

Abstract
UL9 is a multifunctional protein essential for herpes simplex virus type 1 (HSV-1) replication in vivo. UL9 is a member of the superfamily II helicases and exhibits helicase and origin-binding activities. It is thought that UL9 binds the origin of replication and unwinds it in the presence of ATP and the HSV-1 single-stranded DNA (ssDNA)-binding protein. We have previously characterized the biochemical properties of mutants in all helicase motifs except for motif Ia (B. Marintcheva and S. Weller, J. Biol. Chem. 276:6605-6615, 2001). Structural information for other superfamily I and II helicases indicates that motif Ia is involved in ssDNA binding. By analogy, we hypothesized that UL9 motif Ia is important for the ssDNA-binding function of the protein. On the basis of sequence conservation between several UL9 homologs within the Herpesviridae family and distant homology with helicases whose structures have been solved, we designed specific mutations in motif Ia and analyzed them genetically and biochemically. Mutant proteins with residues predicted to be involved in ssDNA binding (R112A and R113A/F115A) exhibited wild-type levels of intrinsic ATPase activity and moderate to severe defects in ssDNA-stimulated ATPase activity and ssDNA binding. The S110T mutation targets a residue not predicted to contact ssDNA directly. The mutant protein with this mutation exhibited wild-type levels of intrinsic ATPase activity and near wild-type levels of ssDNA-stimulated ATPase activity and ssDNA binding. All mutant proteins lack helicase activity but were able to dimerize and bind the HSV-1 origin of replication as well as wild-type UL9. Our results indicate that residues from motif Ia contribute to the ssDNA-binding and helicase activities of UL9 and are essential for viral growth. This work represents the successful application of an approach based on a combination of bioinformatics and structural information from related proteins to deduce valuable information about a protein of interest.
The UL9 gene is essential for herpes simplex virus type 1 (HSV-1) replication in vivo (5, 9). UL9 is a multifunctional protein, exhibiting intrinsic and single-stranded DNA (ssDNA)-stimulated ATPase activity, 3′ to 5′ helicase activity, ssDNA-binding and origin-binding activities (8, 15). It is thought that UL9 binds the HSV-1 origin of replication and unwinds it in the presence of ATP and ICP8, the HSV-1 ssDNA-binding protein. In addition, UL9 interacts with several viral proteins that are essential for HSV-1 replication: ICP8 (6); UL42, the processivity subunit of the viral polymerase (34) and UL8, a subunit of the heteromeric helicase/primase complex (33).
UL9 is a superfamily II (SFII) helicase and possesses seven conserved helicase motifs, positioned within the N-terminal domain of the protein (16). Using site-directed mutagenesis, we have previously shown that conserved residues within helicase motifs I to VI of UL9 are essential for HSV-1 replication in vivo; most of the engineered motif mutants fail to complement the growth of UL9 null virus (32). Consistent with the genetic data, the UL9 helicase motif mutant proteins exhibit defects in ATPase and helicase activity in vitro (30). On the other hand, they resemble the wild-type protein in their abilities to dimerize and bind the HSV-1 origin of replication (30). The C terminus of UL9 (amino acids 535 to 851) forms a domain responsible for the recognition of the HSV origins of replication (1, 3), and it is believed that this domain is responsible for targeting UL9 to the origins during the initiation of DNA replication. Once bound at the origin, however, UL9 is expected to unwind the origin and translocate along ssDNA, an activity that requires the ability to bind ssDNA (37). The UL9 residues responsible for ssDNA binding have not been mapped.
The identification of ssDNA-binding residues in helicases of SFI and SFII has been facilitated by the availability of structural data on several SFI and SFII helicases. Despite the enormous variation in primary sequence, both superfamilies share similar structures, including the presence of two RecA-like domains, composed of parallel β sheets, surrounded by α helices (10, 19, 22, 37, 38). In both superfamilies, the conserved helicase motifs are found in similar positions along a cleft between the two RecA-like domains comprising an ATP-binding site. The structures of Rep with ssDNA (21), PcrA with partially dsDNA (40), and the NS3 helicase with oligo(dU8) (20) were also solved. In all three structures, single-stranded nucleic acid was observed to bind in a cleft at the top of the RecA-like domains, oriented perpendicularly to the ATP-binding site, and in both superfamilies, residues from helicase motif Ia are seen to contact ssDNA (20-22, 40). The DNA-protein interactions are mostly with the phosphodiester backbone of the nucleic acid, as expected for a nonspecific interaction with single-stranded nucleic acid (20-22, 40).
Important residues of the HSV-1 UL9 helicase motifs were mapped genetically (32) before any helicase structures were available, and helicase motifs I to VI were shown to play roles in ATP hydrolysis or the coupling of ATPase and helicase activities (30). The large size and difficulty in expressing large amounts of functional UL9 protein have hampered efforts to obtain structural data on this protein. In this work we have taken advantage of the conserved nature of the helicase structure to make the prediction that the UL9 helicase motif Ia is involved in the ssDNA-binding activity of the enzyme. On the basis of amino acid sequence conservation in several UL9 homologs within the Herpesviridae family and distant homology with helicases with solved structure, we designed specific mutations in motif Ia and tested them genetically and biochemically. Our results indicate that residues from motif Ia contribute to the ssDNA-binding and helicase activities of UL9 and are essential for viral growth. This study demonstrates the power of bioinformatics and the application of structural information from related proteins to make and test predictions about the structure and function of a protein for which no structural information is currently available. This type of analysis will become increasingly important for functional studies of proteins of pathogens such as HSV-1. Furthermore, this study confirms the conserved nature of helicase structure and function in diverse members of helicase SFI and SFII. We have shown that predictions about helicase structure and function based on solved structures can be tested and extended in systems such as HSV-1 which are genetically and biochemically manipulable.
MATERIALS AND METHODS
Reagents and materials.
All restriction enzymes were from New England Biolabs. The pFastBac vector was purchased from Gibco BRL. Supplemented Grace's medium and penicillin-streptomycin were purchased from Gibco BRL; fetal calf serum was from Gemini Bio-products, Inc.
Viruses and cells.
HSV-1 KOS strain was used as a wild-type virus in plaque reduction assay. Hr94 (26), a UL9 LacZ insertion mutant, was used for the transient complementation assay. Vero cells (American Type Culture Collection) were used for all transfection-based assays. The 2B-11 cell line (26), a UL9 null virus permissive cell line, was used to determine the titer of the viral progeny in the transient complementation assay. All mammalian cell lines were maintained in Dulbecco modified Eagle medium as described previously (41).
Spodoptera frugiperda (Sf21) insect cells were maintained in Grace's medium supplemented with 10% (vol/vol) fetal calf serum, 0.1 mg of streptomycin per ml, and 100 U of penicillin per ml. Escherichia coli DH5α cells were used for plasmid amplification. DH10Bac competent cells (Gibco BRL) were used for bacmid packaging and propagation.
Western blot analysis.
Western blot analysis was performed as previously described (4). Anti-UL9 primary antibodies recognizing the C-terminal residues 841 to 851 (R250, a kind gift from M. Challberg, National Institutes of Health) or the N-terminal 35 amino acids of UL9 (17B) (27) were used to detect UL9.
