

Abstract
Noise, or random fluctuations, in gene expression may produce variability in cellular behavior. To measure the noise intrinsic to eukaryotic gene expression, we quantified the differences in expression of two alleles in a diploid cell. We found that such noise is gene-specific and not dependent on the regulatory pathway or absolute rate of expression. We propose a model in which the balance between promoter activation and transcription influences the variability in messenger RNA levels. To confirm the predictions of our model, we identified both cis- and trans-acting mutations that alter the noise of gene expression. These mutations suggest that noise is an evolvable trait that can be optimized to balance fidelity and diversity in eukaryotic gene expression.
The stochastic, or random and probabilistic, nature of chemical reactions may create variation in an identical population of cells (1). The reactions underlying gene expression involve small numbers of molecules (e.g., transcription factors, DNA, and mRNAs) and may therefore exhibit stochastic fluctuations that could generate population variation when phenotypic diversity would be advantageous or could act as a theoretical obstacle when fidelity in cellular behavior is required (2, 3).
To measure the stochasticity of eukaryotic gene expression, we implemented the dual-reporter technique, developed in Escherichia coli (4, 5), in the budding yeast Saccharomyces cerevisiae (6). We constructed diploid yeast strains that express both cyan and yellow fluorescent proteins (CFP and YFP) from identical promoters, integrated at the same locus on homologous chromosomes (Fig. 1A). Two types of noise are distinguished in our analysis: intrinsic noise attributable to stochastic events during gene expression, and extrinsic noise due to any existing cellular heterogeneity that affects gene expression or to stochastic events in upstream signal transduction (5). For each population of cells, we calculated the variability in terms of two metrics: the noise, defined as the standard deviation divided by the mean, which we present to convey the magnitude of variability as a percentage of the level of gene expression; and the noise strength, or variance divided by the mean, which we use for our analysis because it is independent of population mean for a single stochastic process (supporting online text).
Fig. 1.
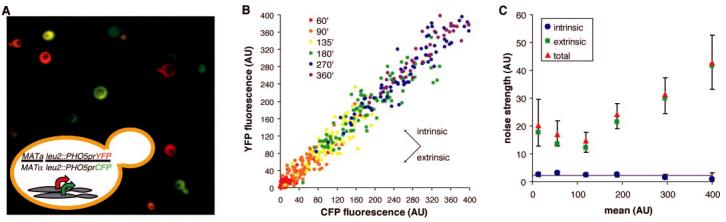
Separation of intrinsic and extrinsic noise for the PHO5 promoter. (A) A false-color overlay of YFP (red) and CFP (green) fluorescence micrographs from a diploid yeast strain that expresses YFP and CFP from identical promoters at homologous loci, as diagrammed in the inset. (B) Scatter plots showing CFP and YFP values for each cell (solid circles) during a time course of PHO5 induction by phosphate starvation. Populations from different time points (in minutes) are indicated with different colors. Extrinsic noise is manifested as scatter along the diagonal and intrinsic noise as scatter perpendicular to the diagonal. AU, arbitrary units of fluorescence. (C) Total, extrinsic, and intrinsic noise strength as functions of population mean for (B). The solid line represents expectations for a single stochastic process, and error bars represent bootstrap values (6).
We induced the expression of CFP and YFP from the budding yeast PHO5 promoter and measured the fluorescence of single cells in random subpopulations at multiple times after induction (Fig. 1B). The total noise of gene expression from the PHO5 promoter was dominated by the contribution from extrinsic factors (Fig. 1C); the intrinsic noise strength, although larger than the error of measurement in our system (supporting online text), represented only between 2% and 20% of the total noise strength. For a second promoter, GAL1, the intrinsic noise strength represented less than 3% of the total noise strength (fig. S1). We conclude that the stochasticity of gene expression is not necessarily reflected by measurements of total noise that employ single-reporter techniques.
