

Abstract
Some poxviruses and their mammalian hosts encode homologous proteins that bind interleukin-18 (IL-18) with high affinity and inhibit IL-18-mediated immune responses. MC54L, the IL-18 binding protein of the human poxvirus that causes molluscum contagiosum, is unique in having a C-terminal tail of nearly 100 amino acids that is dispensable for IL-18 binding. When recombinant MC54L was expressed and purified via a C-terminal six-histidine tag, a shorter fragment was detected in addition to the full-length protein. This C-terminal fragment resulted from the cleavage of MC54L by cellular furin, as it was greatly diminished when furin was specifically inhibited or when a furin-deficient cell line was used for expression. Furthermore, the N- and C-terminal fragments of MC54L were generated by cleavage of the recombinant protein with furin in vitro. The furin cleavage site was mapped within a 32-amino-acid segment that is C terminal to the IL-18 binding domain. Full-length MC54L, but not the N-terminal IL-18 binding fragment, bound to cells and to purified heparin and other glycosaminoglycans that are commonly found on the cell surface and in the extracellular matrix. MC54L bound to heparin with a nanomolar Kd and could simultaneously bind to IL-18. Their different glycosaminoglycan and cell binding properties may allow the long and short forms of MC54L to inactivate IL-18 near the site of infection and at more distal locations, respectively.
Molluscum contagiosum virus (MCV) and variola virus are the sole members of the poxvirus family that use humans as exclusive natural hosts (8). Variola virus belongs to the Orthopoxvirus genus and until recently caused smallpox, an acute infection with a high mortality rate (9). MCV, the only member of the Molluscipoxvirus genus, causes small, benign skin lesions in children and young adults and more extensive disease only when there is a concurrent immunodeficiency such as AIDS (10). Even in immunocompetent individuals, however, the virus-filled skin lesions frequently persist for many months with few signs of inflammation, suggesting local immune suppression. Several potential immune evasion proteins were discovered after the complete MCV genome sequence was determined (17). One of these is an interleukin-18 (IL-18) binding protein (IL-18BP) that inhibits the gamma interferon-inducing activity of IL-18 (25).
IL-18 is a proinflammatory cytokine that enhances innate and acquired immunity and protects against microbial infections and tumors in murine models (6). Excessive IL-18 activity, however, is associated with some autoimmune and inflammatory diseases (13). Regulation of IL-18 activity is mediated by soluble IL-18BPs (14, 24). MC54L, an MCV homolog of mammalian IL-18BP, binds IL-18 with a nanomolar Kd and inhibits the gamma interferon-inducing activity of IL-18 in a dose-dependent manner (22, 25). Functional homologs of IL-18BP are also present in orthopoxviruses, including vaccinia virus, cowpox virus, and ectromelia virus (1, 3, 19), and as yet uncharacterized homologs are encoded by variola virus and members of other poxvirus genera. Deletion of the IL-18BP gene from ectromelia virus decreased virus replication and elevated natural killer cell activity in infected mice, indicating that these proteins contribute to defense against the host immune system (1).
The IL-18 binding sites of human IL-18BP and MC54L were determined through site-directed mutagenesis and quantitative binding studies (22, 23). Despite the relatively low overall sequence identity between MC54L and human IL-18BP, their IL-18 binding sites are almost identical. MC54L, however, is unique among poxvirus and mammalian IL-18BPs in having a C-terminal tail that is almost 100 amino acids long and is entirely dispensable for IL-18 binding (22). Our present studies show that the protein is secreted in two forms: a full-length form that binds cell surface glycosaminoglycans with high affinity through the C-terminal tail and a furin-cleaved form consisting solely of the IL-18 binding domain (IL-18BD). The long and short forms of MC54L may inhibit IL-18 near the site of infection and at more distal locations, respectively.
MATERIALS AND METHODS
Viruses and cells.
An attenuated vaccinia virus encoding full-length MC54L was described previously (22, 25). Recombinant vaccinia viruses encoding MC54L with deletions of residues 142 to 173 or 140 to 235 were constructed for this project. Coding sequences for MC54LΔ(142-173) and MC54LΔ(140-235) were subcloned from previously described expression constructs into the pMC02 transfer vector (4), and the resulting plasmids were transfected into BS-C-1 cells that were infected with vSC20, a highly attenuated growth factor-deficient vaccinia virus mutant (2). The recombinant viruses were selected for loss of thymidine kinase (11) and screened for the synthesis of β-glucuronidase (4). PgsA-745 (7) and LoVo (20) cells were purchased from the American Type Culture Collection (Manassas, Va.). Human foreskin fibroblasts, derived from a healthy donor, were a gift of Alison McBride.
