

Abstract
Prion diseases are fatal neurodegenerative disorders of animals and humans that are characterized by the conversion of the host-encoded prion protein (PrP) to an abnormal isoform. In several species, including humans, polymorphisms in the gene encoding the PrP protein tightly control susceptibility of individuals toward this disease. In the present study we show that Rov cells expressing an ovine PrP allele (VRQPrP) associated with high susceptibility of sheep to scrapie were very sensitive to sheep prion transmission and replicated the agent to high titers. In contrast, we did not find any evidence of infection when Rov cells expressed similar levels of a PrP variant (ARRPrP) linked to resistance. Our data provide the first direct evidence that natural PrP polymorphisms may affect prion susceptibility by controlling prion replication at the cell level. The study of how PrP polymorphisms influence the genetic control of prion propagation in cultured Rov cells may help elucidate basic mechanisms of prion replication.
Transmissible spongiform encephalopathies (TSEs) are fatal degenerative disorders of the central nervous system, which naturally affect animals and humans (for a review, see reference 8). TSEs are associated with the posttranslational conversion of the host-encoded prion protein (PrP) to a conformationally altered form (PrPsc). The causative infectious agents, or prions, are thought to be PrPsc itself or a precursor of it (for reviews, see references 22 and 30).
In several species, including mice, sheep, and humans, susceptibility to prion diseases is tightly controlled by the host. The major genetic determinants controlling the length of the incubation period are polymorphisms of Prnp, the PrP-encoding gene. In humans, homozygosity for methionine at the polymorphic residue 129 is associated with high susceptibility to the new variant form of Creutzfeldt-Jakob disease (14), whereas homozygosity for valine is overrepresented among early cases of iatrogenic Creutzfeldt-Jakob disease in the United Kingdom (7). In sheep, variations at three amino acid positions of PrP are predominantly linked to scrapie susceptibility (for a review, see reference 17). The 136V154R171Q allele (V, R, and Q stand for valine, arginine, and glutamine, respectively) is associated with extremely high susceptibility to scrapie: in naturally infected flocks, sheep homozygous for VRQPrP are almost always affected with scrapie. In contrast, the 136A154R171R variant (A for alanine) is associated with resistance to the clinical disease. No clinical scrapie case has been reported in hundreds of sheep homozygous for ARRPrP from naturally infected flocks in Europe and the United States, and a single case was reported in Japan (12). Experimental challenge of VRQ- and ARR-encoding sheep with ovine and bovine prions further substantiated the dramatic opposite effects of these two alleles on disease susceptibility, since sheep homozygous for ARRPrP could not be infected by either agent (11). Although the link between specific alleles of PrP and susceptibility to the disease has been well documented during the last decade, especially in sheep, how a few amino acid substitutions can so profoundly affect prion pathogenesis is largely unknown. The development of new experimental models may help elucidate the mechanisms underlying the effects of these polymorphisms.
We recently reported that the stable transfection of a tetracycline-regulatable ovine PrP gene in a rabbit epithelial cell line resulted in the efficient propagation of a natural sheep scrapie agent (28). This scrapie cell culture model, named Rov, represented a unique opportunity to investigate the effect of PrP polymorphisms on the susceptibility to prion infection. In the present study, we examined 2 PrP alleles (VRQPrP and ARRPrP) with opposite disease linkage and show that these alleles also have strikingly opposite effects on sheep prion replication in cultured Rov cells.
MATERIALS AND METHODS
Isolation of Rov cells expressing ARR and VRQ PrP variants.
The complete coding sequence of the ARR variant of ovine PrP was PCR cloned from sheep genomic DNA in the pTRE plasmid (Clontech) and verified by DNA sequencing. Transfection of RK13 cells and selection of clones for inducible expression of the specified ovine PrP variant were as described previously (28).
Scrapie strain.
The PG127 isolate originally used to infect Rov cells is from a naturally scrapie-affected sheep (28). This isolate was transmitted to Prnp0/0 transgenic mice (TgOv) expressing the VRQ allele of ovine PrP (29), and the resulting infectious material was used in the present study. Brain homogenate and extracts from infected cells were prepared as described previously (28). When indicated, infectious brain homogenate and cell extracts were diluted in homogenate from healthy brain and in 5% glucose solution containing 5% bovine serum albumin (BSA), respectively.