Heterogeneity in a number of factors that affect gene expression may underlie extrinsic noise, including heterogeneity in cell size and shape, cell cycle stage, or gene-specific signaling. We tested if extrinsic noise factors could be eliminated by flow cytometry to isolate subpopulations of cells that are homogeneous in size and shape. We found that, although diminished by this process, extrinsic noise predominated relative to intrinsic noise in these subpopulations (fig. S2A). Similarly, neither correction for individual cellular volume nor segregation by cell cycle stage (6) resulted in more than a ∼25% decrease in extrinsic noise (fig. S2B). To distinguish extrinsic noise that is gene-specific from a global, nonspecific source, we examined the correlation between CFP and YFP fluorescence in a strain that expresses CFP from one promoter and YFP from a second, distinctly regulated promoter. Expression from the PHO84 and GAL1 promoters was correlated, with a R2 value of 0.88 (fig. S3A); additionally, expression from the PHO84 and ADH1 promoters was correlated (R2 = 0.93) (fig. S3B). Therefore, the majority of extrinsic noise in these cases is not promoter-specific and will cause gene products in a cell to be maintained in constant relative concentration.
To characterize stochasticity in eukaryotic gene expression further, we measured at different rates of gene expression the intrinsic noise strength of the PHO5 and PHO84 promoters, which are regulated by the same transcriptional activator, and the GAL1 promoter (6). The GAL1 and PHO84 promoters display a low level of intrinsic noise strength that does not substantially vary with changes in the rate of gene expression (Fig. 2, A and B). In contrast to the other promoters, PHO5 has a larger intrinsic noise strength that decreases with increasing rate of gene expression (Fig. 2C); at maximal expression, PHO5 displays less than half the intrinsic noise strength that it does at a low expression rate. This decrease in intrinsic noise strength as the rate of gene expression increases is not sensitive to the stimulus used to induce PHO5 expression (Fig. 2C, inset). We conclude that noise intrinsic to gene expression is promoter-specific and does not depend absolutely on the rate of expression, the induction stimulus, or the identity of the sequence-specific transcriptional activator.
Fig. 2.

(A) Intrinsic noise strength as a function of rate of expression for GAL1 (left) and a scatter plot of GAL1-expressing cells at maximal induction (right). To produce different rates of expression, cells were induced with different galactose concentrations. (B) Intrinsic noise strength as a function of rate of expression for PHO84 and a scatter plot of PHO84-expressing cells at maximal induction. Cells were induced with different phosphate concentrations. (C) Intrinsic noise strength as a function of rate of expression for PHO5 and a scatter plot of PHO5-expressing cells at the maximal level of induction by phosphate starvation. Cells were induced with various levels of chemical inhibition of an upstream kinase (left) or with various organic phosphate concentrations (inset). The dashed line indicates the intrinsic error of measurement, and error bars represent standard deviations. Scatter plots contain cells from multiple time points.
A previous model of noise generation in gene expression predicted that noise strength would not vary with a change in the rate of mRNA production (7, 8), similar to our observations of GAL1 and PHO84. This model does not predict the noise profile of the PHO5 promoter, which is known to be regulated by a promoter transition step that is upstream of and independent of transcription (9). We constructed a model of stochastic gene expression, elaborating on previous models (5, 10-13), that incorporates two distinct promoter states: an inactive state not permissive for transcription and an active state that is competent for transcription (Fig. 3A).
Fig. 3.

General stochastic model of gene activation and expression. (A) Schematic of reactions with stochastic rate constants of production (k) and degradation (γ). Ø indicates the null product of degradation. a, active DNA; m, mRNA; p, protein. (B) Three cases, where the relative size of the arrows indicates the relative magnitude of the constants within each case. (C) The effect on intrinsic noise strength of changing the promoter activation rate to change the steady-state mean of expression for case I (orange □), case II (violet □), and case III (cyan □). (D to F) The effect on intrinsic noise strength of changing promoter activation (green □), transcriptional efficiency (red ○), and translational efficiency (purple △) to change the steady-state mean of expression for (D) case I, (E) case II, and (F) case III. For (C) to (F), the solid lines display the predicted values, and the open symbols are averages from stochastic simulations (6).
