


Abstract
Poly (ADP-ribose) polymerase (PARP-1), ATM and DNA-dependent protein kinase (DNA-PK) are all involved in responding to DNA damage to activate pathways responsible for cellular survival. Here, we demonstrate that PARP-1−/− cells are sensitive to the ATM inhibitor KU55933 and conversely that AT cells are sensitive to the PARP inhibitor 4-amino-1,8-napthalamide. In addition, PARP-1−/− cells are shown to be sensitive to the DNA-PK inhibitor NU7026 and DNA-PKcs or Ku80 defective cells shown to be sensitive to PARP inhibitors. We believe PARP inhibition results in an increase in unresolved spontaneous DNA single-strand breaks (SSBs), which collapse replication forks and trigger homologous recombination repair (HRR). We show that ATM is activated following inhibition of PARP. Furthermore, PARP inhibitor-induced HRR is abolished in ATM, but not DNA-PK, inhibited cells. ATM and DNA-PK inhibition together give the same sensitivity to PARP inhibitors as ATM alone, indicating that ATM functions in the same pathways as DNA-PK for survival at collapsed forks, likely in non-homologous end joining (NHEJ). Altogether, we suggest that ATM is activated by PARP inhibitor-induced collapsed replication forks and may function upstream of HRR in the repair of certain types of double-strand breaks (DSBs).
INTRODUCTION
Poly (ADP-ribose) polymerase 1 (PARP-1) is an abundant nuclear protein that binds to a DNA single-strand break (SSB) and catalyses the formation of PAR polymers on itself and other acceptor proteins (1). PAR formation is suggested to be important to protect DNA breaks, alter chromatin structure and to attract DNA repair proteins to the site of damage (1,2). PARP-1 is involved in base excision repair (BER) (3), where interactions between PARP-1 and BER enzymes, such as polymerase β (4) and XRCC1 (5) point to a direct role for PARP-1. PARP-1 has also been associated with homologous recombination (HR), as lack of PARP-1 leads to increased sister chromatid exchange and micronuclei formation (3,6). It does not however appear to be directly required for the process per se, as Rad51 foci form normally in PARP-1 deficient cells and an I-SceI induced DNA double-strand break (DSB) can be repaired normally in PARP inhibited cells (7).PARP-1 is also thought to be involved in a DNA-PK/Artemis/XRCC4/ligase IV independent pathway of DSB end joining repair (8). Here PARP-1 functions with the XRCC1/DNA ligase III complex and as such could be part of a back up pathway for non-homologous end joining (NHEJ). Despite these roles PARP-1 is not essential for cellular survival or prevention of tumourigenesis since PARP-1 knockout mice are viable, fertile and do not develop early onset tumours (9).
Presumably due to its function in various DNA repair processes, PARP inhibition sensitises cells to multiple DNA-damaging agents, such as ionising radiation (IR) (10–12), topoisomerase I inhibitors (13,14) and monofunctional DNA-alkylating agents (11,14–16). PARP inhibitors therefore have potential as chemotherapeutics, when used in combination with many DNA-damaging anti-cancer drugs or radiation (17). Such combination therapy may be particularly important in the treatment of tumours with acquired resistance (18,19). In addition to their role in combined anti-cancer therapies we and others have recently reported that PARP inhibitors alone are efficient to treat tumours which are deficient in HR, such as BRCA2 defective cancers (20,21). We reported that PARP-1 is important for cellular survival in the absence of homologous recombination (HR) and demonstrated that this is likely to be due to the role of PARP-1, together with XRCC1, in SSB repair. We proposed that unresolved spontaneous SSBs in PARP inhibited cells collapse replication forks forming DSBs which require homologous recombination repair (HRR) to continue replication.
Here we examine cellular survival following PARP inhibition in the absence or following inhibition of proteins also known to be involved in the DNA DSB response, ATM and DNA-dependent protein kinase (DNA-PK). We show that survival is decreased when PARP-1 and ATM, DNA-PKcs or Ku80 are absent or inhibited. In addition, PARP inhibition causes ATM activation and PARP inhibitor-enhanced HR is abolished when ATM is inhibited. Altogether, our data suggest that ATM is required for HRR at collapsed replication forks produced following PARP inhibition.
MATERIALS AND METHODS
Chemicals
The PARP inhibitor, 4-Amino-1,8-naphthalimide (22), was a gift from Dr. Nicola Curtin (University of Newcastle, UK) and the ATM inhibitor, KU55933 (23), and the DNA-PK inhibitor, NU7026 (24), were kindly donated by Dr Graeme Smith (KuDOS Pharmaceuticals Ltd, Cambridge, UK). All were dissolved in DMSO and stored at −20°C. Drugs alone or in combination were added to cell cultures so that the final DMSO concentrations did not exceed 0.2%.
Cells and cell culture
The A11 and A19 cell lines were a kind gift from Zhao-Qi Wang (Lyon, France), the AA8 and V3-3 cell lines were provided by Larry Thompson (Livermore, CA), pEBS-YZ5 and AT221JE-T/pEBS AT cells were generously supplied by Yoshi Shiloh (Tel Aviv, Isreal), CHO-K1 and XRS6 were kindly donated by Penny Jeggo (Sussex, UK), GK41 cells were helpfully supplied by C. Smythe (Sheffield, UK).