Generation of motif Ia mutants.
Point mutations in the conserved helicase motif Ia were generated by two-step primer PCR (11). The following mutagenic primers were used: (i) MIaST oligonucleotide (5′-GGACACGAGTGTACTAGTCGTCTCCTGTCGTCGG-3′), introducing the S110T substitution and an SpeI diagnostic restriction site; (ii) MIaRA oligonucleotide (5′-GCTCGTCTCCTGTGCTCGATCGTTTACCCAGACCCAG-3′), introducing the R112A substitution and a PvuI diagnostic restriction site; and (iii) MIaRA/FA oligonucleotide (5′-CGTTCTCCTGTCGTGCGAGTGCTACACAAACGCTAGCGACGCGG-3′), introducing the R113A and F115A substitutions and a NheI diagnostic site. The mutations resulting in amino acid changes are shown in bold type. The silent changes leading to the appearance of diagnostic restriction sites are underlined. The MIa-p6-downstream oligonucleotide (5′-GATCGAAGCTTAGGGCCACGGTTAC-3′) was used as a downstream outside oligonucleotide, and the MIa-p6-upstream oligonucleotide (5′-GCGGAGTCGGGAGATCCTCTGGG-3′) was used as an upstream outside oligonucleotide to generate the S110T and R122A mutations, using the p6UL9-119b plasmid (32) as a template. The resulting PCR products were digested with NheI and AscI and subcloned into pFastBac-UL9-WT plasmid, replacing the corresponding wild-type fragment (30). The PCR product bearing the R113A and F115A mutations (R113A/F115A) was generated using pFastBac-UL9-WT plasmid as a template and the 5′PCL-pFastBac oligonucleotide as an upstream outside oligonucleotide. The resulting PCR product was digested with BamHI and AscI and subcloned into pFastBac-UL9-WT, replacing the corresponding wild-type fragment. The presence of the engineered mutations was confirmed by restriction digestion and DNA sequencing. pFastBac BamHI/EcoRI fragments encoding wild-type and mutant UL9 genes were subcloned into the pCDNA1 vector (Invitrogen) under a cytomegalovirus promoter for in vivo studies in mammalian cells.
Transient transfection-complementation assay.
Vero cells in 60-mm2 plates at 50% confluency were transfected with 2 μg of the plasmid of interest using Lipofectamine Plus (Gibco), according to the manufacturer's recommendations. Eighteen hours posttransfection, cells were superinfected with the UL9 null virus, hr94, at a multiplicity of infection of 3 PFU/cell. Viral progeny were harvested at 48 h posttransfection, and the titers of the virus on 2B-11 cells were determined. Each experiment was repeated three times, and the mean complementation index was calculated.
Plaque reduction assay.
Vero cells in 50% confluent 60-mm2 plates were cotransfected with wild-type HSV-1 infectious DNA and the plasmid of interest (ratio 1:10) using Lipofectamine Plus (Gibco), according to the manufacturer's recommendations. Three hours posttransfection, the plates were overlaid with methylcellulose and incubated at 34°C until plaques were easily visible. Plaques were stained with crystal violet and counted. Each experiment was performed three times. The mean number of plaques and standard deviation were calculated.
Generation of recombinant baculoviruses expressing UL9.
Recombinant baculoviruses (Autographa californica nuclear polyhedrosis baculoviruses) expressing wild-type or mutant UL9 were generated using the pFastBac (Gibco BRL) commercial system according to the manufacturer's instructions.
UL9 expression and purification.
Eighty 225-cm2 flasks containing 50% confluent monolayers of Sf21 cells were infected with the recombinant baculovirus of interest, and the cells were harvested at 48 h postinfection. The proteins were extracted and purified as described previously (30). Briefly, UL9 was precipitated from high-salt nuclear extracts with ammonium sulfate at 50% saturation. The precipitate was resuspended in buffer B (20 mM HEPES, 1 mM EDTA) containing 0.25 M NaCl and 10% glycerol and dialyzed overnight. The dialyzed sample was loaded on an SP Sepharose (Pharmacia) column, and the bound protein to the column was eluted with a 0.25 to 1 M linear gradient of NaCl. Fractions containing UL9 were pooled and loaded on a UnoS column (Bio-Rad), and the protein bound was eluted as described above. UnoS fractions containing UL9 were concentrated using a Millipore concentrator with a 50-kDa cutoff and loaded on a Superose 12HR 10/30 gel filtration column (Pharmacia). In all chromatography steps, the column fractions were monitored by the Bradford protein assay and Western blotting with anti-UL9 antibodies. The protein concentration was determined by the Bradford colorimetric assay (7). Bovine serum albumin (BSA) was used for the preparation of a standard curve for protein concentration. All solutions used for protein purification contained 1 mM dithiothreitol (DTT), 5 mM benzamidine, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 10 mM sodium bisulfite or protease inhibitor cocktail (Sigma).
Limited proteolysis.
Limited proteolysis using proteinase K was performed as described previously (30). Briefly, a Sephadex G75 gel filtration column was used to remove protease inhibitors used during protein purification. Fractions containing UL9 were identified with the Bradford protein assay and analyzed further. A proteolytic reaction time course (15, 30, and 60 min) was performed on ice. Each reaction mixture contained 30 μg of UL9 protein and proteinase K at a concentration of 0.01 mg/ml. The reaction was stopped by the addition of PMSFto a final concentration of 10 mM. Samples were precipitated with trichloroacetic acid (10% [vol/vol] final concentration), and proteins were precipitated on ice for 30 min. The samples were spun down, washed with ice-cold ethanol, air dried, and dissolved in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer (0.001% bromophenol blue, 200 mM Tris-HCl [pH 8.8], 100 mM DTT, 2% SDS, 10% glycerol). The proteolytic fragments were resolved on 10% acrylamide gels and analyzed by Western blotting with anti-UL9 antibodies.
ATPase assay.
ATPase activity was assayed by measuring the release of free phosphate by the malachite green-ammonium molybdate colorimetric assay as described previously (23, 30). M13 ssDNA (20 μM final concentration in nucleotides) was used to assay ssDNA-stimulated ATPase activity. ATPase reactions were performed in HEPES-based buffer (20 mM HEPES [pH 7.6], 1 mM DTT, 5 mM ATP, 5 mM MgCl2, 10% glycerol, 0.5 mg of BSA per ml, 0.2 M NaCl) for 30 min at 37°C, and UL9 at a concentration of 200 nM was used.
Helicase assay.
The helicase activity of wild-type and mutant UL9 was monitored using a partially double-stranded helicase substrate with a 23-nucleotide 5′ ssDNA overhang as described previously (30), except that the annealed substrate was separated from the free oligonucleotide using a Biogel column.
Origin-binding assay.