We distinguish among three kinetic mechanisms of promoter transcriptional activation (Fig. 3B). In case I, the activation step is infrequent relative to transcription and the active promoter state is stable (, where ka is the rate of promoter activation, γa is the rate of promoter inactivation, and km is the rate of transcription). This could correspond to a promoter that is activated by a slow chromatin-remodeling step in which positioned nucleosomes are removed from the DNA and that is slowly inactivated by replacement of the nucleosomes. In case II, the activation step is infrequent relative to transcription and the active promoter state is unstable , corresponding to a relatively infrequent but rapidly reversible activation step such as nucleosomal sliding or prokaryotic promoter DNA looping. In case III, the activation step is frequent relative to transcription and the activated promoter is highly unstable . The third case is equivalent to the previously proposed prokaryotic model (8) and could represent rapid activator binding-dissociation reactions in which transcription occurs only for a fraction of the binding events.
We calculated the solution of the noise strength equation in terms of the stochastic kinetic constants of the model (supporting online text) and also performed stochastic simulations to approximate our experiments in which each intrinsic noise strength measurement is the average of multiple time points of finite cellular populations (6). We examined how varying the kinetic constants in the model to change the steady-state mean of gene expression affects the intrinsic noise strength for the three different cases (Fig. 3, C to F). The noise strength profile of PHO5 (Fig. 2C) is similar to the predictions made for case I when the promoter activation rate is changed (Fig. 3C). Therefore, we hypothesized that noise generation at PHO5 is dependent on the rate of a slow upstream promoter transition. In the inactive state, the PHO5 promoter displays positioned nucleosomes (14); upon binding of the Pho4 transcription factor to upstream activating sequences UAS1 and UAS2, chromatin-remodeling complexes are recruited (15, 16) and catalyze removal of nucleosomes from the promoter region (17, 18). Mutation of these UAS sites causes a defect in the disruption of positioned nucleosomes during activation (9). As predicted by the model (case I, decreasing ka), the two PHO5 UAS mutant promoters had increased intrinsic noise strength compared to the wild-type PHO5 promoter (Fig. 4A). This observation is consistent with a model in which chromatin remodeling is the upstream stochastic promoter transition for the PHO5 promoter.
Fig. 4.

Mutational analysis of the PHO5 promoter. (A) Intrinsic noise strength and rate of expression of wild-type, UASm1, and UASm2 PHO5 promoter variants at maximal induction (top) and a scatter plot for the wild type (○) and UASm1 (red ◆) (bottom). The intrinsic noise of the UASm1 promoter is 77% at a mean of 26 AU, compared to 43% intrinsic noise at a mean of 29 AU for the wild-type promoter. (B) Intrinsic noise strength and rate of expression of the PHO5 promoter at maximal induction in the wild-type background or in strains that lack SNF6, ARP8, or GCN5 (top) and a scatter plot for the wild type (○) and snf6Δ (red ◆) (bottom). The intrinsic noise of the snf6Δ strain is 73% at a mean of 35 AU. (C) Intrinsic noise strength and rate of expression of wild-type and TATA mutant PHO5 promoters at maximal induction (top) and a scatter plot for the wild type (○) and TATA-C2 (red ◆) (bottom). The intrinsic noise of the TATA-C2 promoter is 15% at a mean of 25 AU. Error bars represent standard deviations. Scatter plots contain cells from multiple time points.