Transmission of sheep prion to Rov cells and PrPsc detection.
Inoculation of Rov cells and isolation of sedimentable, proteinase K (PK)-resistant, Rov-derived PrPsc were as described previously (28). Western blots were stained either with 2D6 (25) or 4F2 (18) monoclonal antibodies (MAbs), as indicated.
Mouse bioassay.
The bioassays were performed on the tg301 line of TgOv mice expressing the VRQ allele of ovine PrP and nullizygous for the mouse Prnp gene, as described previously (29). Briefly, animals were inoculated intracerebrally with 20 μl of inoculum and examined for neurologic disease every 2 days and then daily when clinical signs of scrapie were detected.
Immunostaining of PrP on living Rov cells.
Immunofluorescence analysis on living Rov cells expressing the VRQ or the ARR allele of PrP was performed at 4°C, with the MH44 anti-PrP polyclonal antibody (20), as described previously (28).
RESULTS
Rov cells as a candidate system to study the effect of PrP polymorphisms on prion infection.
Rov cells were obtained by transfecting the VRQ allele of ovine PrP in the RK13 epithelial cell line. Previously, we have shown that doxycycline-mediated expression of the VRQPrP in the Rov9 clone resulted in the efficient replication of the sheep scrapie agent in the exposed cultures (28). Additional transfected cell clones expressing VRQPrP have now been obtained, and nine of them (including the already-described Rov9) were exposed to a strain of sheep prions propagated in a VRQPrP animal (see Materials and Methods). The success of these transmissions was assessed by immunodetection of Rov-derived PrPsc, one passage postinoculation (p.i.). Figure 1A (lanes 1 to 5) shows the relative levels of normal PrP in five inoculated VRQ-Rov clones, and Fig. 1B (lanes 1 to 5) shows the amounts of abnormal PrP in the corresponding PK-digested cell lysates. Abnormal PrP was readily detected in all inoculated VRQ-Rov clones. Since PrPsc from residual inoculum is not detected under these experimental conditions (28; see also Fig. 1B, lanes 6 to 9), the presence of cell-derived PrPsc indicated that all VRQ-Rov clones have been successfully infected. However, a marked difference in the amount of PrPsc accumulated in the different VRQ-Rov clones was observed (Fig. 1B, lanes 1 to 5) that was found not to correlate with the level of PrP expression (Fig. 1A, lanes 1 to 5). The different VRQ-Rov clones might synthesize slightly different subsets of PrP glycoforms, which in turn could influence prion replication. The availability of MAbs recognizing distinct subsets of PrP glycoforms (V. Beringue and S. Hawke, unpublished data) should allow investigating this possibility. These infected cultures were expanded for 2 months. Accumulation of PrPsc in each of the individual cultures was evidenced at each passage (data not shown) until the sixth passage (Fig. 1C, lanes 1 to 5), indicating that all of these Rov clones were stably infected. Similar results were obtained with the four other VRQ-Rov clones (data not shown).
FIG. 1.

Analysis of normal and abnormal PrPs in Rov clones expressing the VRQ or the ARR allele of ovine PrP after exposition to sheep prion. Transmission of sheep prion obtained from a VRQPrP animal to nine Rov clones was tested by incubating confluent monolayers expressing the VRQ (lanes 1 to 5) or the ARR (lanes 6 to 9) PrP allele with infectious brain homogenate for 2.5 days. The inoculum was removed, and the cells were washed and passaged 2.5 days later. Cultures were then subcultured once a week at a one-fourth dilution. Parts of the cultures were lysed and analyzed for PrP at either one (A and B) or six (C) passages p.i.. Lane 1, Rov9; lane 2, RovC79; lane 3, RovA7; lane 4, RovA1; lane 5, Rov5; lane 6, RovG2; lane 7, RovE2; lane 8, RovG4; lane 9, RovE5. Some lanes were not loaded. (A) Normal PrP in the different clones at one passage p.i. Proteins (the equivalent of 25 μg) were methanol precipitated, and PrP was analyzed by immunoblotting with 4F2 MAb directed against the NH2 terminus region of PrP. Since most of the Rov-derived PrPsc undergoes a proteolytic cleavage that removes a portion of the NH2 terminus region (unpublished observation), accumulated PrPsc in Rov cells cannot be detected by 4F2 MAb. (B and C) Abnormal PrP was isolated from PK-digested cell lysates (the equivalent of 250 μg of proteins) at one (B) or six (C) passages p.i. The level of PK resistance of PrPsc generated in infected VRQ-Rov cells is high enough to sustain digestion with high amounts (50 μg/ml) of PK (28). However, in these experiments, low concentrations of PK (ca. 5 μg/ml) were used to detect any abnormal PrP with possible low levels of PK resistance. Immunoblots were stained with 2D6 MAb raised against the COOH terminus region of PrP.