All cell lines in this study were grown in DMEM with 10% Fetal bovine serum and penicillin (100 U/ml) and streptomycin sulphate (100 µg/ml) at 37°C under an atmosphere containing 5% CO2. In addition the SPD8 cell line was grown in the presence of 5 µg/ml 6-thioguanine.
Recombination assay
A total of 1.5 × 106 cells were inoculated into 100 mm dishes in media 4 h prior to a 24 h treatment with drugs as indicated or hydroxyurea (HU) as a positive control. After treatments, the cells were rinsed three times with PBS and 10 ml media added before allowing the cells to recover for 48 h. After recovery, cells were released by trypsinisation and counted. HPRT+ revertants were selected by plating 3 × 105 treated cells per dish in the presence of HAsT (50 µM hypoxanthine, 10 µM L-azaserine, and 5 µM thymidine). To determine cloning efficiency, two dishes were plated with 500 cells each. The colonies obtained were stained with methylene blue in methanol (4 g/l), following 7 (in the case of cloning efficiency) or 10 (for reversion) days of incubation.
Toxicity assay
A total of 500 or 1000 cells were plated in triplicate onto 100 mm dishes 4 h prior to treatment with combinations of drugs as indicated. Twelve days later, when colonies could be observed, they were fixed and stained with methylene blue in methanol (4 g/l). Colonies consisting of more than 50 cells were subsequently counted. Each colony was assumed to represent one cell surviving from the original 500 and surviving fraction for each dose calculated.
Western blotting
A total of 2 × 106 cells were plated onto 100 mm dishes and grown for 24 h. Cells were left untreated or treated with 100 µM PARP inhibitor 4-amino-1,8-naphthalimide for times indicated prior to lysis in RIPA buffer in the presence of 1× protease and phosphatase inhibitor cocktails (sigma). An aliquot of 50 µg total protein was then run on a 6 or 10% SDS–PAGE gel and transferred to Hybond ECL membrane (Amersham Pharmacia) using a semi-dry transfer cell (Bio-Rad). This membrane was blocked in 5% milk for 1 h and immunoblotted with rabbit polyclonal antibodies against phospho-Ser 1981 ATM (Rockland 1:1000), total ATM (Oncogene 1:500), phospho-Ser 345 Chk1 (Cell Signaling 1:1000), phospho-Thr 68 Chk2 (Cell Signaling 1:1000), total Chk2 (Cell Signaling 1:1000) and β-actin (sigma 1:2000) proteins in 5% milk overnight. Anti-rabbit peroxidase conjugate (Cell Signaling 1:1000) was used as secondary antibody and immunoreactive protein was visualized using ECL reagents (Amersham Pharmacia) following manufacturer's instructions.
RESULTS
ATM and DNA-PK promote survival in PARP-1 defective or inhibited cells
PARP-1 mouse knockouts are viable and fertile suggesting that PARP-1 is not essential (3,25,26). However, double knockouts of PARP-1 with either ATM or Ku80 are embryonic lethal (27,28). In contrast, knocking out DNA-PKcs in PARP-1−/− mice is not lethal (29). Instead, such double-knockout mice revert the severe combined immune defect normally associated with a DNA-PKcs knockout (29). Here we tested if chemically inhibiting ATM in the absence of PARP-1 or inhibiting PARP-1 in the absence of ATM increased toxicity to each inhibitor in mammalian cells. We found that PARP-1 defective mouse embryonic fibroblast (MEF) cells (A11) were more sensitive to the ATM inhibitor KU55933 than A19 wild-type cells (Figure 1A). Also, ATM defective AT221JE-T/pEBS cells were more sensitive to the PARP inhibitor (4-amino-1,8-naphthalimide) than cells corrected for the AT phenotype by the introduction of the wild-type ATM cDNA (pEBS-YZ5) (Figure 1B). This suggests that ATM is important to cellular survival following PARP inhibition.
Figure 1.

Absence of PARP-1 leads to increased sensitivity to ATM inhibition and vice versa. (A) Survival fraction of A19 (WT) and A11 (PARP-1−/−) MEFs following treatment for 10 days with increasing doses of the ATM inhibitor KU55933. (B) Survival fraction of AT221JE-T/pEBS (pEBS-ATM defective) and pEBS-YZ5 (YZ5—corrected for the AT phenotype by wild-type ATM cDNA) following treatment for 12 days with increasing doses of the PARP inhibitor 4-amino-1,8-napthalamide. The means (symbol) and standard deviations (error bar) from at least three experiments are depicted.
Similar results were obtained with DNA-PK. A11 cells were hypersensitive to the DNA-PK inhibitor NU7026 (Figure 2A), and conversely cells defective in DNA-PK (V3-3) were hypersensitive to PARP inhibition (Figure 2B). In addition XRS6 cells deficient in Ku80 were more sensitive than the relevant wild-type cell line (CHO-K1). As Ku80 and DNA-PK interact and both play a role in NHEJ, the sensitivity of V3-3 and XRC6 is most likely explained by the absence of this pathway. These data suggest a role for NHEJ in survival of PARP inhibited cells. It is interesting to note that Ku80 deficient cells were more sensitive to PARP inhibition than DNA-PK deficient cells. This is consistent with embryonic lethality associated with PARP-1−/− Ku80−/− double-knockout mice (28), and survival of PARP-1−/− DNA-PKcs−/− double-knockout mice (29).
