

Abstract
We have recently reported that female mice are protected to a much greater extent from the injurious effects of reduced-size liver ischemia and reperfusion (RSL+I/R) than are males by an estrogen-dependent mechanism. The objective of this study was to examine the possibility that the protective effect observed in female mice depends on the up-regulation and/or activation of endothelial cell NO synthase (eNOS). Anesthetized female and male wild-type or eNOS-deficient C57BL/6 mice were subjected to 70% liver ischemia for 45 min followed by resection of the remaining 30% nonischemic lobes and reperfusion of ischemic tissue. Survival was monitored daily, whereas liver injury was quantified by using serum alanine aminotransferase determinations and histopathology. Hepatic eNOS mRNA, protein, and enzymatic activity were determined in male and female mice subjected to RSL+I/R. We found that liver injury was reduced and survival increased in female mice compared with males. This protective effect correlated with significant increases in hepatic eNOS message levels and enzyme activity but not protein expression compared with males subjected to the surgery. Furthermore, Nω-nitro-l-arginine methyl ester-treated or eNOS-deficient female mice responded to RSL+I/R with dramatic increases in liver injury and 100% mortality within 2 days of surgery. Finally, we found that pravastatin pretreatment significantly attenuated hepatocellular injury and increased survival of male mice, which was associated with enhanced expression of eNOS message. We conclude that the protective effect afforded female mice is due to the activation of hepatic eNOS activity and enhanced NO production.
Hepatic resection in conjunction with ischemia and reperfusion (I/R) is a common occurrence in resectional surgery as well as reduced-size or split liver transplantation (1, 2). Reduced-size liver (RSL) transplantation is a relatively recent surgical alternative to whole liver transplantation, which is being applied to expand the depleted donor organ pool (1, 2). Although RSL transplantation surgery has been relatively successful, there remain substantial risks involved for both donors and recipients, with a significant number of patients developing liver failure after this type of procedure. Because patients typically develop delayed liver failure within 3–7 days of surgery, it is thought that the I/R induced by crossclamping the portal venous and hepatic arterial blood supply and the subsequent graft preservation process result in hepatocellular injury, which may impair liver regeneration and lead to primary graft dysfunction and failure (2). Indeed, recent work by Selzner et al. (3) has shown that I/R impairs the regenerative capacity of the liver, although a protective strategy was not identified or proposed.
It is becoming increasingly appreciated that gender differences exist in susceptibility to and mortality from a variety of vascular diseases, including vascular occlusive disease, hypertension, stroke, shock, and atherosclerosis (4–6). In addition, retrospective clinical studies suggest that donor and/or recipient gender may influence the outcome of whole liver transplantation. For example, a recent study dealing with the long-term survival outcomes of a large cohort of liver transplant recipients in the U.S. has shown that patient survival was significantly better in female recipients than in males (7). Furthermore, it has been reported that female patients with hepatocellular carcinoma have better survival and lower tumor recurrence rates after surgical resection, although the underlying mechanism(s) remain to be elucidated (8, 9).
We have recently reported remarkable sexual dimorphism in a mouse model of RSL+I/R (10). We demonstrated that female mice are protected to a much greater extent from the injurious effects of RSL+I/R than are male mice via a 17β-estradiol (estrogen)-dependent mechanism. Furthermore, we found that treatment of male mice with estrogen reduces liver injury and enhances survival of these mice after RSL+I/R (10). The mechanisms by which endogenous or exogenous estrogen protects the liver from the injurious effects of RSL+I/R have not been identified. Estrogen, however, is known to have dramatic effects on the expression and activity of the endothelial cell isoform of NO synthase (eNOS). Estrogen rapidly induces the release of NO from eNOS via a nongenomic yet estrogen receptor-α (ER-α)- and Ca2+-dependent manner (11–15). Several studies have demonstrated the presence of plasmalemmal caveolae-associated ER-α, which binds estrogen and mediates Ca2+ uptake and “activation” of eNOS via serine/threonine kinase Akt-activation and involved the translocation of eNOS from the plasma membrane to perinuclear region in the cytosol (11, 14). In addition, estrogen/ER-α transcriptionally activates the expression of eNOS in endothelial cells via the more classical genomic mechanisms (16–19). Because we have recently demonstrated that hepatic eNOS plays an important role in limiting I/R-induced liver injury in vivo in a model of full-size liver I/R injury (20, 21), we hypothesized that the protective mechanisms observed in females subjected to RSL+I/R may depend on the differential up-regulation and/or activation of hepatic eNOS in female mice subjected to the surgery.
Materials and Methods
Animals.