A double-membrane filter-binding assay was performed as described previously (30). The substrate for the specificity assay was prepared by MspI digestion of p-100-1, an OriS-containing plasmid and subsequent labeling of the resulting fragments with [32P]dCTP by a Klenow polymerase reaction (36). Nitrocellulose membranes (BA85; Schleicher & Schuell) (pore size, 0.45 μm) and DEAE-cellulose membranes (NA 45, Schleicher & Schuell) (pore size, 0.45 μm) were prepared as described previously (42), and the binding reactions were filtered through the double membrane. The DNA bound to each of the membranes was eluted as described previously (30) and precipitated with ice-cold ethanol. The precipitated DNA was dissolved in water, resolved on native acrylamide gels, and visualized by autoradiography. The ability of wild-type and mutant UL9 proteins to bind the origin of replication was monitored by a double-membrane dot blot filter-binding assay (42). A gel-purified MspI fragment of p-100-1 plasmid containing OriS, labeled as described above, was used as the DNA substrate. After filtration, the nitrocellulose and DEAE-cellulose membranes were air dried, and the amount of DNA retained was quantitated with a PhosphorImager.
ssDNA-binding assay.
The ssDNA-binding assay was performed under conditions almost identical to those used for ATPase and helicase assay. UL9 and M13 circular ssDNA were incubated for 30 min at 37°C in HEPES-based buffer (20 mM HEPES [pH 7.6], 5 mM DTT, 5 mM ATP, 10% glycerol, 0.5 mg of BSA per ml, 0.2 M NaCl). The reaction was stopped by the addition of DNA electrophoresis loading buffer (36) and loaded immediately on a 1% TAE (Tris-acetate-EDTA)-agarose gel, containing 0.001 mg of ethidium bromide per ml. The DNA substrate, circular M13 ssDNA, was purified as described previously (36).
RESULTS
Mutation design and in vivo properties of the motif Ia mutants.
Mutations in UL9 helicase motifs I, II, III, IV, V, and VI were previously introduced on the basis of sequence conservation in UL9 and other SFII helicases (32). However, to date, the function of UL9 motif Ia has not been addressed experimentally. The sequence alignment of UL9 helicase motif Ia with UL9 homologs from other herpesviruses revealed high levels of sequence identity and conservation throughout the entire family (Fig. 1A), suggesting that it is important for UL9 function.
FIG. 1.

Helicase motif Ia sequence homology with Herpesviridae homologs of UL9 and helicases with solved structures. (A) Sequence alignment of UL9 helicase motif Ia with the closest UL9 homologs in the Herpesviridae family. Identical residues (white letters on a black background), conserved residues (black letters on gray background), and nonconserved residues (black letters on white background) are shown. Abbreviations: B, bovine; C, cercopithecine (simian varicella virus); E, equine; G, gallid; M, meleagrid (turkey); MDV, Marek's disease virus; PRV, pseudorabies virus; VZV, varicella-zoster virus. (B) Sequence alignment of UL9 motif Ia with the motif Ia sequences of SFII (NS3 [20]) and SFI (PcrA [40] and Rep [21]) helicases with solved structures. The residues seen to contact directly ssDNA in the crystal structures of the corresponding helicases are underlined. The numbers at the beginning and end of each sequence refer to the first and last amino acid from the corresponding motif Ia sequence. The single mutations generated (slashes) and the residues mutated in the double mutant generated (asterisks) are indicated. These two sequence alignments were prepared using Conserved Domains Database (29), which is accessible online (http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml).
Recently, motif Ia was found to directly contact ssDNA in the crystal structures of several SFI (Rep and PcrA) and SFII (NS3) helicases in complex with ssDNA-containing substrates (20, 21, 40). Despite the fact that SFI and SFII helicases have only limited homology, their structures are almost superimposable (10, 22, 37, 38). Furthermore, when a sequence alignment based on structural characteristics was performed, the residues directly contacting ssDNA align perfectly (20). We took advantage of the availability of the structural information and aligned motif Ia from UL9 with Ia motifs from helicases with solved structures (Fig. 1B). Initially, UL9 was aligned to the NS3 helicase, a SFII helicase, using PSI-BLAST (2), and the alignment of NS3 helicase motif Ia to motif Ia of Rep and PcrA, published by Kim et al. (20) was used. A similar alignment can be automatically generated using Conserved Domains Database (29), which performs alignments of the protein of interest with homologs with solved structure (Fig. 1B). We chose to mutate the following residues: (i) serine 110, corresponding to a position oriented toward the ATP-binding cleft and not toward the ssDNA-binding cleft (therefore, S110 is not predicted to contact ssDNA); (ii) arginine 112, arginine 113, and phenylalanine 115 corresponding to positions oriented toward or in direct contact with single-stranded nucleic acid residues in the crystal structures of Rep (21), PcrA (40), and NS3 (20) complexes with DNA or RNA. Thus, the R112A and R113A/F115A mutations are predicted to affect the ssDNA-binding function of UL9, whereas the S110T mutation is not.
The functional significance of the engineered mutations was tested in a transient transfection-complementation assay. Vero cells were transfected with pcDNA1 plasmids encoding wild-type and mutant versions of the UL9 gene and subsequently superinfected with hr94, the UL9 null virus. The titers of viral progeny were determined on the UL9 permissive cell line 2B-11. Figure 2A shows the complementation indices calculated from this experiment. All three mutants had a complementation index below 1; therefore, they failed to complement the growth of UL9 null virus. As expected, wild-type UL9 complemented the growth of hr94. The possibility that the failure to complement was due to the absence of mutant protein was ruled out by transient transfection-Western blot analysis. Figure 2B shows that all three mutants synthesize full-length UL9, readily recognizable by anti-UL9 antibody generated against the last 10 amino acids of the protein.
FIG. 2.

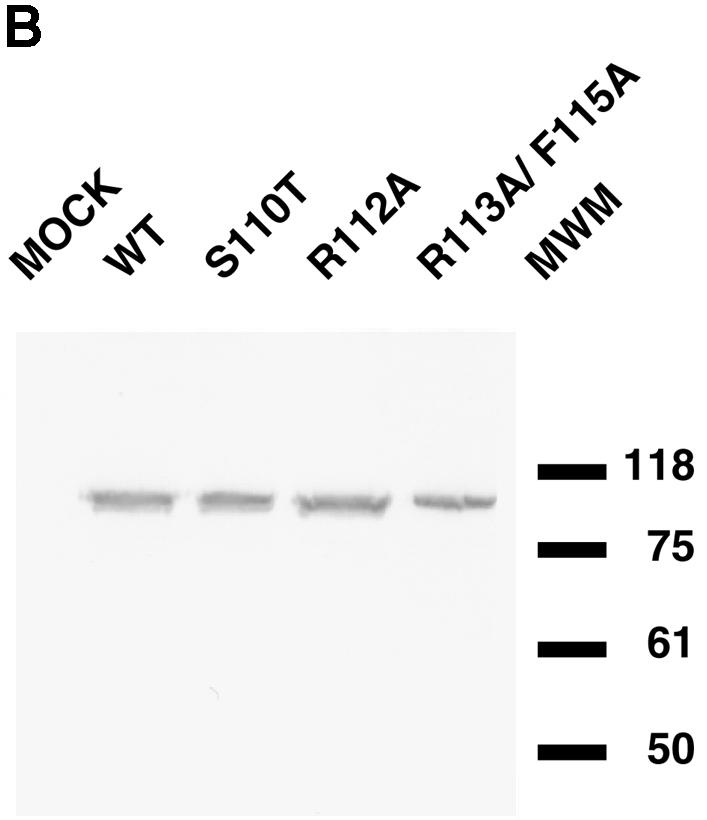
Mutations in UL9 helicase motif Ia fail to complement the growth of UL9 null virus hr94. (A) Complementation indices for wild-type (WT) and mutant UL9 proteins. The transient transfection- complementation assay was performed as described in Materials and Methods. The complementation indices were calculated as a ratio of the viral titer from cells transfected with the wild-type or mutant version of UL9 gene to the viral titer observed when empty plasmid was transfected. The mean complementation index (means shown above the bars) and standard deviation (plotted as error bars) were calculated from three independent experiments. (B) Western blot analysis of the steady-state UL9 protein levels in cells transfected with wild-type (WT) or mutant UL9. The positions (in kilodaltons) of New England Biolabs prestained molecular size markers (MWM) are shown on the right.