Figure 2.

Absence of PARP-1 leads to increased sensitivity to DNA-PK inhibition and vice versa. (A) Survival fraction of A19 (WT) and A11 (PARP-1−/−) MEFs following treatment for 10 days with increasing doses of the DNA-PK inhibitor NU7026. (B) Survival fraction of AA8 (WT) and V3-3 (DNA-PKcs deficient) following treatment for 10 days with increasing doses of the PARP inhibitor 4-amino-1,8-napthalamide. (C) Survival fraction of CHO-K1 (WT) and XRS6 (Ku80 deficient) following treatment for 10 days with increasing doses of the PARP inhibitor 4-amino-1,8-napthalamide. The means (symbol) and standard deviations (error bar) from at least three experiments are depicted.
The wild-type Chinese hamster ovary cell line AA8 was then treated with different combinations of ATM, DNA-PK and PARP inhibitors (Figure 3). When both ATM and DNA-PK were inhibited the cells were more sensitive to PARP inhibition than non-treated or DNA-PK alone inhibited cells. However ATM and DNA-PK inhibited cells had a similar sensitivity to PARP inhibitors as ATM alone inhibited cells (Figure 3). As ATM alone inhibited cells show similar sensitivity to ATM/DNA-PK inhibited cells, they are likely to function on the same pathway for survival following PARP inhibition. However as DNA-PK/ATM inhibited cells display much greater sensitivity than DNA-PK alone inhibited cells, other ATM dependent processes are likely to contribute further to survival following PARP inhibition. After 10 days exposure to either drug the toxicity to PARP inhibition is more pronounced than at 24 h (compare Figure 5C to 3). This suggests that both drugs are active for >24 h. At 24 h (when both are presumed still active), ATM inhibitor had a more severe toxicity than the DNA-PK inhibitor thus the difference in toxicity is not just due to differences in stability but due to a biological effect. Both the ATM inhibitor (23) and the DNA-PK inhibitor (24) were used at doses known to fully inhibit their respective enzyme activity.
Figure 3.

Inhibition of ATM and DNA-PK increases sensitivity to PARP inhibition compared to DNA-PK inhibition alone but not compared to ATM inhibition alone. Survival fraction of AA8 (WT) cells following treatment for 10 days with/without 10 µM KU55933 (ATM inhibitor), 10 µM NU7026 (DNA-PK inhibitor) or both in combination with increasing doses of the PARP inhibitor 4-amino-1,8-napthalamide. The means (symbol) and standard deviations (error bar) from at least three experiments are depicted.
The ATM kinase is activated following PARP inhibition
As PARP inhibition combined with ATM inhibition seemed to be detrimental to cells we investigated whether ATM was activated upon PARP inhibition. Following a 4 h treatment with PARP inhibitors alone phosphorylation of S1981 on ATM was detected; phosphorylation then decreased but was still apparent after 24 h treatment (Figure 4A). This suggests that one or more of the functions downstream of activated ATM are required for survival following PARP inhibition. Both Chk1 and Chk2 are known downstream targets of ATM (30). Here, we found that only Chk2 was phosphorylated in response to PARP inhibition (Figure 4B).
Figure 4.

PARP inhibition activates ATM. (A) Western blot for ATM phospho serine 1981 and ATM control following PARP inhibition for the times indicated. (B) Western blot for CHK1 phospho serine 345, CHK2 phospho threonine 68 and total CHK2 and actin controls following PARP inhibition or 0.5 mM HU treatment for 24 h.
ATM is required for PARP inhibitor-induced homologous recombination repair
Lack of PARP-1 in cells causes increased sister chromatid exchange, RAD51 foci and micronuclei formation (3,6,31), suggesting increased levels of HR. In addition, we have previously demonstrated that PARP inhibitors cause an increase in γH2AX foci formation, and suggested that these form when the increased number of SSBs following PARP inhibition, collapse into DSBs during replication (20). It has been shown that such collapsed replication forks require HR for repair (32,33). Here we investigated HR using a cell line (SPD8) (34)that contains a partial duplication of the hypoxanthine guanine phosphoribosyl transferase (hprt) gene which leads to expression of non-functional HPRT protein. HR between the duplicated regions reverts the hprt gene to wild-type and can be selected for in HAsT media; colonies formed following selection are therefore indicative of HR (34). Treatment with the PARP inhibitor 4-amino-1,8-napthalamide caused an increase in HPRT positive colonies, confirming that PARP inhibition triggers HRR. Treatment with ATM or DNA-PK inhibitors alone made no difference to HR levels (Figure 5). However, co-treatment with the ATM inhibitor prevented the PARP inhibitor-induced increase in HR back to non-treated background levels (Figure 5A). DNA-PK inhibition made no difference to PARP inhibitor-induced HR (Figure 5B). The cloning efficiency of cells following each treatment was also determined (Figure 5C) and this was taken into consideration when calculating the recombination frequencies. Thus the decrease in PARP inhibitor-induced HR seen when ATM is also inhibited is not due to a difference in survival following treatment.