Nonviral expression plasmids.
Full-length MC54L was amplified by PCR with a Clontech Advantage-GC cDNA PCR kit, which allowed accurate amplification of GC-rich templates (Clontech, Palo Alto, Calif.). The PCR product was then fused in frame with DNA encoding a flexible linker, a biotinylation site, and a six-histidine tag in pYX45 by using the NheI and BamHI sites as previously described (23).
Protein expression and purification.
Ten roller bottles containing monolayers of BS-C-1 cells were infected with recombinant vaccinia virus encoding MC54L at approximately 10 infectious units per cell. Three hours after infection, the medium in each roller bottle was replaced with 30 ml of serum-free Opti-mem (Invitrogen, Carlsbad, Calif.). The furin inhibitor dec-RVKR-cmk (Bachem, King of Prussia, Pa.) was added to the medium in half of the bottles to a final concentration of 50 μM. After approximately 30 h, the medium with or without dec-RVKR-cmk was harvested separately and incubated overnight at 4°C with 3 ml of Ni-nitrilotriacetic acid agarose (Qiagen, Valencia, Calif.). The beads were then packed into a column and washed with 15 mM imidazole in phosphate-buffered saline (PBS) containing 150 mM NaCl. The recombinant protein was eluted with 250 mM imidazole in PBS. Protein concentrations were determined by the Bradford assay with bovine serum albumin (BSA) as the standard. The purity and mass of the full-length MC54L protein were estimated with the Kodak 1D Image Analysis Software (Eastman Kodak, Rochester, N.Y.) after sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and Coomassie blue staining. MC54L proteins with the deletions Δ(142-173) and Δ(140-235) were expressed and purified similarly. For protein expression in 293T cells, 10 six-well plates were transfected with 2 μg of plasmid per well by using Lipofectamine (Invitrogen, Carlsbad, Calif.) and following the manufacturer's protocol. After overnight incubation, the medium in each well was replaced with 1.2 ml of serum free Opti-mem. The furin inhibitor dec-RVKR-cmk, at a 50 μM final concentration, was added to the medium in 5 of the 10 plates, and the same amount of fresh dec-RVKR-cmk was added to the medium after another day of incubation. For all transfected cells, the medium was harvested approximately 3 days after the start of transfection and incubated for 5 h at 4°C with 0.5 ml of Ni-nitrilotriacetic acid beads. The beads was packed into a column and washed with 15 mM imidazole in PBS containing 150 mM NaCl. The recombinant protein was eluted with 250 mM imidazole in PBS.
For detection of recombinant MC54L proteins in Western blots, a monoclonal antibody (MAb) against four consecutive histidines (Qiagen) was used as the primary antibody.
Furin digestion.
Recombinant MC54L protein was dialyzed overnight against a buffer containing 50 mM Tris-HCl (pH 7.5), 10 mM CaCl2, and 100 mM NaCl and incubated with approximately 0.1 U of recombinant furin per μl (New England Biolabs, Beverly, Mass.) at 30°C for 3 h.
Heparin-agarose binding.
Recombinant MC54L proteins were incubated with 30 μl of heparin-agarose (Gibco-BRL, Gaithersburg, Md.) in the presence of 0.2% BSA, various concentrations of NaCl, and heparin (Fisher Scientific, Fair Lawn, N.J.) at room temperature for 2 h. The heparin-agarose was then washed three times with 0.2% BSA in PBS and one time with PBS. The proteins that bound to heparin-agarose were eluted with 30 μl of SDS-PAGE loading buffer.
Surface plasmon resonance assay.
Biotinylated albumin and albumin-heparin (Sigma, St. Louis, Mo.) were captured on a streptavidin-coated BIAcore SA chip (BIAcore, Piscataway, N.J.). The chip was then washed several times with injections of 10 mM glycine (pH 1.5) to remove any loosely bound materials.