In order to test if the motif Ia mutants were transdominant, they were tested in a plaque reduction assay. In this in vivo assay, wild-type infectious DNA was cotransfected with excess plasmid encoding wild-type or mutant versions of the UL9 protein (28, 39). The number of plaques observed when the infectious DNA was transfected alone was normalized (set at 100). When a plasmid encoding wild-type UL9 was cotransfected with the wild-type infectious DNA alone, the normalized plaque number decreased moderately (Fig. 3), whereas the plaque numbers were reduced significantly when plasmids encoding motif I, II, and VI were used. These mutants were thus classified as transdominant. The cotransfection of plasmids containing motif Ia mutants with wild-type infectious DNA resulted in plaque numbers lower than those for the wild type and comparable to those observed for motif I (Fig. 3); therefore, these mutants are considered to be transdominant.
FIG. 3.

UL9 helicase motif Ia mutants are transdominant. Wild-type infectious DNA was cotransfected with 10× molar excess of plasmid encoding wild-type (WT) or mutant UL9, and the number of plaques was counted. The number of plaques observed when infectious DNA was transfected by itself was normalized (set at 100). Each experiment was repeated three times, and the mean normalized plaque number and the standard deviation were calculated. pCDNA3-UL9-K87A plasmid bearing motif I mutation was previously shown to be transdominant (28). In the last lane on the right, “=” refers to transfection with wild-type infectious DNA alone.
The UL9 motif Ia mutant proteins are able to dimerize and exhibit an overall conformation similar to that of the wild type.
In order to biochemically characterize the motif Ia mutants, recombinant baculoviruses carrying the genes for wild-type and mutant UL9 variants were constructed and expressed in Sf21 insect cells. Mutant and wild-type proteins were purified by a combination of ammonium sulfate precipitation and chromatography as described in Materials and Methods. The purification scheme resulted in protein preparations suitable for biochemical studies (Fig. 4).
FIG. 4.

Purified UL9 wild-type and mutant proteins. Wild-type (WT) and mutant UL9 proteins were purified from Sf21 insect cells infected with recombinant baculoviruses as described in Materials and Methods. Two micrograms of each protein was resolved on a SDS-9% polyacrylamide gel and stained with Coomassie brilliant blue. When larger amounts of protein were used, a contaminating band of approximately 40 kDa was observed. It was identified as a baculovirus protein by microsequencing (30). Previous results indicated that this baculovirus protein is not expected to interfere with the biochemical assays performed in this paper. The positions (in kilodaltons) of New England Biolabs prestained molecular size markers (MWM) are shown to the right of the gel.
Limited proteolysis with proteinase K, followed by Western blot analysis with anti-UL9 antibodies, showed that the wild-type and mutant proteolytic patterns are very similar (Fig. 5). This indicates that the overall conformation of UL9 is not significantly altered by the engineered mutations. Thus, the failure of the motif Ia mutants to complement the growth of UL9 null virus is most likely due to functional defects in UL9 mutant proteins and not to global conformational changes.
FIG. 5.

The UL9 helicase motif Ia mutations do not alter the overall conformation of the protein. Wild-type (WT) and mutant UL9 proteins were subjected to limited proteolysis with proteinase K for time intervals of 0, 15, 30, and 60 min (indicated by the thickness of the black triangle above the lanes). Western blot analysis was used to monitor the N- and C-terminal fragments during the reaction. (A) Western blot analysis with anti-UL9 antibody R250, recognizing the C terminus of UL9. (B) Western blot with anti-UL9 antibody 17B, recognizing the N terminus of UL9. The positions (in kilodaltons) of Gibco prestained molecular size markers are shown to the right of the gels.
The ability of motif Ia mutant proteins to dimerize was tested by gel filtration. Wild-type and mutant UL9 proteins were found to elute from the Superose HR 10/30 column as a wide peak centered around a position corresponding to a molecular mass of 180 kDa (Fig. 6). This result is consistent with our previous reports (30) and those of other research groups (8, 15). Thus, as was found previously for UL9 helicase motifs I to VI, motif Ia is most likely not involved in UL9 dimerization, providing support for the idea that residues outside the helicase motifs are involved in protein-protein interactions (21).
FIG. 6.
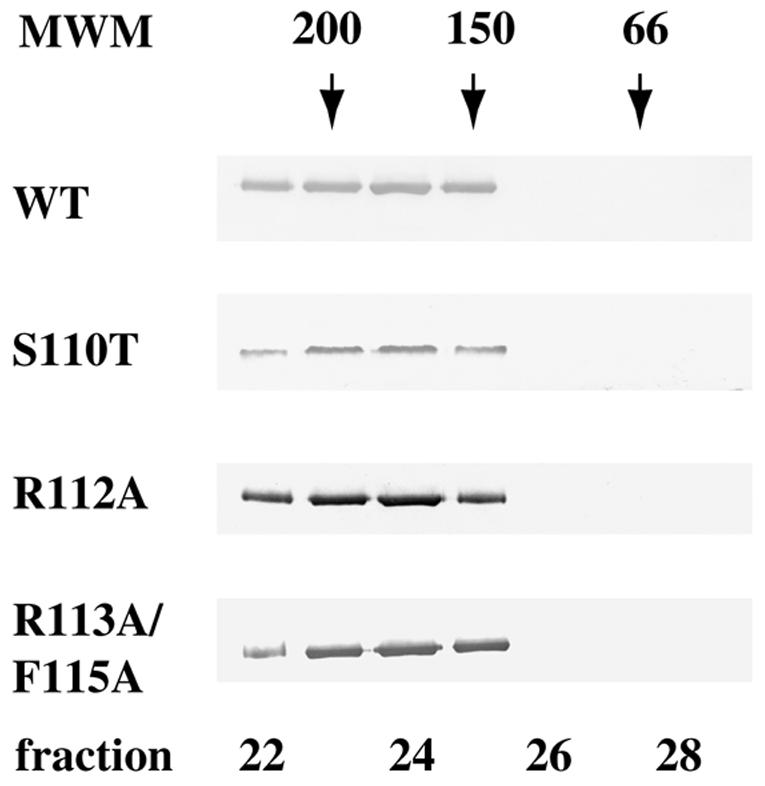
Mutations in helicase motif Ia do not alter the dimerization state of UL9 protein. A 100 nM solution of UL9 protein was analyzed by gel filtration using a Superose 12HR column (Pharmacia). Fifty 0.5-ml fractions were collected, and a 15-μl portion of each fraction was subjected to SDS-PAGE and Western blot analysis with anti-UL9 rabbit polyclonal serum R250. Wild-type (WT) and mutant UL9 proteins peak in fractions 23 to 25, corresponding to a dimeric state. The Western blots were developed for a short period of time in order to visualize only the peak fractions; however, with longer exposures, some trailing material was observed (up to fraction 27). The positions of gel filtration molecular mass markers (MWM) (Sigma) (amylase [200 kDa], alcohol dehydrogenase [150 kDa], and BSA [66 kDa]) are depicted above the Western blot strips.