Figure 5.

ATM inhibition prevents PARP inhibitor-induced HR. (A and B) Recombination frequency in hprt gene following treatment for 24 h with/without 10 µM KU55933 (ATM inhibitor), 10 µM NU7026 (DNA-PK inhibitor), 100 µM 4-amino-1,8-napthalamide (PARP inhibitor), 0.5 mM HU or combinations of the above. (C) Cloning efficiencies (% of control) of the same cells. The means (symbol) and standard deviations (error bar) from at least three experiments are depicted.
A kinase dead dominant negative ATR does not affect sensitivity to PARP inhibitors
ATR primarily signals at stalled replication forks (35) and may therefore be implicated in signalling from PARP inhibitor-induced DNA damage. However we did not see activation of Chk1, the main downstream target of ATR (36), following PARP inhibition (Figure 4B). We tested the sensitivity of an inducible ATR kinase dead dominant mutant cell line (GK41) to PARP inhibition and found that upon expression of the dominant dead kinase the sensitivity to PARP inhibitors although slightly increased was not significantly altered from wild-type levels (Figure 6). As a positive control the sensitivity of kinase dead dominant expressing cells to HU was tested; as reported earlier (37) they were more sensitive than non-expressing cells (data not shown). These data suggest that ATR may not play a large role in survival from PARP inhibitor-induced DNA damage.
Figure 6.
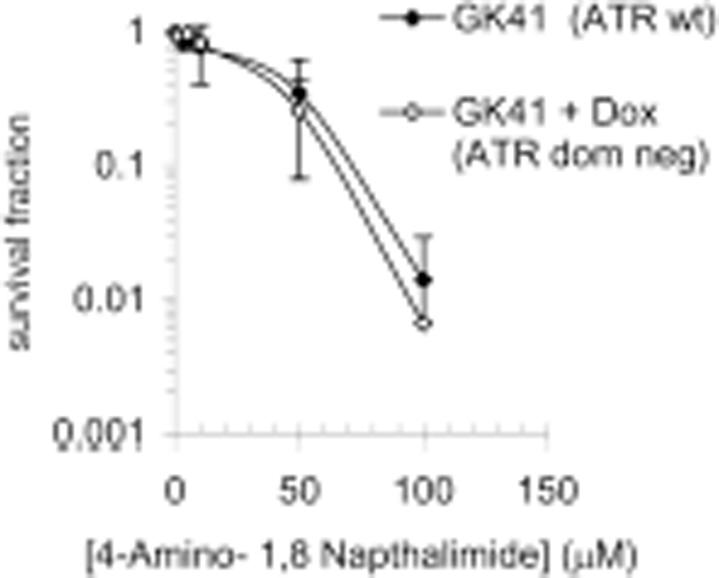
A kinase dead dominant negative ATR does not alter sensitivity to PARP inhibition. Survival fraction of GK41 cells following treatment for 10 days with increasing doses of the PARP inhibitor 4-amino-1,8-napthalamide in the presence or absence of 1.5 µg/ml doxycycline to induce expression of kinase dead dominant negative ATR. The means (symbol) and standard deviations (error bar) from at least three experiments are depicted.
DISCUSSION
The ATM−/− and PARP-1−/− double-knockout mice are embryonic lethal (27), suggesting that a functional relationship exists between the two proteins. In agreement with this we show that AT cells are hypersensitive to a PARP inhibitor and vice versa that PARP-1−/− cells are hypersensitive to a specific ATM inhibitor, KU55933. In addition, we find that ATM is phosphorylated following PARP inhibition and that the downstream target Chk2 is also phosphorylated.
Lack or inhibition of PARP-1 has been shown to increase γH2AX foci formation, this increase is thought to reflect the collapse of unresolved spontaneous SSBs into DSBs at replication forks (20). A collapsed replication fork is therefore likely to be the substrate that activates ATM in PARP inhibited cells [it is established that ATM is activated by DSBs (38)]. In support of this, ATM is reported to be activated by the topoisomerase I inhibitor camptothecin (39), which stabilizes SSBs that then collapse into DSBs at replication forks (40).
SSBs, when collapsed into DSBs at replication forks, are the major lesions triggering spontaneous HR (32). Previously we demonstrated that the HR pathway becomes essential for survival following inhibition or loss of PARP-1 (20). In our model, we suggest that loss of repair of spontaneous SSBs in PARP inhibited cells results in collapsed forks that trigger HRR (Figure 7). As ATM is also required for survival in PARP inhibited cells, there might be a relationship between ATM and HR. A controlling role for ATM in HR has been suggested (41) and we previously showed that ATM is required for HRR at slowed replication forks (37). Here, we find that PARP inhibition-induced HRR does not occur when ATM is inhibited (Figure 5A). This suggests that an ATM response is necessary for and may function upstream of HR during the repair of PARP inhibitor-induced collapsed forks (Figure 7). Thus, the sensitivity of AT and PARP-1 defective cells to PARP and ATM inhibitors, respectively, is likely to be explained by a loss of HRR. However, ATM defective or inhibited cells do not show the same degree of sensitivity to PARP inhibitors as do BRCA2 defective cells (20), suggesting that some degree of HRR may proceed at collapsed forks even in the absence of ATM.