Age-matched (7- to 10-wk-old, 16- to 23-g) male and female C57BL/6 wild-type and eNOS-deficient (eNOS−/−) mice were purchased from The Jackson Laboratory. All experimental procedures complied with the Guide for the Care and Use of Laboratory Animals (22), approved by the Council of the American Physiological Society, and with federal and state regulations.
Animal Model of Reduced Size Liver I/R.
Fasted (16–18 h) mice were anesthetized with intramuscular ketamine (150 mg/kg) and xylazine (7.5 mg/kg) and subjected to RSL+I/R as described (10). Briefly, after the midline laparotomy, 70% liver ischemia was achieved by occluding the hepatic arterial and portal venous blood supply to the left lateral and median lobes by using a microaneurysm clip. Mice were then given an i.p. dose of heparin (200 units/kg) to prevent blood coagulation. After 45 min of ischemia, the clip was removed, and the nonischemic lobes were excised, leaving the remaining two reperfused lobes. Sham-operated control mice were treated in an identical fashion without hepatectomy or vascular clamping. The abdominal cavity was closed, and the animals were allowed to recover with free access to food and water. Mice were then observed daily until day 8 postsurgery to assess survival or were euthanized at different time points postsurgery, and liver tissues and blood samples were obtained for analysis.
l-NAME and/or Pravastatin Treatment.
eNOS was inhibited in vivo by using the NOS inhibitor Nω-nitro-l-arginine methyl ester (l-NAME (Sigma) 15 mg/kg, i.v. 15 min before surgery) (23). To investigate the effects of pravastatin, a 3-hydroxy-3-methylglutaryl CoA reductase inhibitor, on animal survival, liver injury, and eNOS expression, male mice were treated with 1, 2, or 5 mg/kg pravastatin (i.p.) in PBS or PBS vehicle 24 and 6 h before surgery.
Blood and Tissue Analyses.
Serum alanine aminotransferase (ALT) levels were determined by using commercially available reagents (Sigma). In addition, liver tissue was removed, weighed, and then snap frozen in liquid nitrogen and stored at −70°C for RNA isolation or stored in 10% PBS-buffered formalin for histopathological analysis.
Hepatic eNOS mRNA, Enzyme Activity, and Protein Expression.
eNOS message expression.
Hepatic eNOS mRNA was determined by using semiquantitative RT-PCR 1 h after surgery. Total RNA was isolated from liver tissue by using Trizol reagent (GIBCO/BRL). One microgram of RNA was reverse-transcribed to complementary DNA by using MuLV Reverse Transcriptase (Applied Biosystems) and amplified by using the following primers for the mouse eNOS gene: sense, 5′-GCAGAAGAGTCCAGCGAACA-3′; antisense, 5′-GGCAGCCAAACACCAAAGTC-3′. Thermal cycle conditions were 94°C for 30 sec, 58°C for 30 sec, and 72°C for 30 sec, for a total of 30 cycles. The final cycle was followed with a 5-min incubation at 72°C. PCR amplification of a housekeeping gene (GAPDH) was performed by using the same cDNA reaction. RT-PCR products were viewed by ethidium bromide staining and analyzed by densitometry by using an Alpha Innotech gel documentation system (San Leandro, CA). eNOS mRNA expression was illustrated by determining the ratio of band intensity of eNOS and GAPDH and presented as relative values to controls.
NOS activity measurements.
The l-NAME inhibitable conversion of l-arginine to l-citruline in liver extracts was used to determine NOS activity by using a minor modification of the method of Shah et al. (24). Briefly, livers were homogenized in lysis buffer (50 mM Tris⋅HCl/0.1 mM EDTA/0.1 mM EGTA/0.1% SDS/1% Nonidet P-40/0.1% deoxycholic acid, pH 7.5). Homogenates were applied to a Dowex AG 50WX-8 resin (Sigma). Samples were incubated at 37°C for 20 min in a reaction buffer containing 1 mM NADPH, 0.1 μM calmodulin, 2.5 mM CaCl2, 30 μM tetrahydrobiopterin, 50 μM Nω-hydroxy-nor-l-arginine, and 10 μM l-[14C]arginine. To determine NOS activity, duplicate samples were incubated in the presence or absence of 2 mM l-NAME. The reactions were terminated by the addition of a 1:1 stop buffer (20 mM Hepes/2 mM EDTA/2 mM EGTA, pH 5.5): Dowex AG 50WX-8 slurry. Radiolabeled cpm of l-citrulline generated were measured, and l-NAME-inhibited NOS activity was determined.
Western Blot Analysis.
Liver tissue was homogenized in lysis buffer used for eNOS activity assay. Protein quantification was performed by using BCA reagent (Pierce). Total liver lysates (100 μg) were separated by SDS/PAGE on a 7.5% acrylamide gel, and protein was electroblotted onto nitrocellulose membrane. Membranes were stained with Ponceau S to confirm equal protein loading and transfer between lanes. Membranes were blocked in Tris-buffered saline with 0.1% Tween-20 and incubated with eNOS mAb (Transduction Laboratories, San Diego). Detection of eNOS was performed by using enhanced chemiluminescence (Amersham Pharmacia Biosciences).