Motif Ia mutant proteins exhibit defects in ssDNA-stimulated ATPase and helicase activities.
The ATPase activity of wild-type and motif Ia mutant proteins was measured as described in Materials and Methods. The rate constants for intrinsic and ssDNA-stimulated ATPase activity were calculated from time course experiments and plotted using Kaleidagraph (Fig. 7). Consistent with previous results (15, 30), wild-type UL9 hydrolyzes ATP with a rate constant of 3 min−1 in the absence of ssDNA and with a rate constant of 27 min−1 in the presence of ssDNA under the experimental conditions described in Materials and Methods. All motif Ia mutants were found to exhibit intrinsic ATPase activity very similar to wild-type levels (Fig. 7B). The relevant rate constants are 5 min−1 for the S110T mutant, 2 min−1for the R112A mutant, and 3 min−1 for the R113A/F115A double mutant.
FIG. 7.

Mutations in helicase motif Ia affect the ssDNA-stimulated ATPase activity but not the intrinsic ATPase activity of UL9. (A) A time course of intrinsic and ssDNA-stimulated ATPase activity of wild-type and mutant UL9 proteins. Circular M13 ssDNA (20 μM concentration in nucleotides) was used as a DNA effector. The reactions were performed in the presence (closed symbols) and absence (open symbols) of ssDNA. Symbols: squares, wild-type UL9; circles, S110T mutant; diamonds, R112A mutant; triangles, R113A/F115A double mutant. The means from three independent experiments are shown. (B) Plot of the rate constants for intrinsic ATPase activity (gray bars) and ssDNA-stimulated ATPase activity (black bars). The mean kcat (minute−1) values and standard deviations (error bars) were calculated from three independent experiments. WT, wild-type UL9.
The levels of ssDNA-stimulated ATPase activity of all mutants were somewhat lower than that of the wild type. The S110T mutant exhibited approximately 75% of wild-type activity (kcat, 20 min−1), and a moderate defect was observed for the R112A mutant, which exhibited approximately 50% of wild-type activity (kcat, 13 min−1). A more severe defect was observed for the R113A/F115A double mutant: its ATPase activity was not stimulated by ssDNA at all (kcat, 3 min−1 in the presence and absence of ssDNA) under the experimental conditions used. The inability of ssDNA to stimulate the ATPase activity of the R113A/F115A double mutant provides the first indication that motif Ia may be involved in ssDNA binding, supporting our original hypothesis. S110 is a residue expected to be oriented toward the ATP-binding cleft, and a mutant bearing the S110T mutation at this position retains significant intrinsic and ssDNA-stimulated ATPase activities. In contrast, mutations in the residues predicted to contact ssDNA show moderate or severe defects in ssDNA-stimulated ATPase activity.
An in vitro unwinding assay was used to examine the effects of the helicase motif Ia mutations on the helicase activity of UL9. None of the mutants tested exhibited any helicase activity (Fig. 8). This result correlates with the failure of these mutants to complement the growth of UL9 null virus and provides support for the idea that UL9 does indeed unwind the HSV-1 origin of replication.
FIG. 8.

UL9 motif Ia mutant proteins lack helicase activity. Wild-type (WT) and mutant UL9 proteins were purified from insect cells and tested for helicase activity as described in Materials and Methods. A helicase reaction mixture without UL9 protein was used as a reference for the mobility of the annealed substrate, and a helicase reaction boiled for 10 min before loading was used as a reference for the mobility of the unwound oligonucleotide. Each helicase reaction mixture contained 1 nM helicase substrate and 200 nM UL9 protein, and each reaction was performed under the same conditions as for the ATPase assay (see Materials and Methods). After 30-min incubation, the reaction mixtures were resolved on 8% native polyacrylamide gels and visualized by autoradiography.
Mutations in the helicase motif Ia do not interfere with the origin-specific DNA-binding activity of UL9.
The effects of the helicase motif Ia mutations on the origin-binding function of UL9 were tested using a double-membrane filter-binding assay. In this assay, protein-DNA complexes are retained on the first membrane, the nitrocellulose membrane, whereas free DNA passes through and binds to the DEAE-cellulose membrane. Thus, the counts per minute for DNA fragments recovered from the nitrocellulose membrane represent the bound DNA, and the counts per minute for DNA fragments recovered from the DEAE-cellulose membrane represent unbound DNA. Our results show that none of the characterized mutations interferes with the specificity of the origin binding (Fig. 9A) or with the ability of UL9 to bind OriS (Fig. 9B). To test the specificity of the binding, MspI-digested p-100-1 plasmid (Fig. 9A, lane 1) was used as a substrate in the filter-binding reaction. It was found that only the OriS-containing fragment was exclusively retained on the nitrocellulose membrane by the wild-type and mutant UL9 proteins (Fig. 9A, lanes 6 to 9), despite the presence of multiple labeled plasmid fragments of similar length. The origin-binding abilities of wild-type and mutant proteins were compared at protein concentrations of 1, 4, and 10 nM. These concentrations were shown previously to be in the linear part of the binding isotherm of wild-type UL9 to an OriS-containing DNA fragment (7). It is expected that in this region of the binding isotherm, minimal changes in protein concentration would result in maximal effects on DNA binding. At all concentrations, wild-type and mutant UL9 proteins showed similar abilities to bind OriS (Fig. 9B), leading to the conclusion that mutations in helicase motif Ia do not affect the ability of UL9 to bind the dsDNA fragments containing the origin of replication.
FIG. 9.


Mutations in helicase motif Ia do not interfere with UL9 origin-binding activity. (A) The ability of wild-type (WT) and mutant UL9 proteins to bind OriS was tested by a double-membrane filter-binding assay, performed as described in Materials and Methods. Lanes 2 to 5 were loaded with DNA eluted from the DEAE-cellulose membrane (unbound DNA). Lanes 6 to 9 were loaded with DNA eluted from the nitrocellulose membrane (bound DNA). Lane 1 shows the input DNA. Lanes 2 to 5 and 6 to 9 were loaded with equal counts per minute and therefore do not reflect the ability of each protein species to bind OriS. (B) Comparison of the OriS-binding abilities of wild-type (WT) and mutant UL9. The percent bound DNA in the wild-type reaction was set at 100% (thick black line). Binding reactions with 1 nM (gray bars), 4 nM (black bars), and 10 nM (hatched bars) enzyme are shown. The percentage of bound DNA is defined as follows: 100 × (cpm of the nitrocellulose membrane)/(cpm of the nitrocellulose membrane + cpm of the DEAE − cellulose membrane), where cpm is the counts per minute.