Figure 7.

Model for ATM activation following PARP inhibition. PARP inhibition results in more collapsed replication forks, probably because of an inability to efficiently repair endogenous SSBs (20). HR is the most important pathway for repair of collapsed replication forks in mammalian cells (32,33) and loss of this pathway results in lethality following PARP inhibition (20). Our data suggests that the HRR pathway involves an ATM signal, which would explain the increased sensitivity to PARP inhibitors in AT cells. In addition our data also imply that a second pathway involving DNA-PK and ATM is required in survival following PARP inhibition, this is most likely to be NHEJ.
While DSBs associated with replication forks are primarily repaired with HRR, NHEJ does have an overlapping role in the repair (33). Thus NHEJ may also become important for cellular survival following PARP inhibition and subsequent replication fork collapse. This then could explain why we see decreased cellular survival in response to PARP inhibitors when DNA-PKcs or Ku80 are absent or inhibited. It is interesting to note that specific inhibition of PARP inhibits DNA-PK activity and vice versa (42) suggesting that PARP-1 can modulate the efficiency of DSB repair in more than one way effecting both HR and NHEJ.
ATM and DNA-PK inhibition together gave the same sensitivity to PARP inhibitors as ATM alone, but an increased sensitivity compared to DNA-PK inhibitor alone. These data indicate that ATM and DNA-PK function on the same pathway and that ATM has additional functions to DNA-PK in cellular survival following PARP inhibition. It is reported that in response to IR ATM phosphorylates Artemis which then process a subset of non-ligatable DNA double-strand ends with damaged termini prior to NHEJ repair involving DNA-PK (43). Our data, putting ATM and DNA-PK in the same survival pathway following PARP inhibition, raise the possibility that ATM signals to Artemis so that the DNA ends at a collapsed fork can be repaired by NHEJ. Indeed all the substrates of endonucleolytic action by Artemis:DNA-PKcs have been shown to contain transitions of double- and single-stranded DNA (44). Decreased survival in ATM inhibited cells upon PARP inhibition may therefore, in part, be due to a lack of NHEJ. The sensitivity of ATM inhibited cells may be due to its contribution to both NHEJ and HR. The relatively small increase in sensitivity upon DNA-PK inhibition probably reflects the relatively small proportion of DSBs at collapsed replication forks which are repaired by NHEJ as opposed to the majority which are resolved by HR (33).
The absence of Chk1 signal in response to PARP inhibition and the lack of significant increase in sensitivity to PARP inhibitors in cells expressing an ATR kinase dead dominant mutant were unexpected given that ATR normally signals from lesions which impair replication fork progression (35). ATR is activated when it binds to regions of ssDNA which are coated with RPA (45). Maybe chemically inhibiting PARP leaves it irreversibly bound to DNA ends protecting from ssDNA formation. Alternately PARP bound to DNA may prevent RPA from binding and thus ATR from signalling. In a third scenario the lesion formed by PARP inhibition may not trigger ATR. This seems less likely as CPT induced damage (which we believe produces a similar lesion to PARP inhibition) triggers ATR/Chk1 signalling which protects from Topo-1 poison induced cell killing (46,47). This surprising lack of a role for ATR following PARP inhibition will be the subject of further investigation.
Our data reveals a relationship between PARP-1 and ATM, and DNA-PK which maybe of value when considering the therapeutic potential of inhibitors in the treatment of cancer. Mutations in the ATM gene have been found in T-cell prolymphocytic leukaemia (48,49), mantle cell lymphoma (50,51), and B-cell chronic lymphocytic leukaemia (52–55). Such cases would be ideal candidates for PARP inhibitor therapy, as the cells containing the ATM mutation would be expected to be more sensitive to PARP inhibitors than the surrounding ATM proficient tissue and thus the side effects usually seen with classical cytotoxic anti-cancer drugs minimal.
In summary, our data demonstrate that ATM is activated by PARP inhibition, most likely at collapsed replication forks, and is required for and may function upstream of HRR and possibly NHEJ in the repair of certain types of DSBs.
Acknowledgments
The authors wish to thank Drs Graeme Smith, Nicola Curtin, Zhao-Qi Wang, Larry Thompson, Yoshi Shiloh and Penny Jeggo for materials. The Yorkshire Cancer Research provided financial support for this investigation. The Open Access publication charges for this article were waived by Oxford University Press.
Conflict of interest statement. None declared.