Prion titer associated with infected VRQ-Rov extract was determined by endpoint titration in transgenic mice expressing the ovine VRQPrP (TgOv mice), previously shown to be highly sensitive to sheep scrapie (29). Infected VRQ-Rov cultures, grown for 3.5 months (14 passages p.i.), accumulated ca. 2 LD50 (50% lethal doses) per cell (Table 1). In a second set of experiments, we determined the sensitivity of Rov cells to transmission of sheep prion by exposing VRQ-Rov cells to serial dilutions of the ovine strain of prion and looking for PrPsc accumulation at five passages p.i. Under these conditions, transmission was still detectable after inoculation with a 10−3 dilution of 10% brain homogenate, corresponding to 12.5 μg of infectious brain (Fig. 2). For comparison, from 2 to 20 μg of Chandler- or RML-infected brain material is required to detect infection of permissive murine N2a and GT1 cells (3, 19). The relative sensitivity of Rov cells is therefore roughly similar to that of these two widely used neuronal cell models. Finally, transmission of sheep prion to VRQ-Rov cells was found to be very reliable. All of the different VRQ-Rov clones tested so far (n = 9) were readily infected, and all transmission attempts to the Rov9 clone (>50) have been successful, based on the biochemical detection of abnormal PrP accumulation at the very first passage after inoculation (i.e., within 2 weeks p.i.).
TABLE 1.
Bioassay in TgOv mice of VRQ- and ARR-expressing Rov cells challenged with sheep prion
| Ovine PrP allele | Infected culturea
|
Mean survival time (n/n0)b ± SD | ||
|---|---|---|---|---|
| Clone | Passage p.i. | Dilution | ||
| VRQ | Rov9 | 14 | 1/106 | 89 ± 2.4 (5/5) |
| VRQ | Rov9 | 14 | 1/107 | 104.5 ± 2.1 (2/5) |
| VRQ | Rov9 | 14 | 1/108 | >300 (0/5) |
| VRQ | RovA7 | 8 | 1/1 | 57.3 ± 0.73 (4/4) |
| ARR | RovE2 | 8 | 1/1 | >420 (0/5) |
| ARR | RovG4 | 8 | 1/1 | >450 (0/7) |
| None | NAc | 8 | 1/1 | >420 (0/6) |
Mice were inoculated intracerebrally with 20 μl of cell extracts from infected Rov or parental RK13 cultures at the indicated passage postinoculation, either undiluted (3 × 106 cells per mouse) or diluted as indicated. Two distinct infected VRQ- and ARR-Rov clones were bioassayed. The titer of infected Rov9 culture, determined according to Kärber's method, was 2.65 LD50 per cell (i.e., 106.9 LD50 in 3 × 106 cells).
Mean days to the death ± SEM; n/n0, number of terminally ill/number of inoculated animals.
NA, not applicable.
FIG. 2.

Sensitivity of VRQ-expressing Rov cells to sheep prion. Rov cells (the Rov9 clone) were exposed to sheep prion (2.5% [wt/vol] brain homogenate) at the indicated dilutions for 7 days. Accumulated PrPsc was detected at five passages p.i. with 2D6 MAb.
These data showed that expression of the VRQ allele of PrP conferred to Rov cells sensitivity to low doses of sheep prion and consistently allowed stable and efficient replication of the agent.
Rov cells expressing the ARR allele are resistant to infection with sheep prion.