The R112A and R113A/F115A mutations exhibit defects in ssDNA binding.
To directly test the hypothesis that motif Ia is involved in the ssDNA-binding activity of UL9, a ssDNA-binding assay was performed. It is expected that the UL9 protein with the S110T mutation, designed in a residue predicted to be oriented toward the ATP-binding cleft and to not contact ssDNA, would exhibit little or no defect in ssDNA binding. In contrast, UL9 proteins with the R112A or R113A/ F115A mutations, designed in residues predicted to contact ssDNA are expected to exhibit defects in ssDNA-binding activity. Increasing amounts of wild-type and mutant UL9 proteins were incubated with M13 circular ssDNA, and the mobility of the protein/DNA complexes was monitored on ethidium bromide-stained agarose gels. At a 100 nM protein concentration, only the wild type and the S110T mutant were able to bind ssDNA, as judged by the appearance of a gel-shifted band with a mobility lower than that of the free DNA (Fig. 10A, compare lanes 2 [wild type] and 3 [S110T] with lane 1 [free DNA]). The gel-shifted bands were diffuse, suggesting that UL9/ssDNA complexes are unstable and/or heterogeneous. Another line of evidence suggesting instability and/or heterogeneity is the appearance of some smearing in the lanes with protein/DNA complexes. At this concentration, R112A and R113A/F115A mutants did not bind DNA. At higher protein concentrations (Fig. 10B, C, and D), the shifted bands show decreased mobility, as expected for protein/DNA complexes of higher molecular weight. At a 200 nM concentration of the R112A and R113A/F115A proteins, the sharp bands of the free DNA become diffuse with almost no change in mobility (Fig. 10B, lanes 4 and 5), suggesting that a limited amount of DNA/protein complexes was formed. Well-defined gel-shifted bands for these mutants are seen at the 400 nM concentration (Fig. 10C). The band corresponding to the R113A/F115A/ssDNA complex migrates slightly faster that the one corresponding to the R112A/ssDNA complex (Fig. 10C, compare lanes 4 and 5), suggesting that the double mutation affects the ssDNA-binding properties of UL9 more drastically than the R112A mutation. The last point is confirmed in Fig. 10D at 800 nM protein concentration. The R113A/F115A mutant is the only one which still generated a gel shift, whereas the wild type and the other two mutants generated complexes unable to enter the gel. At 1,200 nM, all UL9 variants produce complexes unable to enter the gel (data not shown). A Western blot with anti-UL9 antibody was performed in order to confirm that UL9 was present in the observed protein/ssDNA complexes (Fig. 11). A well-defined band containing UL9 was seen in all binding reactions, as the mobility of the band decreased with the increase of the UL9 concentration. The Western blot pattern correlates well with the pattern seen on the ethidium bromide-stained gel (Fig. 11, compare lanes 2, 3, and 4 in both panels). UL9 by itself was barely able to enter the gel (Fig. 11A, lane 5), consistent with the high pI of the protein (pI = 8.24). In summary, the ssDNA-binding experiments confirmed the hypothesis that conserved residues from helicase motif Ia are involved in the ssDNA-binding activity of UL9. Since M13 ssDNA was used in this assay, we cannot rule out the possibility that secondary structure of the M13 substrate contributes to the observed binding. M13 ssDNA was chosen for consistency between the ATPase, helicase, and ssDNA-binding assays used in this work.
FIG. 10.

Analysis of the ssDNA-binding properties of wild-type and mutant UL9 proteins. ssDNA-binding reaction mixtures containing 100 (A), 200 (B), 400 (C), and 800 (D) nM mutant or wild-type (WT) UL9 protein were resolved on 1% agarose gels and stained with ethidium bromide. The gel-shifted bands are marked with white asterisks.
FIG. 11.

UL9/ssDNA complexes are detected by Western blotting. ssDNA-binding reaction mixtures containing 400, 800, and 1,000 nM UL9 protein (indicated by the thickness of the black triangle above the gel) with R113/F115A mutations and containing DNA (+) were resolved on a 1% agarose gel (B), electrotransferred to an Immobilon P membrane, and probed with an anti-UL9 antibody (A). The positions of free ssDNA (three asterisks) and UL9 alone (two asterisks) are indicated at the side of the gel. The positions of UL9/ssDNA complexes are indicated by the single asterisks in the gel.
It is interesting that although the engineered motif Ia mutants exhibited defects in ssDNA-binding activity, their ability to bind the double-stranded HSV-1 origin of replication was similar to that of the wild type. This finding is consistent with the notion that the N-terminal domain of UL9 harbors all functions attributable to a helicase (including ssDNA binding), whereas the C-terminal domain harbors the dsDNA origin-binding function (1, 3).
DISCUSSION
Helicases are a heterogeneous group of enzymes involved in practically every molecular process in the cell involving nucleic acids. Despite their sequence diversity, all SFI and SFII helicases have similar structures and functionally conserved motifs shown to be important for ATP binding and hydrolysis (motifs I and II), ssDNA binding (motifs Ia, III, IVa, and V of SFI and motifs Ia, IV, and V of SFII) and coupling the ATPase, helicase, and ssDNA activities (motifs III to VI) (19). The accumulation of structural data for several helicases in complex with ssDNA-containing substrates indicated that different helicases interact with ssDNA in a similar fashion and that helicase motif Ia harbors residues which directly contact ssDNA. We have hypothesized that similar to other helicases, UL9 helicase motif Ia is involved in the ssDNA-binding function of the enzyme, and we found genetic and biochemical evidence supporting our hypothesis. This work reports the successful use of the combination of bioinformatics and structural information originating from distant homologs to deduce information about the function of a protein of interest. This analysis has resulted in the identification of residues important for the ssDNA-binding function of UL9, a key player in replication initiation of the ubiquitous human pathogen HSV-1.
On the basis of distant homology with helicases with solved structure, we designed mutations in three residues (R112, R113, and F115), corresponding to residues seen to contact ssDNA or to be oriented toward the ssDNA-binding cleft, and one residue (S110), corresponding to a residue seen to be oriented toward the ATP-binding cleft. We showed genetically that these residues are essential for UL9 function in vivo. The functional significance of the mutated residues was characterized biochemically. The R112A and R113A/F115A mutants exhibited no helicase activity and moderate to severe defects in ssDNA-stimulated ATPase activity and ssDNA binding, whereas their intrinsic ATPase activity was unaltered. In contrast, the S110T mutant exhibited intrinsic and ssDNA-stimulated ATPase and ssDNA-binding activities similar to those of the wild type. All three mutants exhibited unaltered ability to dimerize or to bind specifically the origin of replication. Surprisingly, the S110T mutant did not exhibit any helicase activity, despite near wild-type levels of ATPase and ssDNA-binding activities. Thus, it appears that the S110T mutant may have a defect in coupling ATPase and helicase activities. Mutants with similar phenotypes, exhibiting wild-type or greater levels of ATPase activity but lacking helicase activity, have been previously described for the E. coli UvrB (35) and HSV-1 UL5 (17) helicases. In summary, our results show that as predicted, helicase motif Ia is involved in the ssDNA-binding function of UL9 and this motif is indispensable for UL9 helicase function. The absence of helicase activity correlates with the failure of motif Ia mutants to complement the growth of UL9 null virus. The results presented here further support the importance of UL9 helicase function for HSV-1 replication (30, 31).