REFERENCES
- 1.Lindahl T., Satoh M.S., Poirier G.G., Klungland A. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem. Sci. 1995;20:405–411. doi: 10.1016/s0968-0004(00)89089-1. [DOI] [PubMed] [Google Scholar]
- 2.D'Amours D., Desnoyers S., D'Silva I., Poirier G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- 3.de Murcia J.M., Niedergang C., Trucco C., Ricoul M., Dutrillaux B., Mark M., Oliver F.J., Masson M., Dierich A., LeMeur M., Walztinger C., Chambon P., de Murcia G. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl Acad. Sci. USA. 1997;94:7303–7307. doi: 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caldecott K., Aoufouchi S., Johnson P., Shall S. XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular ‘nick- sensor’ in vitro. Nucleic Acids Res. 1996;24:4387–4394. doi: 10.1093/nar/24.22.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masson M., Niedergang C., Schreiber V., Muller S., Menissier-de Murcia J., de Murcia G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell Biol. 1998;18:3563–3571. doi: 10.1128/mcb.18.6.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z.Q., Stingl L., Morrison C., Jantsch M., Los M., Schulze-Osthoff K., Wagner E.F. PARP is important for genomic stability but dispensable in apoptosis. Genes Dev. 1997;11:2347–2358. doi: 10.1101/gad.11.18.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schultz N., Lopez E., Saleh-Gohari N., Helleday T. Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids. Res. 2003;31:4959–4964. doi: 10.1093/nar/gkg703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Audebert M., Salles B., Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 2004;279:55117–55126. doi: 10.1074/jbc.M404524200. [DOI] [PubMed] [Google Scholar]
- 9.Conde C., Mark M., Oliver F.J., Huber A., de Murcia G., Menissier-de Murcia J. Loss of poly(ADP-ribose) polymerase-1 causes increased tumour latency in p53-deficient mice. EMBO J. 2001;20:3535–3543. doi: 10.1093/emboj/20.13.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arundel-Suto C.M., Scavone S.V., Turner W.R., Suto M.J., Sebolt-Leopold J.S. Effects of PD 128763, a new potent inhibitor of poly(ADP-ribose) polymerase, on X-ray-induced cellular recovery processes in chinese hamster V79 Cells. Radiation Res. 1991;126:367–371. [PubMed] [Google Scholar]
- 11.Bowman K.J., White A., Golding B.T., Griffin R.J., Curtin N.J. Potentiation of anti-cancer agent cytotoxicity by the potent poly(ADP-ribose) polymerase inhibitors NU1025 and NU1064. Br. J. Cancer. 1998;78:1269–1277. doi: 10.1038/bjc.1998.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weltin D., Holl V., Hyun J.W., Dufour P., Marchal J., Bischoff P. Effect of 6(5H)-phenanthridinone, a poly (ADP-ribose)polymerase inhibitor, and ionizing radiation on the growth of cultured lymphoma cells. Int. J. Radiat. Biol. 1997;72:685–692. doi: 10.1080/095530097142843. [DOI] [PubMed] [Google Scholar]
- 13.Bowman K.J., Newell D.R., Calvert A.H., Curtin N.J. Differential effects of the poly (ADP-ribose) polymerase (PARP) inhibitor NU1025 on topoisomerase I and II inhibitor cytotoxicity in L1210 cells in vitro. Br. J. Cancer. 2001;84:106–112. doi: 10.1054/bjoc.2000.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delaney C.A., Wang L.Z., Kyle S., White A.W., Calvert A.H., Curtin N.J., Durkacz B.W., Hostomsky Z., Newell D.R. Potentiation of temozolomide and topotecan growth inhibition and cytotoxicity by novel poly(adenosine diphosphoribose) polymerase inhibitors in a panel of human tumor cell lines. Clin. Cancer Res. 2000;6:2860–2867. [PubMed] [Google Scholar]
- 15.Tentori L., Portarena I., Graziani G. Potential clinical applications of poly(ADP-ribose) polymerase (PARP) inhibitors. Pharmacol. Res. 2002;45:73–85. doi: 10.1006/phrs.2001.0935. [DOI] [PubMed] [Google Scholar]
- 16.Boulton S., Pemberton L.C., Porteous J.K., Curtin N.J., Griffin R.J., Golding B.T., Durkacz B.W. Potentiation of temozolomide-induced cytotoxicity: a comparative study of the biological effects of poly(ADP-ribose) polymerase inhibitors. Br. J. Cancer. 1995;72:849–856. doi: 10.1038/bjc.1995.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Curtin N.J. PARP inhibitors for cancer therapy. Expert Rev. Mol. Med. 2005;7:1–20. doi: 10.1017/S146239940500904X. [DOI] [PubMed] [Google Scholar]
- 18.Curtin N.J., Wang L.Z., Yiakouvaki A., Kyle S., Arris C.A., Canan-Koch S., Webber S.E., Durkacz B.W., Calvert H.A., Hostomsky Z., et al. Novel poly(ADP-ribose) polymerase-1 inhibitor, AG14361, restores sensitivity to temozolomide in mismatch repair-deficient cells. Clin. Cancer Res. 2004;10:881–889. doi: 10.1158/1078-0432.ccr-1144-3. [DOI] [PubMed] [Google Scholar]