Rov clones expressing the ARRPrP allele associated with sheep resistance to the clinical disease were generated. Four clones expressing the ARR allele were selected. They expressed this PrP variant at levels similar to VRQ-PrP in VRQ-Rov cells (data not shown; see also Fig. 1A) and allele-specific glycosylation pattern differences were not detected through Western blot analysis in the present study (Fig. 1A and data not shown). All of the four ARR-Rov clones were exposed to the strain of sheep prion used for VRQ-Rov infection. As described above, cultures were tested for the presence of normal and abnormal PrPs, at one passage p.i. (Fig. 1A, lanes 6 to 9; Fig. 1B, lanes 6 to 9, respectively). In contrast to the situation observed with VRQ-Rov cells, no PrPsc could be detected in inoculated ARR-expressing clones (Fig. 1B, lanes 6 to 9). Each week, these cultures were passaged and analyzed for PrPsc. No abnormal PrP could be observed at six passages p.i. (Fig. 1C, lane 6 to 9) or before (data not shown), indicating that production of PrPsc in inoculated ARR-Rov cells was not simply delayed. Extracts from two of the challenged ARR-Rov clones were bioassayed in TgOv mice along with a VRQ-Rov clone infected and passaged in parallel. No infectivity could be demonstrated in the ARR-Rov cultures (Table 1). The fact that each of the nine VRQ-Rov clones but none of the four ARR-Rov could be infected strongly argues against the possibility that the inhibitory effect might be mediated by cell-clone differences unrelated to the sequence of PrP.
There is evidence that normal PrP first transits through the plasma membrane before being converted to abnormal PrP (2, 6). We verified that the ARRPrP was expressed at the cell surface of Rov cells as shown previously for the VRQPrP (28). Immunofluorescence analysis of living cells showed that both ARRPrP and VRQPrP were present at the outer membrane (Fig. 3). Therefore, the nonpermissiveness of ARR-Rov cells cannot be attributed to a defect of PrP expression at the cell surface.
FIG. 3.
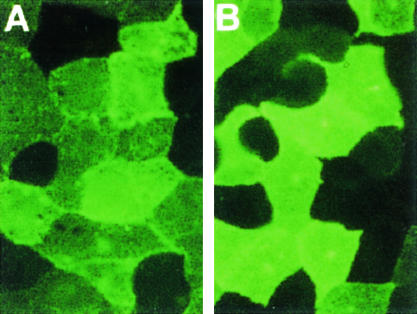
Immunostaining of PrP in Rov clones expressing the VRQ or the ARR variant of ovine PrP. Confluent monolayers of living cells expressing the VRQ (clone C79) (A) or the ARR (clone E2) (B) PrP allele were labeled with an anti-ovine PrP polyclonal antibody (MH44) and then with fluorescein-conjugated immunoglobulin G second antibodies. No staining was observed when primary antibodies were omitted. Magnification, ×500.
DISCUSSION
In this study, with sheep scrapie as an experimental model, we demonstrated that natural polymorphisms of the PrP protein can control prion propagation in permissive cultured cells. Rov cells were highly susceptible to sheep prion when expressing the VRQ allele of PrP. Infected VRQ-Rov cultures produced high titers of sheep prion (ca. 2 LD50 per cell), a value that compared favorably with titers obtained in two widely used cellular models of mouse prions infection, i.e., neuroblastoma N2a (0.004 LD50 per cell [5]) and neuronal GT1 (0.06 LD50 per cell [26]) cell lines. The efficient prion propagation in Rov cells could explain why subcloning of the inoculated cultures was unnecessary to assess positive transmission by Western blotting at the very first passage p.i. and to achieve a high percentage (at least 30% [28]) of infected cells. This may explain at least in part why the transmission of sheep prion to VRQ-Rov cells was found to be extremely reliable. Permissive N2a and GT1 cell lines are composed of cells with different susceptibilities to infection with the RML strain of murine prions (3, 9), and these marked differences were independent of the level of normal PrP expression (9). In contrast, we found no evidence for the presence of VRQ-expressing cell clones resistant to infection. Prion replication might need some essential cellular factor(s) absent in a significant number of N2a and GT1 cells but present in most of the Rov cells or the postulated factor may be missing in Rov cells too, where it would not be required for the replication of the ovine strain studied.