Multiple chromatin-remodeling complexes, including SWI/SNF, INO80, and SAGA, participate in remodeling at the PHO5 promoter (15, 16, 19, 20). To test further the hypothesis that chromatin remodeling is the stochastic promoter activation step, we examined the noise strength of the maximally induced PHO5 promoter in yeast strains that lack single components of these three chromatin-remodeling complexes (Fig. 4B). The deletion of components of SWI/SNF (snf6Δ), INO80 (arp8Δ), or SAGA (gcn5Δ) each resulted in increased intrinsic noise strength, consistent with the predictions of the noise model. There were substantial differences in the noise strength among the mutants, which may reflect different roles of these complexes in the promoter transition process.
The TATA element of the PHO5 promoter is required for efficient transcription but dispensable for chromatin remodeling (9). To confirm the prediction that noise strength should scale with the efficiency of a transcription step downstream of promoter activation, we measured, at maximal induction, the noise strength of a series of PHO5 promoters with various TATA box sequences (Fig. 4C). As predicted, the mutant TATA box promoters displayed decreasing noise strength with a decreasing rate of gene expression. These observations suggest that our model represents a useful framework for the rationalization of noise generation at the PHO5 promoter.
Our results support a model for noise generation that is applicable to both eukaryotic and prokaryotic promoters, in which the relative rates of activation, deactivation, and transcription determine variability in mRNA levels. Our noise measurements and model contradict the previous assertion that noise generation in gene expression is fundamentally different between prokaryotes and eukaryotes (13). Rather, we assert that the diverse mechanisms of gene regulation in these systems fall into three general categories of noise profiles, dependent upon the relative rates of promoter reactions. Two promoters can produce the same mean mRNA population with different noise characteristics: a promoter that undergoes frequent activation steps followed by inefficient transcription will produce a cellular population with little variability, whereas a promoter that undergoes infrequent activation steps followed by efficient transcription can display large differences from cell to cell. We have identified simple sequence mutations in the PHO5 promoter that exemplify these two extremes: the UASm1 and TATA-A1T6 variants have similar rates of gene expression on a population level but different levels of stochasticity—a more than 30-fold change in intrinsic noise strength. Because the intrinsic noise is altered by small changes in promoter sequence and independent of the absolute rate of gene expression, we speculate that stochasticity is an evolvable characteristic of each eukaryotic gene, determined by both cis- and trans-acting factors.
Stochasticity intrinsic to gene expression has been invoked as a source of phenotypic variation in the lambda phage lysis-lysogeny switch (21), mammalian olfactory neuronal receptor choice (22), and tumor formation in response to transcription factor haploinsufficiency (23). The variability of gene expression due to stochasticity in promoter transitions is predicted to be proportional to the inverse square root of the gene copy number, providing a rationale for the preservation of multiple copies of the same gene and the effects of haploinsufficiency when variability is deleterious. If the majority of extrinsic noise is not gene-specific, stochasticity will create variability in the ratio of one gene product to the other within a population; the preservation of the stoichiometric ratio between gene products may be important for components of multisubunit complexes (24). Additionally, stochasticity may cause variation in the ratio of expression of two alleles with distinct functions, allowing a heterozygous population of cells to display multiple phenotypes, including those of each corresponding homozygote. Such phenotypic variability may be beneficial in a variable environment and may contribute to the phenomenon of hybrid vigor. Stochasticity in gene expression is not necessarily an obstacle to invariant cellular behavior but may constitute an evolvable source of advantageous population diversity.