- 19.Cheng C.L., Johnson S.P., Keir S.T., Quinn J.A., Ali-Osman F., Szabo C., Li H., Salzman A.L., Dolan M.E., Modrich P., Bigner D.D., Friedman H.S. Poly(ADP-ribose) polymerase-1 inhibition reverses temozolomide resistance in a DNA mismatch repair-deficient malignant glioma xenograft. Mol. Cancer Ther. 2005;4:1364–1368. doi: 10.1158/1535-7163.MCT-05-0128. [DOI] [PubMed] [Google Scholar]
- 20.Bryant H.E., Schultz N., Thomas H.D., Parker K.M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N.J., Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 21.Farmer H., McCabe N., Lord C.J., Tutt A.N., Johnson D.A., Richardson T.B., Santarosa M., Dillon K.J., Hickson I., Knights C., Martin N.M., Jackson S.P., Smith G.C., Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 22.Schlicker A., Peschke P., Burkle A., Hahn E.W., Kim J.H. 4-Amino-1,8-naphthalimide: a novel inhibitor of poly(ADP-ribose) polymerase and radiation sensitizer. Int. J. Radiat. Biol. 1999;75:91–100. doi: 10.1080/095530099140843. [DOI] [PubMed] [Google Scholar]
- 23.Hickson I., Zhao Y., Richardson C.J., Green S.J., Martin N.M., Orr A.I., Reaper P.M., Jackson S.P., Curtin N.J., Smith G.C. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 24.Veuger S.J., Curtin N.J., Smith G.C., Durkacz B.W. Effects of novel inhibitors of poly(ADP-ribose) polymerase-1 and the DNA-dependent protein kinase on enzyme activities and DNA repair. Oncogene. 2004;23:7322–7329. doi: 10.1038/sj.onc.1207984. [DOI] [PubMed] [Google Scholar]
- 25.Wang Z.-Q., Auer B., Stingl L., Berghammer H., Haidacher D., Schweiger M., Wagner E.F. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995;9:509–520. doi: 10.1101/gad.9.5.509. [DOI] [PubMed] [Google Scholar]
- 26.Masutani M., Nozaki T., Nishiyama E., Shimokawa T., Tachi Y., Suzuki H., Nakagama H., Wakabayashi K., Sugimura T. Function of poly(ADP-ribose) polymerase in response to DNA damage: gene-disruption study in mice. Mol. Cell Biochem. 1999;193:149–152. [PubMed] [Google Scholar]
- 27.Menisser-de Murcia J., Mark M., Wendling O., Wynshaw-Boris A., de Murcia G. Early embryonic lethality in PARP-1 Atm double-mutant mice suggests a functional synergy in cell proliferation during development. Mol. Cell Biol. 2001;21:1828–1832. doi: 10.1128/MCB.21.5.1828-1832.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henrie M.S., Kurimasa A., Burma S., Menissier-de Murcia J., de Murcia G., Li G.C., Chen D.J. Lethality in PARP-1/Ku80 double mutant mice reveals physiological synergy during early embryogenesis. DNA Repair (Amst) 2003;2:151–158. doi: 10.1016/s1568-7864(02)00199-4. [DOI] [PubMed] [Google Scholar]
- 29.Morrison C., Smith G.C., Stingl L., Jackson S.P., Wagner E.F., Wang Z.Q. Genetic interaction between PARP and DNA-PK in V(D)J recombination and tumorigenesis. Nature Genet. 1997;17:479–482. doi: 10.1038/ng1297-479. [DOI] [PubMed] [Google Scholar]
- 30.Sancar A., Lindsey-Boltz L.A., Unsal-Kacmaz K., Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 31.Schultz N., Lopez E., Saleh-Gohari N., Helleday T. Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Res. 2003;31:4959–4964. doi: 10.1093/nar/gkg703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saleh-Gohari N., Bryant H.E., Schultz N., Parker K.M., Cassel T.N., Helleday T. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol. Cell Biol. 2005;25:7158–7169. doi: 10.1128/MCB.25.16.7158-7169.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arnaudeau C., Lundin C., Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J. Mol. Biol. 2001;307:1235–1245. doi: 10.1006/jmbi.2001.4564. [DOI] [PubMed] [Google Scholar]
- 34.Helleday T., Arnaudeau C., Jenssen D. A partial hprt gene duplication generated by non-homologous recombination in V79 Chinese hamster cells is eliminated by homologous recombination. J. Mol. Biol. 1998;279:687–694. doi: 10.1006/jmbi.1998.1809. [DOI] [PubMed] [Google Scholar]
- 35.Bartek J., Lukas C., Lukas J. Checking on DNA damage in S phase. Nature Rev. Mol. Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- 36.Abraham R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 37.Bolderson E., Scorah J., Helleday T., Smythe C., Meuth M. ATM is required for the cellular response to thymidine induced replication fork stress. Hum. Mol. Genet. 2004;31:2937–2945. doi: 10.1093/hmg/ddh316. [DOI] [PubMed] [Google Scholar]
- 38.Khanna K.K., Jackson S.P. DNA double-strand breaks: signaling, repair and the cancer connection. Nature Genet. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 39.Kurose A., Tanaka T., Huang X., Halicka H.D., Traganos F., Dai W., Darzynkiewicz Z. Assessment of ATM phosphorylation on Ser-1981 induced by DNA topoisomerase I and II inhibitors in relation to Ser-139-histone H2AX phosphorylation, cell cycle phase, and apoptosis. Cytometry A. 2005;68:1–9. doi: 10.1002/cyto.a.20186. [DOI] [PubMed] [Google Scholar]