The functional significance of motif Ia has been studied genetically and biochemically in a limited number of helicases (12-14, 18, 25). It has been shown genetically that residues of helicase motif Ia are essential for the replication of AcMNPV baculovirus (25), but biochemical studies have not been performed. Motif Ia of CI helicase of plum pox potyvirus maps within a region involved in ssRNA binding, defined by deletion analysis (13, 14).
Some insights about the possible mechanistic significance of the R112 residue are suggested by site-specific mutagenesis of residues in analogous positions in Rep and NS3 helicases. T56 in Rep, which aligns to the R112 position in UL9 (Fig. 1), was reported to participate in a direct hydrogen bond with the phosphodiester backbone of the ssDNA substrate (21). The corresponding NS3 residue (S231) participates in a water-mediated hydrogen bond with a phosphate oxygen from ssDNA (20). Interestingly, when S231 was mutated to alanine, no significant effect on ssRNA binding was seen. The intrinsic ATPase activity of mutant protein was higher than that of the wild type, a moderate reduction of the ssRNA-stimulated ATPase activity and wild-type helicase activity (24). The phenotype appears to be less pronounced than the effects we observed with UL9 with the R112A mutation.
R113 from UL9 corresponds to N57 from Rep and V232 from NS3 (Fig. 1). Structural information (20, 21) indicates that these residues are involved in contacts with the ssDNA backbone, but no mutagenesis studies are available to confirm their impact on the overall function of the corresponding proteins. An interesting phenotype was observed for a mutation in P228 (P228A) of vaccinia virus NPH II helicase, which corresponds to R113A of UL9. Although it exhibited wild-type levels of ssRNA binding and ssDNA-stimulated ATPase, its helicase activity was only 20% of the wild-type level (18). When the neighboring residue R229 was mutated to alanine, the helicase and ssDNA-stimulated ATPase activities were practically abolished, whereas the RNA-binding activity was similar to that of the wild type. In summary, a limited number of genetic studies are available on the function of motif Ia. Residues in similar position are shown to be important for the helicase activity of several helicases, but the effects observed on the single-stranded nucleic acid binding are variable. This may be a consequence of the complex nature of the ssDNA- or RNA-binding activity, as residues from several helicase motifs are believed to contribute to overall binding. The functional significance of each particular residue may be system specific and/or dependent on the nature of the single-stranded nucleic acid substrate and the characteristics of the process in which the corresponding enzyme is involved.
We previously proposed a model to explain the transdominance of UL9 helicase motif mutants (28, 30), and the genetic and biochemical properties of motif Ia mutants provide further support for this model. We have shown that mutations in motifs I, II, and VI exerted a dominant-negative effect on the ability of wild-type viral DNA to form plaques when Vero cells were cotransfected with mutant UL9 genes and wild-type viral infectious DNA. Motif I, II, and VI mutant proteins are able to dimerize and to bind the HSV-1 origin as well as the wild-type protein but lack helicase activity. We suggested that the transdominance of these mutant UL9 proteins was due to the fact that they can still bind the origin of replication effectively and dimerize but cannot initiate viral DNA replication due to their defect in helicase function. In this paper, we show that all three mutations in motif Ia are also transdominant. The biochemical properties of these mutants are similar to the properties of motif I, II, and VI transdominant mutants described previously (28, 30). Thus, the transdominance of the motif Ia mutants is most likely mediated by the same mechanism.
In summary, we hypothesized that helicase motif Ia is involved in the ability of UL9 to bind ssDNA, and the validity of this prediction was confirmed by genetic and biochemical approaches. This analysis has provided valuable information about the function of motif Ia residues of UL9, a key player in the initiation of replication of a ubiquitous human pathogen, HSV-1. Furthermore, this work represents an important proof of the value of a combined bioinformatic-structural biological approach to study proteins for which no structural information is available.
Acknowledgments
We thank all members of the Weller laboratory for helpful discussions of the manuscript. We gratefully acknowledge M. Challberg for providing reagents used in this study.
This investigation was supported in part by Public Health Service grant AI21747.
REFERENCES
- 1.Abbotts, A. P., and N. D. Stow. 1995. The origin-binding domain of the herpes simplex virus type 1 UL9 protein is not required for DNA helicase activity. J. Gen. Virol. 76:3125-3130. [DOI] [PubMed] [Google Scholar]
- 2.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arbuckle, M. I., and N. D. Stow. 1993. A mutational analysis of the DNA-binding domain of the herpes simplex virus type 1 UL9 protein. J. Gen. Virol. 74:1349-1355. [DOI] [PubMed] [Google Scholar]
- 4.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl. 1990. Current protocols in molecular biology. John Wiley & Sons, New York, N.Y.