Supplementary Material
Footnotes
Supporting Online Material http://www.sciencemag.org/cgi/content/full/1098641/DC1 Materials and Methods SOM Text Figs. S1 to S5 References and Notes
References and Notes
- 1.McAdams HH, Arkin A. Trends Genet. 1999;15:65. doi: 10.1016/s0168-9525(98)01659-x. [DOI] [PubMed] [Google Scholar]
- 2.Schroedinger E. What Is Life? Cambridge Univ. Press; Cambridge: 1944. [Google Scholar]
- 3.Rao CV, Wolf DM, Arkin AP. Nature. 2002;420:231. doi: 10.1038/nature01258. [DOI] [PubMed] [Google Scholar]
- 4.Elowitz MB, Levine AJ, Siggia ED, Swain PS. Science. 2002;297:1183. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- 5.Swain PS, Elowitz MB, Siggia ED. Proc. Natl. Acad. Sci. U.S.A. 2002;99:12795. doi: 10.1073/pnas.162041399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Materials and methods are available as supporting material on Science Online
- 7.Ozbudak EM, Thattai M, Kurtser I, Grossman AD, van Oudenaarden A. Nature Genet. 2002;31:69. doi: 10.1038/ng869. [DOI] [PubMed] [Google Scholar]
- 8.Thattai M, van Oudenaarden A. Proc. Natl. Acad. Sci. U.S.A. 2001;98:8614. doi: 10.1073/pnas.151588598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fascher KD, Schmitz J, Horz W. J. Mol. Biol. 1993;231:658. doi: 10.1006/jmbi.1993.1317. [DOI] [PubMed] [Google Scholar]
- 10.Peccoud J, Ycart B. Theor. Pop. Biol. 1995;48:222. [Google Scholar]
- 11.Paulsson J. Nature. 2004;427:415. doi: 10.1038/nature02257. [DOI] [PubMed] [Google Scholar]
- 12.Ko MS. J. Theor. Biol. 1991;153:181. doi: 10.1016/s0022-5193(05)80421-7. [DOI] [PubMed] [Google Scholar]
- 13.Blake WJ, Kaern M, Cantor CR, Collins JJ. Nature. 2003;422:633. doi: 10.1038/nature01546. [DOI] [PubMed] [Google Scholar]
- 14.Almer A, Horz W. EMBO J. 1986;5:2681. doi: 10.1002/j.1460-2075.1986.tb04551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steger DJ, Haswell ES, Miller AL, Wente SR, O'Shea EK. Science. 2003;299:114. doi: 10.1126/science.1078062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barbaric S, Reinke H, Horz W. Mol. Cell. Biol. 2003;23:3468. doi: 10.1128/MCB.23.10.3468-3476.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fascher KD, Schmitz J, Horz W. EMBO J. 1990;9:2523. doi: 10.1002/j.1460-2075.1990.tb07432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boeger H, Griesenbeck J, Strattan JS, Kornberg RD. Mol. Cell. 2003;11:1587. doi: 10.1016/s1097-2765(03)00231-4. [DOI] [PubMed] [Google Scholar]
- 19.Ebbert R, Birkmann A, Schuller HJ. Mol. Microbiol. 1999;32:741. doi: 10.1046/j.1365-2958.1999.01390.x. [DOI] [PubMed] [Google Scholar]
- 20.Barbaric S, Walker J, Schmid A, Svejstrup JQ, Horz W. EMBO J. 2001;20:4944. doi: 10.1093/emboj/20.17.4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arkin A, Ross J, McAdams HH. Genetics. 1998;149:1633. doi: 10.1093/genetics/149.4.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serizawa S, et al. Science. 2003;302:2088. doi: 10.1126/science.1089122. [DOI] [PubMed] [Google Scholar]
- 23.Magee JA, Abdulkadir SA, Milbrandt J. Cancer Cell. 2003;3:273. doi: 10.1016/s1535-6108(03)00047-3. [DOI] [PubMed] [Google Scholar]
- 24.Fraser HB, Hirsh AE, Giaever G, Kumm J, Eisen MB.PLoS Biol 20042e137published online 27 April 2004 (10.1371/journal.pbio.0020137) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.We thank J. Paulsson, D. Wykoff, J. Weissman, H. Li, R. Kishony, D. Schaffer, H. Madhani, W. Marshall, and members of the O'Shea lab for discussion and critical commentary. Supported by the Howard Hughes Medical Institute, NIH grant no. GM51377, and the David and Lucile Packard Foundation (E.K.O.) and by an American Diabetes Association Medical Scholars fellowship and the UCSF Medical Scientist Training Program (J.M.R.)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.