- 40.Strumberg D., Pilon A.A., Smith M., Hickey R., Malkas L., Pommier Y. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol. Cell Biol. 2000;20:3977–3987. doi: 10.1128/mcb.20.11.3977-3987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morrison C., Sonoda E., Takao N., Shinohara A., Yamamoto K., Takeda S. The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J. 2000;19:463–471. doi: 10.1093/emboj/19.3.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Veuger S.J., Curtin N.J., Smith G.C., Durkacz B.W. Effects of novel inhibitors of poly(ADP-ribose) polymerase-1 and the DNA-dependent protein kinase on enzyme activities and DNA repair. Oncogene. 2004;23:7322–7329. doi: 10.1038/sj.onc.1207984. [DOI] [PubMed] [Google Scholar]
- 43.Riballo E., Kuhne M., Rief N., Doherty A., Smith G.C., Recio M.J., Reis C., Dahm K., Fricke A., Krempler A., Parker A.R., Jackson S.P., Gennery A., Jeggo P.A., Lobrich M. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol. Cell. 2004;16:715–724. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 44.Ma Y., Schwarz K., Lieber M.R. The Artemis:DNA-PKcs endonuclease cleaves DNA loops, flaps, and gaps. DNA Repair (Amst) 2005;4:845–851. doi: 10.1016/j.dnarep.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 45.Zou L., Elledge S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 46.Siu W.Y., Lau A., Arooz T., Chow J.P., Ho H.T., Poon R.Y. Topoisomerase poisons differentially activate DNA damage checkpoints through ataxia-telangiectasia mutated-dependent and -independent mechanisms. Mol. Cancer Ther. 2004;3:621–632. [PubMed] [Google Scholar]
- 47.Cliby W.A., Lewis K.A., Lilly K.K., Kaufmann S.H. S phase and G2 arrests induced by topoisomerase I poisons are dependent on ATR kinase function. J. Biol. Chem. 2002;277:1599–1606. doi: 10.1074/jbc.M106287200. [DOI] [PubMed] [Google Scholar]
- 48.Rosenwald A., Chuang E.Y., Davis R.E., Wiestner A., Alizadeh A.A., Arthur D.C., Mitchell J.B., Marti G.E., Fowler D.H., Wilson W.H., Staudt L.M. Fludarabine treatment of patients with chronic lymphocytic leukemia induces a p53-dependent gene expression response. Blood. 2004;104:1428–1434. doi: 10.1182/blood-2003-09-3236. [DOI] [PubMed] [Google Scholar]
- 49.Stilgenbauer S., Schaffner C., Litterst A., Liebisch P., Gilad S., Bar-Shira A., James M.R., Lichter P., Dohner H. Biallelic mutations in the ATM gene in T-prolymphocytic leukemia. Nature Med. 1997;3:1155–1159. doi: 10.1038/nm1097-1155. [DOI] [PubMed] [Google Scholar]
- 50.Stankovic T., Hubank M., Cronin D., Stewart G.S., Fletcher D., Bignell C.R., Alvi A.J., Austen B., Weston V.J., Fegan C., Byrd P.J., Moss P.A.H., Taylor A.M.R. Microarray analysis reveals that TP53- and ATM-mutant B-CLLs share a defect in activating proapoptotic responses after DNA damage but are distinguished by major differences in activating prosurvival responses. Blood. 2004;103:291–300. doi: 10.1182/blood-2003-04-1161. [DOI] [PubMed] [Google Scholar]
- 51.Stankovic T., Weber P., Stewart G., Bedenham T., Murray J., Byrd P.J., Moss P.A., Taylor A.M.R. Inactivation of ataxia telangiectasia mutated gene in B-cell chronic lymphocytic leukaemia. Lancet. 1999;353:26–29. doi: 10.1016/S0140-6736(98)10117-4. [DOI] [PubMed] [Google Scholar]
- 52.Bullrich F., Rasio D., Kitada S., Starostik P., Kipps T., Keating M., Albitar M., Reed J.C., Croce C.M. ATM mutations in B-cell chronic lymphocytic leukemia. Cancer Res. 1999;59:24–27. [PubMed] [Google Scholar]
- 53.Austen B., Powell J.E., Alvi A., Edwards I., Hooper L., Starczynski J., Taylor A.M.R., Fegan C., Moss P., Stankovic T. Mutations in the ATM gene lead to impaired overall and treatment-free survival that is independent of IGVH mutation status in patients with B-CLL. Blood. 2005;106:3175–3182. doi: 10.1182/blood-2004-11-4516. [DOI] [PubMed] [Google Scholar]
- 54.Stankovic T., Stewart G.S., Fegan C., Biggs P., Last J., Byrd P.J., Keenan R.D., Moss P.A.H., Taylor A.M.R. Ataxia telangiectasia mutated-deficient B-cell chronic lymphocytic leukemia occurs in pregerminal center cells and results in defective damage response and unrepaired chromosome damage. Blood. 2002;99:300–309. doi: 10.1182/blood.v99.1.300. [DOI] [PubMed] [Google Scholar]
- 55.Schaffner C., Stilgenbauer S., Rappold G.A., Dohner H., Lichter P. Somatic ATM mutations indicate a pathogenic role of ATM in B-cell chronic lymphocytic leukemia. Blood. 1999;94:748–753. [PubMed] [Google Scholar]