Histology.
Formalin-fixed liver specimens were embedded in JB-4 plastic (Polysciences, Warrington, PA), and 5-μm sections were cut and stained with hematoxylin/eosin. Histological assessment was performed by using liver specimens obtained 20 h after RSL+I/R.
Statistical Analyses.
All values are expressed as mean ± SEM. Statistical significance between two groups of parametric data was evaluated by using an unpaired Student's t test. Survival curves for 7 days were analyzed by using the Kaplan–Meier method, and the differences in survival rates were evaluated by using the generalized Wilcoxon's test. Statistical significance was accepted at P < 0.05.
Results
Sexual Dimorphism and Hepatic eNOS Expression After RSL+I/R.
We have previously shown that female mice are protected from RSL+I/R-induced injury and survive indefinitely after surgery, whereas all male mice die within 5 days after RSL+I/R (10). To test the hypothesis that the up-regulation and/or activation of hepatic eNOS accounts for the gender difference and mediates the protective effect observed in females, we first evaluated hepatic eNOS mRNA expression in both male and female mice 1 h after RSL+I/R or sham-surgery. We found that sham-operated female mice exhibited a 3-fold increase in hepatic eNOS message levels compared with male mice (Fig. 1A). Furthermore, RSL+I/R enhanced further hepatic eNOS message in female mice (but not in male mice), resulting in a 9-fold increase in eNOS message levels in female mice compared with their male counterparts (Fig. 1A). Hepatic eNOS mRNA levels 20 h after RSL+I/R were enhanced to similar levels in both male and female mice (data not shown). It should be noted that neither inducible NOS (iNOS) message nor iNOS protein was expressed any time after RSL+I/R up to 20 h (data not shown). The increase in hepatic eNOS mRNA in females correlated with enzymatic activity. We observed an increase in hepatic eNOS activity in females of ≈2-fold compared with males 3 h after the surgery (Fig. 1B). Interestingly, this increase in enzymatic activity was not due to increases in eNOS protein expression; we found no difference in eNOS protein in male vs. female mice 3 h after RSL+I/R (Fig. 1C).
Fig 1.

(A) Hepatic eNOS message levels from male and female mice 1 h after RSL+I/R. (Upper) eNOS and GAPDH mRNA expression. (Lower) eNOS expression normalized to GAPDH and expressed as relative changes over sham-operated males. *, P < 0.05 compared with RSL+I/R males; **, P < 0.05 compared with sham males. (B) Hepatic NOS activity in males and females 3 h after RSL+I/R. *, P < 0.05 compared with RSL+I/R males (n = six for each group). (C) Hepatic eNOS protein levels in males and females 3 h after RSL+I/R.
Role of eNOS in Survival and Liver Injury After RSL+I/R.
To directly test our hypothesis that hepatic eNOS protects female mice from the injurious effects of the RSL+I/R injury, we subjected eNOS−/− male and female mice to RSL+I/R and assessed survival and liver injury in these mice. We found that 100% of both female and male eNOS−/− mice died within 2 days of surgery (Fig. 2A). In addition to survival, we also quantified and compared serum levels of ALT in both male and female wild-type and eNOS−/− mice 3 h after RSL+I/R. We found that serum ALT levels were significantly greater in male vs. female wild-type mice (Fig. 2B). In addition, we observed further increases in serum ALT levels in eNOS−/− male and female mice compared with their wild-type counterparts (Fig. 2B). This enhancement in serum ALT levels was not significantly different between eNOS−/− male and female mice. Because eNOS−/− mice may compensate for the lack of this enzyme during development, we also examined the role of eNOS by using the NOS inhibitor l-NAME. We found that pretreatment of female mice with l-NAME 15 min before surgery resulted in 100% mortality within 2 days of surgery (Fig. 3A). This increased mortality was associated with a dramatic enhancement in liver injury 3 h postsurgery compared with female mice subjected to the surgery plus vehicle (Fig. 3B).
Fig 2.

(A) Survival of male and female wild-type (wt) and eNOS−/− mice after RSL+I/R. Survival for 7 days was 0% for wild-type males (n = 10) and 100% for wild-type females (n = 9) (P < 0.001 compared with wild-type male mice). All eNOS−/− female mice (n = 6) died within 2 days of RSL+I/R. No eNOS−/− male (n = 6) mice survived beyond 1 day. (B) Serum ALT levels in male and female wild-type (wt) and eNOS−/− mice 3 h after RSL+I/R. *, P < 0.05 and **, P < 0.01.
