data not shown). The exact mechanism of Gα12 to regulate Tec kinase is currently unclear. However, it is interesting to note that thrombin, which can activate the Gα12/13 pathway, has also been reported to activate Tec in platelets (17). It is possible that activated Gα12 may recruit Tec in close proximity of LARG and facilitate the phosphorylation of LARG. Tec is also activated by other stimuli, such as cytokines and growth factors. These signaling pathways will also be able to regulate the Gα12-LARG pathway.
In addition to Tec, the involvement of Pyk2 in the G12/13-RhoGEF pathway has been reported (27). Furthermore, Chikumi et al. (28) recently demonstrated that thrombin stimulation activated nonreceptor tyrosine kinase FAK in HEK293 cells and that activated FAK could phosphorylate PDZ-RhoGEF or LARG but not p115RhoGEF. They also demonstrated the enhancement of Rho activation by coexpression of activated FAK and PDZ-RhoGEF in cells. They proposed that tyrosine phosphorylation of PDZ-RhoGEF or LARG by FAK might be involved in the activation of Rho. However, its biochemical mechanism remained unclear. We demonstrated here that tyrosine phosphorylation of LARG by Tec does not affect its basal RhoGEF activity, but rather changes its regulation by Gα subunits. It is possible that the activity of PDZ-RhoGEF is also regulated by tyrosine phosphorylation. The modulation of the Gα12/13-RhoGEF pathway by tyrosine kinases may be a widely used mechanism for G protein-coupled receptor-mediated Rho activation.
Acknowledgments
We thank Dr. T. Nagase for providing cDNAs for KIAA0380 and KIAA0382 and P. M. Sternweis for the assistance in cloning full-length LARG cDNA. This work was supported in part by the National Institutes of Health and the American Heart Association (T.K.). T.K. is an Established Investigator of the American Heart Association.
Abbreviations
- GEF
guanine nucleotide exchange factor
- LARG
leukemia-associated RhoGEF
- TH
Tec homology
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Hall A. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 2.Whitehead I P, Campbell S, Rossman K L, Der C J. Biochim Biophys Acta. 1997;1332:F1–F23. doi: 10.1016/s0304-419x(96)00040-6. [DOI] [PubMed] [Google Scholar]
- 3.Gohla A, Harhammer R, Schultz G. J Biol Chem. 1998;273:4653–4659. doi: 10.1074/jbc.273.8.4653. [DOI] [PubMed] [Google Scholar]
- 4.Aragay A M, Collins L R, Post G R, Watson A J, Feramisco J R, Brown J H, Simon M I. J Biol Chem. 1995;270:20073–20077. doi: 10.1074/jbc.270.34.20073. [DOI] [PubMed] [Google Scholar]
- 5.Kranenburg O, Poland M, van Horck F P G, Drechsel D, Hall A, Moolenaar W H. Mol Biol Cell. 1999;10:1851–1857. doi: 10.1091/mbc.10.6.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kozasa T, Jiang X, Hart M J, Sternweis P M, Singer W D, Gilman A G, Bollag G, Sternweis P C. Science. 1998;280:2109–2111. doi: 10.1126/science.280.5372.2109. [DOI] [PubMed] [Google Scholar]
- 7.Hart M J, Jiang X, Kozasa T, Roscoe W, Singer W D, Gilman A G, Sternweis P C, Bollag G. Science. 1998;280:2112–2114. doi: 10.1126/science.280.5372.2112. [DOI] [PubMed] [Google Scholar]
- 8.Buhl A M, Johnson N L, Dhanasekaran N, Johnson G L. J Biol Chem. 1996;270:24631–24634. doi: 10.1074/jbc.270.42.24631. [DOI] [PubMed] [Google Scholar]