The reliable transmission of sheep prion, along with the undetectable expression of the endogenous rabbit PrP in these cells (28), makes it feasible to study in-depth relationships between PrP sequence and permissiveness to infection by a reverse genetic approach. As a main result of the present study, we found no evidence of infection in Rov cells expressing the ARRPrP-resistant variant. First, no accumulated PrPsc could be detected in ARR-Rov cells exposed to sheep prion, whereas infection of VRQ-Rov cultures with a 1,000-fold-diluted inoculum led to detectable PrPsc. This indicated that ARRPrP is at least a 1,000-fold less efficient to support prion replication than is VRQPrP. Second, none of the TgOv mice inoculated with challenged ARR-Rov cultures developed the clinical disease. However, the possibility that low levels of infectivity had been transmitted to these mice, leading to a subclinical level of prion replication (16, 23), is presently being investigated by secondary passages. These findings provide the first ex vivo evidence for a direct link between prion replication and natural PrP allelic variation. Since sheep homozygous for ARRPrP are resistant to the various natural strains of sheep scrapie from the field tested so far, it is very unlikely that inhibition of prion replication observed in Rov cells would be restricted to the ovine strain used in the present study. An efficient way to modulate susceptibility to prion disease is to modulate the incubation period by increasing or decreasing the level of PrP expression (10, 21). Since the influence of the PrP genotype on PrP levels in sheep tissues or in particular cell types remains to be established (20), the possibility that PrP polymorphisms could influence the amount of the protein, directly or indirectly, remains open. Our study demonstrating a dramatic effect of PrP polymorphism on prion replication in cells expressing similar levels of different PrP variants implies that PrP polymorphisms may control susceptibility to the disease by conferring distinct functional and/or biochemical properties to the different variants rather than affecting their amounts.
What hypothesis can be made about the cellular mechanisms controlling susceptibility to prion disease through PrP polymorphisms? One possibility could be that some PrP variants might be more prone to be converted to abnormal PrP than others. This notion has been supported by cell-free conversion of sheep PrP alleles. In these assays, guanidium-denatured ovine PrP, purified after solubilization of uninfected cell cultures, was converted to PK-resistant forms upon addition of PrPsc obtained from scrapie-infected sheep brain tissue, and indeed VRQPrP forms were more efficiently converted than ARRPrP (4). However, as no generation of infectivity has been demonstrated in cell-free conversion assays (15), it is unclear if conversion reaction truly represents TSE-agent propagation, i.e., if newly converted abnormal PrP can in turn promote conversion of new PrP molecules, as could be expected for a PrPsc-mediated infectious process. Several lines of evidence indicate that the cellular context of PrP is of crucial importance: association of PrP with detergent-resistant microdomains, or rafts, is important in the formation of PrPsc (for a review, see reference 13), and a recent study raised the possibility that insertion of PrP and PrPsc in the same contiguous membrane might be critical for conversion to occur (1). PrP, however, is not the only player in prion replication. Host factors other than PrP, yet to be identified, are necessary for efficient prion infection (9, 24, 27), and some of them could interact differently with distinct PrP alleles. The possibility that VRQPrP and ARRPrP could be recognized and processed differently by Rov cells deserves attention. It has been reported that recombinant VRQ and ARR PrP variants, produced in bacteria, have distinct biochemical and structural properties (25), the biological significance of which needs to be appreciated in mammalian cells. Therefore, we consider TSE cellular models, such as Rov, to be relevant tools for studying the role of PrP polymorphisms on prion infection and to elucidate why the ARR variant, in interaction with the cellular machinery, does not lead to prion multiplication.
In conclusion, we have shown here that the genetic control of susceptibility to prion disease via PrP polymorphisms can be modeled in a cell culture model of scrapie replication. Our data provide evidence that PrP polymorphisms may act on susceptibility by primarily affecting the level of scrapie replication in the infected animal and that Rov cells represent attractive tools to investigate the underlying mechanisms. More generally, both infected and uninfected Rov cells could be used to study PrP structure-function relationships through a reverse genetic approach.
Acknowledgments
We acknowledge M. F. Madelaine for expert technical assistance, J. Grosclaude and M. Moudjou (INRA, Jouy, France) for the kind gift of 2D6 and MH44 antibodies, respectively, and J. Grassi (CEA, Saclay, France) for providing 4F2 antibodies. We thank V. Beringue, J. A. Gingrich, and C. La Bonnardière for critical reading of the manuscript and B. Nicolas for the artwork.