Fig 3.

(A) Survival of female wild-type mice treated with L-NAME (15 mg/kg) or vehicle before RSL+I/R. Survival for 7 days was 100% for vehicle-treated mice (n = 7), whereas all L-NAME-treated mice (n = 7) died within 2 days of RSL+I/R (P < 0.01 compared with vehicle-treated mice). (B) Serum ALT levels in female mice treated with either L-NAME or vehicle 3 h after RSL+I/R. *, P < 0.05 compared with vehicle-treated controls.
Hepatic histopathology 20 h after RSL+I/R correlated well with serum ALT levels, in that we observed more injury in male vs. female wild-type mice (see Fig. 5, which is published as supporting information on the PNAS web site, www.pnas.org). Interestingly, eNOS−/−- or l-NAME-treated female mice exhibited enhanced liver injury compared with wild-type or vehicle-treated counterparts that was not different from male eNOS−/− mice (see Figs. 5–7, which are published as supporting information on the PNAS web site). In addition, neutrophil infiltration was greater in eNOS−/− female mice than in wild-type females (Figs. 5 and 6). It should be noted that virtually no neutrophil infiltration was observed in wild-type male or female mice subjected to RSL+I/R earlier than 20 h postsurgery (data not shown).
Effect of Pravastatin Treatment on Survival, Liver Injury, and eNOS Expression After RSL+I/R.
It is well known that certain 3-hydroxy-3-methylglutaryl CoA reductase inhibitors increase eNOS expression and activity in vascular endothelial cells in vitro and in vivo (25–27). To evaluate the effects of pravastatin on survival and liver injury in mice subjected to RSL+I/R, we examined 7-day survival and serum ALT levels as well as hepatic eNOS mRNA expression levels after RSL+I/R in vehicle-, pravastatin- (2 mg/kg i.p.), or pravastatin + l-NAME-treated male mice. We found that the 7-day survival was significantly higher in pravastatin mice than in vehicle- or pravastatin + l-NAME-treated mice (Fig. 4A). Furthermore, serum ALT levels were significantly reduced in pravastatin-treated mice compared with vehicle or pravastatin + l-NAME-treated mice (Fig. 4B). We determined that increasing the dose of paravastatin to 5 mg/kg did not provide any additional protection to the mice, whereas decreasing the dose to 1 mg/kg eliminated this protective effect as assessed by serum ALT levels (3,934 ± 600, 2,252 ± 215, 1,370 ± 225, and 1,536 ± 253 for vehicle, 1, 2, and 5 mg/kg-treated mice, respectively; P < 0.05 for 2 and 5 mg/kg compared with vehicle-treated group). The protective effects of 2 and 5 mg/kg but not 1 mg/kg pravastatin correlated with improved 7-day survival (data not shown). In addition, the protective effect of 2 mg/kg pravastatin correlated with enhanced (3.8-fold) hepatic expression of eNOS mRNA in pravastatin-treated mice compared with vehicle-treated mice (Fig. 4C).
Fig 4.

Effect of pravastatin on survival and liver injury in male mice after RSL+I/R. (A) Survival of male mice treated with pravastatin (2 mg/kg), vehicle, or pravastatin plus L-NAME (15 mg/kg). Pravastatin was administered i.p. 24 and 6 h before surgery, whereas L-NAME was given i.v. 15 min before surgery. Survival rates for 7 days were 11% in vehicle-treated mice (n = 9); however, treatment with pravastatin significantly improved the survival rate to 50% (n = 10), whereas mice treated with the combination of L-NAME and pravastatin resulted in 0% survival by day 3 (P < 0.05 compared with vehicle-treated controls). (B) Serum ALT levels in male mice treated with pravastatin, vehicle, or pravastatin + L-NAME 3 h after RSL+I/R. **, P < 0.05 compared with vehicle- or pravastatin + L-NAME-treated mice. (C) Effect of vehicle, pravastatin, or pravastatin + L-NAME on hepatic eNOS expression in male mice after RSL+I/R. *, P < 0.05 compared with vehicle-treated controls.
Discussion
We have recently reported remarkable sexual dimorphism in a mouse model of RSL+I/R (10). We demonstrated that female mice are protected to a much greater extent from the injurious effects of RSL+I/R than are male mice via estrogen-dependent mechanisms. However, the mechanisms by which estrogen protects female mice from the injurious effects of RSL+I/R were not identified. We hypothesized that estrogen may act to enhance expression and/or enzymatic activity of eNOS. Previous work from our laboratory suggested that eNOS is important in limiting the extent of postischemic liver injury in full size liver I/R (20, 21). The results of the current study clearly demonstrate that the protective effect afforded to female mice is associated with increases in hepatic eNOS activity. Indeed, although we demonstrated in previous studies that estrogen was sufficient for the protection in female mice subjected to RSL+I/R (10), data obtained in the present study show that eNOS is required for this protective effect. In addition, we demonstrate, to our knowledge for the first time, that pretreatment of male mice with pravastatin attenuates RSL+I/R-induced injury and increases survival in what appears to be an eNOS-dependent mechanism.