- 9.Crespo P, Schuebel K E, Ostrom A A, Gutkind J S, Bustelo X R. Nature. 1997;385:169–172. doi: 10.1038/385169a0. [DOI] [PubMed] [Google Scholar]
- 10.Schuebel K E, Movilla N, Rosa J L, Bustelo X R. EMBO J. 1998;17:6608–6621. doi: 10.1093/emboj/17.22.6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kato J, Kaziro Y, Satoh T. Biochem Biophys Res Commun. 2000;268:141–147. doi: 10.1006/bbrc.2000.2106. [DOI] [PubMed] [Google Scholar]
- 12.Katoh H, Aoki J, Yamaguchi Y, Kitano Y, Ichikawa A, Negishi M. J Biol Chem. 1998;273:28700–28707. doi: 10.1074/jbc.273.44.28700. [DOI] [PubMed] [Google Scholar]
- 13.Klages B, Brandt U, Simon M I, Schultz G, Offermanns S. J Cell Biol. 1999;144:745–754. doi: 10.1083/jcb.144.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mao J, Xie W, Yuan H, Simon M I, Mano H, Wu D. EMBO J. 1998;17:5638–5646. doi: 10.1093/emboj/17.19.5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang Y, Ma W, Wan Y, Kozasa T, Hattori S, Huang X-Y. Nature. 1998;395:808–813. doi: 10.1038/27454. [DOI] [PubMed] [Google Scholar]
- 16.Mano H. Cytokine Growth Factor Rev. 1999;10:267–280. doi: 10.1016/s1359-6101(99)00019-2. [DOI] [PubMed] [Google Scholar]
- 17.Hamazaki Y, Kojima H, Mano H, Nagata Y, Todokoro K, Abe T, Nagasawa T. Oncogene. 1998;16:2773–2780. doi: 10.1038/sj.onc.1201799. [DOI] [PubMed] [Google Scholar]
- 18.Nagase T, Ishikawa K, Nakajima D, Ohira M, Seki N, Miyajima N, Kotani H, Nomura N, Ohara O. DNA Res. 1997;4:141–150. doi: 10.1093/dnares/4.2.141. [DOI] [PubMed] [Google Scholar]
- 19.Yoshida K, Yamashita Y, Miyazato A, Ohya K, Kitanaka A, Ikeda U, Shimada K, Yamanaka T, Ozawa K, Mano H. J Biol Chem. 2000;275:24945–24952. doi: 10.1074/jbc.M909012199. [DOI] [PubMed] [Google Scholar]
- 20.Kozasa T. In: G Proteins: Analysis of Technique. Manning D R, editor. Boca Raton, FL: CRC; 1999. pp. 23–38. [Google Scholar]
- 21.Ren X D, Schwartz M A. Methods Enzymol. 2000;325:264–272. doi: 10.1016/s0076-6879(00)25448-7. [DOI] [PubMed] [Google Scholar]
- 22.Kourlas P J, Strout M P, Becknell B, Veronese M L, Croce C M, Theil K S, Krahe R, Ruutu T, Knuutila S, Bloomfield C D, Caligiuri M A. Proc Natl Acad Sci USA. 2000;97:2145–2150. doi: 10.1073/pnas.040569197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukuhara S, Murga C, Zohar M, Igishi T, Gutkind J S. J Biol Chem. 1999;274:5868–5879. doi: 10.1074/jbc.274.9.5868. [DOI] [PubMed] [Google Scholar]
- 24.Fukuhara S, Chikumi H, Gutkind J S. FEBS Lett. 2000;485:183–188. doi: 10.1016/s0014-5793(00)02224-9. [DOI] [PubMed] [Google Scholar]
- 25.Fromm C, Coso O A, Montaner S, Xu N, Gutkind J S. Proc Natl Acad Sci USA. 1997;94:10098–10103. doi: 10.1073/pnas.94.19.10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Offermanns S, Mancino V, Revel J-P, Simon M I. Science. 1997;275:533–536. doi: 10.1126/science.275.5299.533. [DOI] [PubMed] [Google Scholar]
- 27.Shi C S, Sinnarajah S, Cho H, Kozasa T, Kehrl J H. J Biol Chem. 2000;275:24470–24476. doi: 10.1074/jbc.M908449199. [DOI] [PubMed] [Google Scholar]
- 28.Chikumi H, Fukuhara S, Gutkind J S. J Biol Chem. 2002;277:12463–12473. doi: 10.1074/jbc.M108504200. [DOI] [PubMed] [Google Scholar]