This work was partially supported by grants from the French government (CI-ESST) and from the European Union (Biotech. PL976064).
REFERENCES
- 1.Baron, G. S., K. Wehrly, D. W. Dorward, B. Chesebro, and B. Caughey. 2002. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrP(Sc)) into contiguous membranes. EMBO J. 21:1031-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borchelt, D. R., A. Taraboulos, and S. B. Prusiner. 1992. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem. 267:16188-16199. [PubMed] [Google Scholar]
- 3.Bosque, P. J., and S. B. Prusiner. 2000. Cultured cell sublines highly susceptible to prion infection. J. Virol. 74:4377-4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bossers, A., P. B. G. M. Belt, G. J. Raymond, B. Caughey, R. de Vries, and M. A. Smits. 1997. Scrapie susceptibility-linked polymorphisms modulate the in vitro conversion of sheep prion protein to protease-resistant forms. Proc. Natl. Acad. Sci. USA 94:4931-4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butler, D. A., M. R. Scott, J. M. Bockman, D. R. Borchelt, A. Taraboulos, K. K. Hsiao, D. T. Kingsbury, and S. B. Prusiner. 1988. Scrapie-infected murine neuroblastoma cells produce protease-resistant prion proteins. J. Virol. 62:1558-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caughey, B., and G. J. Raymond. 1991. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J. Biol. Chem. 266:18217-18223. [PubMed] [Google Scholar]
- 7.Collinge, J., M. S. Palmer, and A. J. Dryden. 1991. Genetic predisposition to iatrogenic Creutzfeldt-Jakob disease. Lancet 337:1441-1442. [DOI] [PubMed] [Google Scholar]
- 8.Collinge, J. 2001. Prion diseases of humans and animals: their causes and molecular basis. Annu. Rev. Neurosci. 24:519-550. [DOI] [PubMed] [Google Scholar]
- 9.Enari, M., E. Flechsig, and C. Weissmann. 2001. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc. Natl. Acad. Sci. USA 98:9295-9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer, M., T. Rulicke, A. Raeber, A. Sailer, M. Moser, B. Oesch, S. Brandner, A. Aguzzi, and C. Weissmann. 1996. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 15:1255-1264. [PMC free article] [PubMed] [Google Scholar]
- 11.Goldmann, W., N. Hunter, G. Smith, J. Foster, and J. Hope. 1994. PrP genotype and agent effects in scrapie: change in allelic interaction with different isolates of agent in sheep, a natural host of scrapie. J. Gen. Virol. 75:989-995. [DOI] [PubMed] [Google Scholar]
- 12.Ikeda, T., M. Horiuchi, N. Ishiguro, Y. Muramatsu, G. D. Kai-Uwe, and M. Shinagawa. 1995. Amino acid polymorphisms of PrP with reference to onset of scrapie in Suffolk and Corriedale sheep in Japan. J. Gen. Virol. 76:2577-2581. [DOI] [PubMed] [Google Scholar]
- 13.Harris, D. A. 1999. Cellular biology of prion diseases. Clin. Microbiol. Rev. 12:429-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hill, A. F., R. J. Butterworth, S. Joiner, G. Jackson, M. N. Rossor, D. J. Thomas, A. Frosh, N. Tolley, J. E. Bell, M. Spencer, A. King, S. Al-Sarraj, J. W. Ironside, P. L. Lantos, and J. Collinge. 1999. Investigation of variant Creutzfeldt-Jakob disease and other human prion diseases with tonsil biopsy samples. Lancet 353:183-189. [DOI] [PubMed] [Google Scholar]
- 15.Hill, A. F., M. Antoniou, and J. Collinge. 1999. Protease-resistant prion protein produced in vitro lacks detectable infectivity. J. Gen. Virol. 80:11-14. [DOI] [PubMed] [Google Scholar]
- 16.Hill, A. F., S. Joiner, J. Linehan, M. Desbruslais, P. L. Lantos, and J. Collinge. 2000. Species-barrier-independent prion replication in apparently resistant species. Proc. Natl. Acad. Sci. USA 97:10248-10253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hunter, N. 1997. PrP genetics in sheep and the applications for scrapie and BSE. Trends Microbiol. 5:331-334. [DOI] [PubMed] [Google Scholar]
- 18.Krasemann, S., M. H. Groschup, S. Harmeyer, G. Hunsmann, and W. Bodemer. 1996. Generation of monoclonal antibodies against human prion proteins in PrP0/0 mice. Mol. Med. 2:725-734. [PMC free article] [PubMed] [Google Scholar]
- 19.Lehmann, S., H. Laude, D. A. Harris, R. I. Carp, D. Vilette, S. Katamine, J.-Y. Madec, and N. Nishida. 2001. Ex vivo transmission of mouse-adapted prion strains to N2a and GT1-7 cell lines, p. 680-686. In K. Iqbal, S. S. Sisodia, and B. Winblad (ed.), Alzheimer disease: advances in etiology, pathogenesis, and therapeutics. John Wiley & Sons, Inc., New York, N.Y.