Although eNOS was originally described as a constitutive enzyme, it has become clear over the past few years that eNOS expression and activity may be regulated by several different biochemical, cellular, and physical stimuli, including estrogen, hypoxia, shear stress, oxidants such as hydrogen peroxide, and cell proliferation (28–30). The mechanisms by which female eNOS activity is enhanced and thus protects the liver from the injurious effects of RSL+I/R are not known. However, our previous studies suggest that it may be mediated by estrogen- as well as hypoxia- and oxidant-dependent mechanisms (10, 28–30). Indeed, it is well known that physiological responses of estrogen may be mediated by genomic and/or nongenomic responses. For example, estrogen may increase NO production through the transcriptional up-regulation of eNOS expression in vascular endothelial cells as well as sinusoidal endothelial cells from liver (16–19). Consistent with these observations, we found that basal eNOS mRNA expression in female mice was ≈3-fold higher than males (Fig. 1A). Furthermore, we observed that eNOS mRNA expression was enhanced further after 45 min of ischemia, resection, and 1-h reperfusion of the reduced size liver such that female eNOS message levels were increased 2.6-fold over their basal levels and 9-fold over males subjected to RSL+I/R (Fig. 1A). The mechanisms responsible for this relatively rapid and dramatic up-regulation of eNOS message in females are not known at the present time, but it is clear that eNOS expression may be enhanced by hypoxia (ischemia) and by specific oxidants such as hydrogen peroxide that are thought to be generated during reperfusion (28–30). The large and significant increases in message levels of eNOS correlated well with increases in enzymatic activity of eNOS. We found that female mice exhibited approximately twice the hepatic NOS activity when compared with males (Fig. 1B). Because RSL+I/R does not induce the expression of inducible NOS in our model (data not shown), we propose that the increase in hepatic NOS activity is most probably due to the increases in eNOS activity. However, our observation that the increase in eNOS activity in females was not due to increased translation of eNOS message to protein was surprising. We found no difference between males and females with respect to eNOS protein expression (Fig. 1C). Taken together, these data suggest a nongenomic mechanism for activation of eNOS and the subsequent protection from liver injury. It is well appreciated that eNOS-derived NO may be rapidly increased by several different mechanisms, including vascular sheer stress, oxidant exposure and estrogen-dependent activation of existing eNOS (13, 14). Indeed, several laboratories have demonstrated the presence of plasmalemmal caveolae-associated estrogen receptor-α that binds estrogen and mediates Ca2+ uptake and “activation” of eNOS. Additional studies demonstrated that this rapid nongenomic production of eNOS-derived NO depends on the serine/threonine kinase Akt activation and involves the translocation of eNOS from the plasma membrane to perinuclear region in the cytosol (13, 14). Studies to assess the role of the Akt in our model of RSL+I/R are currently underway. The cellular source of eNOS in the mouse liver was not assessed in the present study. However, several other laboratories have demonstrated that eNOS appears to be localized exclusively to the sinusoidal endothelial cells in rodent liver (19, 31, 32).
The role of eNOS as a crucial component for the protective effects afforded to female mice was confirmed in studies using l-NAME-treated or eNOS−/− female and male mice. We found that l-NAME-treated or eNOS−/− female mice responded to RSL+I/R with dramatic increases in liver injury and mortality compared with their wild-type controls (Figs. 2 and 3). Because exogenously administered estrogen to eNOS−/− female (or male) mice failed to attenuate liver injury and improve survival (data not shown), we conclude that eNOS activity is required for the protective effects.
Taken together, these data predict that manipulations of wild-type mice to enhance hepatic eNOS may prove beneficial in protecting against RSL+I/R induced injury and mortality. This hypothesis was tested by using pravastatin, a 3-hydroxy-3-methylglutaryl (HMG) CoA reductase inhibitor and known inducer of eNOS expression and activity in vascular endothelial cells. HMG CoA reductase is a key enzyme for cholesterol synthesis in the liver, and its inhibitors (collectively known as “statins”) are widely used clinically to treat individuals with hypercholesterolemia. In addition to their cholesterol-lowering actions, recent studies suggest that statins may also exert their cardiovascular-protective effects by enhancing the expression of eNOS via stabilization of its mRNA (25–27). Although several prospective clinical trials have convincingly demonstrated that statins can effectively lower the incidence of cardiovascular events, their usefulness in the treatment of different liver disorders has not been extensively studied. Several recent investigations, however, have demonstrated that statins possess hepatoprotective properties after hepatectomy or liver transplantation in rats (33, 34). Accordingly, we found that pravastatin significantly reduced injury and increased survival after RSL+I/R that was associated with enhanced eNOS message expression (Fig. 4 A and B). Thus, we provide, to our knowledge for the first time, evidence that pravastatin also exerts a hepatoprotective effect in a mouse model of RSL+I/R.