- 5.Blumel, J., and B. Matz. 1995. Thermosensitive UL9 function is required for early stages of herpes simplex virus type 1 DNA synthesis. J. Gen. Virol. 76:3119-3124. [DOI] [PubMed] [Google Scholar]
- 6.Boehmer, P. E., and I. R. Lehman. 1993. Physical interaction between the herpes simplex virus 1 origin-binding protein and single-stranded DNA-binding protein ICP8. Proc. Natl. Acad. Sci. USA 90:8444-8448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 8.Bruckner, R. C., J. J. Crute, M. S. Dodson, and I. R. Lehman. 1991. The herpes simplex virus 1 origin binding protein: a DNA helicase. J. Biol. Chem. 266:2669-2674. [PubMed] [Google Scholar]
- 9.Carmichael, E. P., M. J. Kosovsky, and S. K. Weller. 1988. Isolation and characterization of herpes simplex virus type 1 host range mutants defective in viral DNA synthesis. J. Virol. 62:91-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caruthers, J. M., and D. B. McKay. 2002. Helicase structure and mechanism. Curr. Opin. Struct. Biol. 12:123-133. [DOI] [PubMed] [Google Scholar]
- 11.Colosimo, A., Z. Xu, G. Novelli, B. Dallapiccola, and D. C. Gruenert. 1999. Simple version of “megaprimer” PCR for site-directed mutagenesis. BioTechniques 26:870-873. [DOI] [PubMed] [Google Scholar]
- 12.Dillingham, M. S., P. Soultanas, P. Wiley, M. R. Webb, and D. B. Wigley. 2001. Defining the roles of individual residues in the single-stranded DNA binding site of PcrA helicase. Proc. Natl. Acad. Sci. USA 98:8381-8387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernandez, A., and J. A. Garcia. 1996. The RNA helicase CI from plum pox potyvirus has two regions involved in binding to RNA. FEBS Lett. 388:206-210. [DOI] [PubMed] [Google Scholar]
- 14.Fernandez, A., S. Lain, and J. A. Garcia. 1995. RNA helicase activity of the plum pox potyvirus CI protein expressed in Escherichia coli. Mapping of an RNA binding domain. Nucleic Acids Res. 23:1327-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fierer, D. S., and M. D. Challberg. 1992. Purification and characterization of UL9, the herpes simplex virus type 1 origin-binding protein. J. Virol. 66:3986-3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gorbalenya, A. E., E. V. Koonin, A. P. Donchenko, and V. M. Blinov. 1989. Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res. 17:4713-4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graves-Woodward, K. L., J. Gottlieb, M. D. Challberg, and S. K. Weller. 1997. Biochemical analyses of mutations in the HSV-1 helicase-primase that alter ATP hydrolysis, DNA unwinding, and coupling between hydrolysis and unwinding. J. Biol. Chem. 272:4623-4630. [DOI] [PubMed] [Google Scholar]
- 18.Gross, C. H., and S. Shuman. 1998. The nucleoside triphosphatase and helicase activities of vaccinia virus NPH-II are essential for virus replication. J. Virol. 72:4729-4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hall, M. C., and S. W. Matson. 1999. Helicase motifs: the engine that powers DNA unwinding. Mol. Microbiol. 34:867-877. [DOI] [PubMed] [Google Scholar]
- 20.Kim, J. L., K. A. Morgenstern, J. P. Griffith, M. D. Dwyer, J. A. Thomson, M. A. Murcko, C. Lin, and P. R. Caron. 1998. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure 6:89-100. [DOI] [PubMed] [Google Scholar]
- 21.Korolev, S., J. Hsieh, G. H. Gauss, T. M. Lohman, and G. Waksman. 1997. Major domain swiveling revealed by the crystal structures of complexes of E. coli Rep helicase bound to single-stranded DNA and ADP. Cell 90:635-647. [DOI] [PubMed] [Google Scholar]
- 22.Korolev, S., N. Yao, T. M. Lohman, P. C. Weber, and G. Waksman. 1998. Comparisons between the structures of HCV and Rep helicases reveal structural similarities between SF1 and SF2 superfamilies of helicases. Protein Sci. 7:605-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lanzetta, P. A., L. J. Alvarez, P. S. Reinach, and O. A. Candia. 1979. An improved assay for nanomole amounts of inorganic phosphate. Anal Biochem. 100:95-97. [DOI] [PubMed] [Google Scholar]
- 24.Lin, C., and J. L. Kim. 1999. Structure-based mutagenesis study of hepatitis C virus NS3 helicase. J. Virol. 73:8798-8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu, G., and E. B. Carstens. 1999. Site-directed mutagenesis of the AcMNPV p143 gene: effects on baculovirus DNA replication. Virology 253:125-136. [DOI] [PubMed] [Google Scholar]
- 26.Malik, A. K., R. Martinez, L. Muncy, E. P. Carmichael, and S. K. Weller. 1992. Genetic analysis of the herpes simplex virus type 1 UL9 gene: isolation of a LacZ insertion mutant and expression in eukaryotic cells. Virology 190:702-715. [DOI] [PubMed] [Google Scholar]
- 27.Malik, A. K., L. Shao, J. D. Shanley, and S. K. Weller. 1996. Intracellular localization of the herpes simplex virus type-1 origin binding protein, UL9. Virology 224:380-389. [DOI] [PubMed] [Google Scholar]
- 28.Malik, A. K., and S. K. Weller. 1996. Use of transdominant mutants of the origin-binding protein (UL9) of herpes simplex virus type 1 to define functional domains. J. Virol. 70:7859-7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marchler-Bauer, A., A. R. Panchenko, B. A. Shoemaker, P. A. Thiessen, L. Y. Geer, and S. H. Bryant. 2002. CDD: a database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Res. 30:281-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marintcheva, B., and S. K. Weller. 2001. Residues within the conserved helicase motifs of UL9, the origin-binding protein of herpes simplex virus-1, are essential for helicase activity but not for dimerization or origin binding activity. J. Biol. Chem. 276:6605-6615. [DOI] [PubMed] [Google Scholar]
- 31.Marintcheva, B., and S. K. Weller. 2001. A tale of two HSV-1 helicases: roles of phage and animal virus helicases in DNA replication and recombination. Prog. Nucleic Acid Res. Mol. Biol. 70:77-118. [DOI] [PubMed] [Google Scholar]
- 32.Martinez, R., L. Shao, and S. K. Weller. 1992. The conserved helicase motifs of the herpes simplex virus type 1 origin-binding protein UL9 are important for function. J. Virol. 66:6735-6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McLean, G. W., A. P. Abbotts, M. E. Parry, H. S. Marsden, and N. D. Stow. 1994. The herpes simplex virus type 1 origin-binding protein interacts specifically with the viral UL8 protein. J. Gen. Virol. 75:2699-2706. [DOI] [PubMed] [Google Scholar]
- 34.Monahan, S. J., L. A. Grinstead, W. Olivieri, and D. S. Parris. 1998. Interaction between the herpes simplex virus type 1 origin-binding and DNA polymerase accessory proteins. Virology 241:122-130. [DOI] [PubMed] [Google Scholar]
- 35.Moolenaar, G. F., R. Visse, M. Ortiz-Buysse, N. Goosen, and P. van de Putte. 1994. Helicase motifs V and VI of the Escherichia coli UvrB protein of the UvrABC endonuclease are essential for the formation of the preincision complex. J. Mol. Biol. 240:294-307. [DOI] [PubMed] [Google Scholar]
- 36.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 37.Singleton, M. R., and D. B. Wigley. 2002. Modularity and specialization in superfamily 1 and 2 helicases. J. Bacteriol. 184:1819-1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soultanas, P., and D. B. Wigley. 2000. DNA helicases: “inching forward.” Curr. Opin. Struct. Biol. 10:124-128. [DOI] [PubMed] [Google Scholar]
- 39.Stow, N. D., O. Hammarsten, M. I. Arbuckle, and P. Elias. 1993. Inhibition of herpes simplex virus type 1 DNA replication by mutant forms of the origin-binding protein. Virology 196:413-418. [DOI] [PubMed] [Google Scholar]
- 40.Velankar, S. S., P. Soultanas, M. S. Dillingham, H. S. Subramanya, and D. B. Wigley. 1999. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell 97:75-84. [DOI] [PubMed] [Google Scholar]
- 41.Weller, S. K., K. J. Lee, D. J. Sabourin, and P. A. Schaffer. 1983. Genetic analysis of temperature-sensitive mutants which define the gene for the major herpes simplex virus type 1 DNA-binding protein. J. Virol. 45:354-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong, I., and T. M. Lohman. 1993. A double-filter method for nitrocellulose-filter binding: application to protein-nucleic acid interactions. Proc. Natl. Acad. Sci. USA 90:5428-5432. [DOI] [PMC free article] [PubMed] [Google Scholar]