- 20.Moudjou, M., Y. Frobert, J. Grassi, and C. La Bonnardière. 2001. Cellular prion protein status in sheep: tissue-specific biochemical signatures. J. Gen. Virol. 82:2017-2024. [DOI] [PubMed] [Google Scholar]
- 21.Prusiner, S. B., M. Scott, D. Foster, K. M. Pan, D. Groth, C. Mirenda, M. Torchia, S. L. Yang, D. Serban, G. A. Carlson, P. C. Hoppe, D. Westaway, and S. DeArmond. 1990. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 63:673-686. [DOI] [PubMed] [Google Scholar]
- 22.Prusiner, S. B. 1998. Prions. Proc. Natl. Acad. Sci. USA 117:421-434. [Google Scholar]
- 23.Race, R., A. Raines, G. J. Raymond, B. Caughey, and B. Chesebro. 2001. Long-term subclinical carrier state precedes scrapie replication and adaptation in a resistant species: analogies to bovine spongiform encephalopathy and variant Creutzfeldt-Jakob disease in humans. J. Virol. 75:10106-10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raeber, A. J., A. Sailer, I. Hegyi, M. A. Klein, T. Rulicke, M. Fischer, S. Brandner, A. Aguzzi, and C. Weissmann. 1999. Ectopic expression of prion protein (PrP) in T lymphocytes or hepatocytes of PrP knockout mice is insufficient to sustain prion replication. Proc. Natl. Acad. Sci. USA 96:3987-3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rezaei, H., D. Marc, Y. Choiset, M. Takahashi, G. Hui Bon Hoa, T. Haertle, J. Grosclaude, and P. Debey. 2000. High yield purification and physico-chemical properties of full-length recombinant allelic variants of sheep prion protein linked to scrapie susceptibility. Eur. J. Biochem. 267:2833-2839. [DOI] [PubMed] [Google Scholar]
- 26.Schätzl, H. M., L. Laszlo, D. M. Holtzman, J. Tatzelt, S. J. DeArmond, R. I. Weiner, W. C. Mobley, and S. B. Prusiner. 1997. A hypothalamic neuronal cell line persistently infected with scrapie prions exhibits apoptosis. J. Virol. 71:8821-8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Telling, G. C., M. Scott, J. Mastrianni, R. Gabizon, M. Torchia, F. E. Cohen, S. J. DeArmond, and S. B. Prusiner. 1995. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 83:79-90. [DOI] [PubMed] [Google Scholar]
- 28.Vilette, D., O. Andréoletti, F. Archer, M. F. Madelaine, J. L. Vilotte, S. Lehmann, and H. Laude. 2001. Ex vivo propagation of infectious sheep scrapie agent in heterologous epithelial cells expressing ovine prion protein. Proc. Natl. Acad. Sci. USA 98:4055-4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vilotte, J. L., S. Soulier, R. Essalmani, M. G. Stinnakre, D. Vaiman, L. Lepourry, J. C. Da Silva, N. Besnard, M. Dawson, A. Buschmann, M. Groschup, S. Petit, M. F. Madelaine, S. Rakatobe, A. Le Dur, D. Vilette, and H. Laude. 2001. Markedly increased susceptibility to natural sheep scrapie of transgenic mice expressing ovine PrP. J. Virol. 75:5977-5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weissmann, C. 1996. The Ninth Datta Lecture. Molecular biology of transmissible spongiform encephalopathies. FEBS Lett. 389:3-11. [DOI] [PubMed] [Google Scholar]