The precise mechanisms by which enhanced eNOS-derived NO protects female mice from the injurious effects of RSL+I/R remain to be identified. One possible mechanism may involve the antioxidant properties of eNOS-derived NO (35, 36). For example, data obtained from several different laboratories suggest that the acute postischemic hepatocellular injury may be mediated directly or indirectly by reactive oxygen species (ROS) generated during the early reperfusion period (37). It is now well appreciated that the initial phase of hepatic I/R injury is characterized by Kupffer cell activation with the release of ROS, including superoxide [O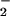 and hydrogen peroxide (H2O2)] (37–39). This process may require activation of a redox-related enzyme NADPH oxidase. Interestingly, estrogen has been shown to reduce the expression of Kupffer cell-associated NADPH oxidase, and this inhibition may partially account for our findings that hepatic eNOS expression in female or estrogen-treated male animals is enhanced and remains elevated for several hours after RSL+I/R (40). In addition, it is known that NO production and/or bioavailability are reduced in postischemic tissue (41). Because NO is known to inhibit ROS-mediated reactions, it has been suggested that the protective effects in a variety of cardiovascular disorders and diseases are due to the ability of NO to detoxify ROS such as O
and hydrogen peroxide (H2O2)] (37–39). This process may require activation of a redox-related enzyme NADPH oxidase. Interestingly, estrogen has been shown to reduce the expression of Kupffer cell-associated NADPH oxidase, and this inhibition may partially account for our findings that hepatic eNOS expression in female or estrogen-treated male animals is enhanced and remains elevated for several hours after RSL+I/R (40). In addition, it is known that NO production and/or bioavailability are reduced in postischemic tissue (41). Because NO is known to inhibit ROS-mediated reactions, it has been suggested that the protective effects in a variety of cardiovascular disorders and diseases are due to the ability of NO to detoxify ROS such as O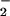 , OH⋅, and/or ferryl hemoprotein (35). Another mechanism by which increased eNOS-derived NO may protect in our model of RSL+I/R is vasodilation and enhanced perfusion of the postischemic remnant tissue via a cGMP-dependent mechanism (13, 14).
, OH⋅, and/or ferryl hemoprotein (35). Another mechanism by which increased eNOS-derived NO may protect in our model of RSL+I/R is vasodilation and enhanced perfusion of the postischemic remnant tissue via a cGMP-dependent mechanism (13, 14).
Other mechanisms by which eNOS-derived NO may protect the liver from RSL+I/R-induced injury may be via inhibition of platelet aggregation and adhesion as well as attenuation of endothelium–leukocyte interactions, all of which may contribute to reducing hepatic I/R injury (42). Full-size liver I/R is characterized by a late-phase injury (>6 h) involving a complex inflammatory pathway that culminates with hepatic accumulation of neutrophils. This process includes surface expression of endothelial cell adhesion molecules, proinflammatory cytokines, and liver-directed chemokines, all of which contribute to neutrophil adhesion to the vascular wall and transmigration into the liver tissue. In our model of RSL+I/R, however, little or no neutrophil accumulation was observed in wild-type male or female mice within the first 20 h of reperfusion. However, we did observe large and significant neutrophil infiltration in eNOS−/− male and female animals, suggesting that eNOS-derived NO may play an important role in limiting neutrophil adhesion and/or emigration into the tissue, thereby limiting hepatocellular injury in these mutant mice.
Finally, increased production of eNOS-derived NO may protect female mice after RSL+I/R by protecting the liver from the injurious effects of the growth-promoting cytokines TNF-α and IL-6, thereby enhancing liver angiogenesis and regeneration. Indeed, Rai et al. (43) suggest that NO is critical for normal restoration of liver mass after partial hepatectomy by preventing or limiting the proapoptotic activity of TNF-α. In summary, data obtained in the present study demonstrate, to our knowledge for the first time, that the protective effects afforded female mice subjected to RSL+I/R surgery critically depend on the presence of eNOS. These results may have important implications for the design of new therapeutic strategies for protecting the tissue from the injurious effects of liver resectional and/or transplantation surgery.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (DK43785) and the Arthritis Center of Excellence at Louisiana State University Health Sciences Center (Shreveport).
Abbreviations
I/R, ischemia and reperfusion
l-NAME, Nω-nitro-l-arginine methyl ester
NOS, NO synthase
eNOS, endothelial cell NOS
RSL, reduced-size liver
ALT, alanine aminotransferase
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Keeffe E. B. (2001) Gastroenterology 120, 749-762. [DOI] [PubMed] [Google Scholar]
- 2.Strasberg S. M., Lowell, J. A. & Howard, T. K. (1999) Liver Transpl. Surg. 5, 437-450. [DOI] [PubMed] [Google Scholar]
- 3.Selzner M., Camargo, C. A. & Clavien, P. A. (1999) Hepatology 30, 469-475. [DOI] [PubMed] [Google Scholar]
- 4.Hayward C. S., Kelly, R. P. & Collins, P. (2000) Cardiovasc. Res. 46, 28-49. [DOI] [PubMed] [Google Scholar]
- 5.Dubey R. K. & Jackson, E. K. (2001) J. Appl. Physiol 91, 1868-1883. [DOI] [PubMed] [Google Scholar]
- 6.Mendelsohn M. E. & Karas, R. H. (1999) N. Engl. J. Med. 340, 1801-1811. [DOI] [PubMed] [Google Scholar]
- 7.Jain A., Reyes, J., Kashyap, R., Dodson, S. F., Demetris, A. J., Ruppert, K., Abu-Elmagd, K., Marsh, W., Madariaga, J., Mazariegos, G., et al. (2000) Ann. Surg. 232, 490-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee C. C., Chau, G. Y., Lui, W. Y., Tsay, S. H., King, K. L., Loong, C. C., Hshia, C. Y. & Wu, C. W. (2000) Hepatogastroenterology 47, 446-449. [PubMed] [Google Scholar]
- 9.Ng I. O., Ng, M. & Fan, S. T. (1997) Am. J. Gastroenterol. 92, 1355-1358. [PubMed] [Google Scholar]
- 10.Harada H., Pavlick, K. P., Hines, I. N., Hoffman, J. M., Bharwani, S., Gray, L., Wolf, R. E. & Grisham, M. B. (2001) J. Appl. Physiol. 91, 2816-2822. [DOI] [PubMed] [Google Scholar]
- 11.Mendelsohn M. E. (2000) Circ. Res. 87, 956-960. [DOI] [PubMed] [Google Scholar]
- 12.Chen Z., Yuhanna, I. S., Galcheva-Gargova, Z., Karas, R. H., Mendelsohn, M. E. & Shaul, P. W. (1999) J. Clin. Invest. 103, 401-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haynes M. P., Sinha, D., Russell, K. S., Collinge, M., Fulton, D., Morales-Ruiz, M., Sessa, W. C. & Bender, J. R. (2000) Circ. Res. 87, 677-682. [DOI] [PubMed] [Google Scholar]
- 14.Hisamoto K., Ohmichi, M., Kurachi, H., Hayakawa, J., Kanda, Y., Nishio, Y., Adachi, K., Tasaka, K., Miyoshi, E., Fujiwara, N., et al. (2001) J. Biol. Chem. 276, 3459-3467. [DOI] [PubMed] [Google Scholar]
- 15.Rubanyi G. M., Freay, A. D., Kauser, K., Sukovich, D., Burton, G., Lubahn, D. B., Couse, J. F., Curtis, S. W. & Korach, K. S. (1997) J. Clin. Invest. 99, 2429-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kleinert H., Wallerath, T., Euchenhofer, C., Ihrig-Biedert, I., Li, H. & Forstermann, U. (1998) Hypertension 31, 582-588. [DOI] [PubMed] [Google Scholar]
- 17.MacRitchie A. N., Jun, S. S., Chen, Z., German, Z., Yuhanna, I. S., Sherman, T. S. & Shaul, P. W. (1997) Circ. Res. 81, 355-362. [DOI] [PubMed] [Google Scholar]
- 18.Nuedling S., Kahlert, S., Loebbert, K., Doevendans, P. A., Meyer, R., Vetter, H. & Grohe, C. (1999) Cardiovasc. Res. 43, 666-674. [DOI] [PubMed] [Google Scholar]
- 19.Sakamoto M., Uen, T., Nakamura, T., Hashimoto, O., Sakata, R., Kin, M., Ogata, R., Kawaguch, T., Torimura, T. & Sata, M. (2001) J. Hepatol. 34, 858-864. [DOI] [PubMed] [Google Scholar]
- 20.Kawachi S., Hines, I. N., Laroux, F. S., Hoffman, J., Bharwani, S., Gray, L., Leffer, D. & Grisham, M. B. (2000) Biochem. Biophys. Res. Commun. 276, 851-854. [DOI] [PubMed] [Google Scholar]
- 21.Hines I. N., Harada, H., Bharwani, S., Pavlick, K. P., Hoffman, J. M. & Grisham, M. B. (2001) Biochem. Biophys. Res. Commun. 284, 972-976. [DOI] [PubMed] [Google Scholar]
- 22.Institute for Laboratory Animal Research, National Research Council, (1996) Guide for the Care and Use of Laboratory Animals (National Academy Press, Washington, DC).
- 23.Liu P., Xu, B., Spokas, E., Lai, P. S. & Wong, P. Y. (2000) Shock 13, 217-223. [DOI] [PubMed] [Google Scholar]
- 24.Shah V., Cao, S., Hendrickson, H., Yao, J. & Katusic, Z. S. (2001) Am. J. Physiol. 280, G1209-G1216. [DOI] [PubMed] [Google Scholar]
- 25.Laufs U., La, F., V, Plutzky, J. & Liao, J. K. (1998) Circulation 97, 1129-1135. [DOI] [PubMed] [Google Scholar]
- 26.Lefer A. M., Scalia, R. & Lefer, D. J. (2001) Cardiovasc. Res. 49, 281-287. [DOI] [PubMed] [Google Scholar]
- 27.Davignon J. & Laaksonen, R. (1999) Curr. Opin. Lipidol. 10, 543-559. [DOI] [PubMed] [Google Scholar]
- 28.Arnet U. A., McMillan, A., Dinerman, J. L., Ballermann, B. & Lowenstein, C. J. (1996) J. Biol. Chem. 271, 15069-15073. [DOI] [PubMed] [Google Scholar]
- 29.Cai H., Davis, M. E., Drummond, G. R. & Harrison, D. G. (2001) Arterioscler. Thromb. Vasc. Biol. 21, 1571-1576. [DOI] [PubMed] [Google Scholar]
- 30.Drummond G. R., Cai, H., Davis, M. E., Ramasamy, S. & Harrison, D. G. (2000) Circ. Res. 86, 347-354. [DOI] [PubMed] [Google Scholar]
- 31.Shah V., Haddad, F. G., Garcia-Cardena, G., Frangos, J. A., Mennone, A., Groszmann, R. J. & Sessa, W. C. (1997) J. Clin. Invest. 100, 2923-2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yokomori H., Oda, M., Ogi, M., Kamegaya, Y., Tsukada, N. & Ishii, H. (2001) Liver 21, 198-206. [DOI] [PubMed] [Google Scholar]
- 33.Kakkis J. L., Ke, B., Dawson, S., Maggard, M., Si, M., Kaldas, F., Cai, W., Shau, H., Seu, P., Sauri, H., et al. (1997) J. Surg. Res. 69, 393-398. [DOI] [PubMed] [Google Scholar]
- 34.Zivna H., Zivny, P., Palicka, V. & Simakova, E. (2002) Nutrition 18, 51-55. [DOI] [PubMed] [Google Scholar]
- 35.Grisham M. B., Jourd'heuil, D. & Wink, D. A. (1999) Am. J. Physiol. 276, G315-G321. [DOI] [PubMed] [Google Scholar]
- 36.Wink D. A., Miranda, K. M., Espey, M. G., Pluta, R. M., Hewett, S. J., Colton, C., Vitek, M., Feelisch, M. & Grisham, M. B. (2001) Antioxid. Redox. Signal. 3, 203-213. [DOI] [PubMed] [Google Scholar]
- 37.Jaeschke H. (1991) Chem. Biol. Interact. 79, 115-136. [DOI] [PubMed] [Google Scholar]
- 38.Jaeschke H. (1999) Hepatology 30, 1527-1528. [DOI] [PubMed] [Google Scholar]
- 39.Schauer R. J., Bilzer, M., Kalmuk, S., Gerbes, A. L., Leiderer, R., Schildberg, F. W. & Messmer, K. (2001) Transplantation 72, 1692-1699. [DOI] [PubMed] [Google Scholar]
- 40.Wagner A. H., Schroeter, M. R. & Hecker, M. (2001) FASEB J. 15, 2121-2130. [DOI] [PubMed] [Google Scholar]
- 41.Lefer A. M. & Lefer, D. J. (1996) Cardiovasc. Res. 32, 743-751. [PubMed] [Google Scholar]
- 42.Kubes P., Kurose, I. & Granger, D. N. (1994) Am. J. Physiol 267, H931-H937. [DOI] [PubMed] [Google Scholar]
- 43.Rai R. M., Lee, F. Y., Rosen, A., Yang, S. Q., Lin, H. Z., Koteish, A., Liew, F. Y., Zaragoza, C., Lowenstein, C. & Diehl, A. M. (1998) Proc. Natl. Acad. Sci. USA 95, 13829-13834. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}