For kinetic experiments, various concentrations of partially purified full-length MC54L or HB-EGF (Sigma) were injected in a running buffer containing 0.01 M HEPES (pH 7.4), 0.15 M NaCl, 3 mM EDTA, and 0.1% Tween 20. A total of 250 μl of the proteins was injected at a flow rate of 50 μl/min. Dissociation was monitored for 5 min, followed by two injections of 5 M NaCl and one injection of 10 mM glycine (pH 1.5) to regenerate the surface.
Sensorgrams were analyzed with BIAevaluation software (BIAcore). To correct for refractive index changes, the binding responses generated in the control surface (biotin-albumin) were subtracted from the responses generated in the surface with immobilized biotin-albumin-heparin. The binding data from the injection of five different concentrations of the proteins were globally fitted to a one-to-one binding model. Analyses with the same concentration series were repeated four times.
For competition binding studies, 2 nM MC54L with or without competing glycosaminoglycans was injected over an albumin-heparin-coated SA chip for 10 min at 20 μl/min. After 10 min of injection, the responses approached equilibrium. Heparan sulfate from bovine kidney, chondroitin sulfate A from bovine trachea, chondroitin sulfate B from porcine intestinal mucosa, chondroitin sulfate C from shark cartilage (Sigma), and heparin (Fisher Scientific) were each reconstituted with BIAcore running buffer. Recombinant murine IL-18 was purchased from R&D Systems (Minneapolis, Minn.).
Cell binding.
A 48-well plate with or without confluent cells was blocked at 4°C for 1 h with medium containing 10% fetal calf serum and for 1 h with medium containing 1% BSA and then incubated for an additional 2 h with recombinant MC54L protein in medium containing 10% fetal calf serum. After removal of the medium, the wells were washed three times with 1% BSA in PBS and six times with PBS at 4°C. SDS-PAGE loading buffer (60 μl) was added to the cells or empty wells. Half of the lysed cells and 1/10 of the medium were analyzed separately by SDS-PAGE and Western blotting.
RESULTS
Processing of MCV IL-18BP MC54L by cellular furin.
Because MCV cannot replicate in cultured cells, recombinant vaccinia virus was used as a surrogate poxvirus for cytoplasmic expression of MC54L (24). The recombinant protein with a C-terminal six-histidine tag was secreted from BS-C-1 cells into the medium and purified by metal affinity chromatography. SDS-PAGE revealed full-length MC54L protein, as well as a shorter product, which we initially attributed to nonspecific degradation (24). In subsequent studies with a nonviral expression vector and 293T cells, only short products that failed to bind IL-18 were purified by metal affinity chromatography (shown later). At first, we thought that these small proteins might have been translated from spliced RNAs, which could not have formed with the vaccinia virus cytoplasmic expression system. However, only full-length RNAs were detected in transfected 293T cells by reverse transcription-PCR (data not shown). Moreover, when 293T cells were infected with the recombinant vaccinia virus expressing MC54L, most of the product was also shorter than the full-length protein (data not shown). An alternative explanation was that the full-length MC54L protein was cleaved during passage through the secretory pathway and that the amount of cleavage varied with different cell types and levels of MC54L expression. Inspection of the MC54L sequence supported this idea, as potential cleavage sites for furin, a proprotein convertase that resides in the secretory pathway and on the cell surface, were found (Fig. 1). Residues 158 to 162 of MC54L (Arg-Arg-Arg-Arg-Arg) could comprise two overlapping optimal furin cleavage sites, Arg-Xaa-(Lys/Arg)-Arg, while residues 232 to 235 conform to the minimal furin cleavage site, Arg-Xaa-Xaa-Arg (12).
FIG. 1.

Amino acid sequence of the C-terminal half of MC54L. A diagram of the full-length MC54L open reading frame is shown indicating the signal peptide (SP), the IL-18BD (IL-18 Binding), and the C-terminal tail (Tail) of MC54L. The amino acid sequence of the C-terminal tail is expanded below the diagram. Basic amino acids are in bold, and sequences that conform to the consensus furin cleavage site are underlined.
To gather evidence for furin cleavage of MC54L, we analyzed metal affinity-purified recombinant MC54L protein synthesized in monkey kidney BS-C-1 cells, primary human fibroblasts, and LoVo cells, a human colon carcinoma cell line that is deficient in furin (20). Both full-length MC54L and a smaller fragment were secreted by BS-C-1 cells and human fibroblasts, but only the full-length protein was secreted by furin-deficient LoVo cells (Fig. 2).
FIG. 2.
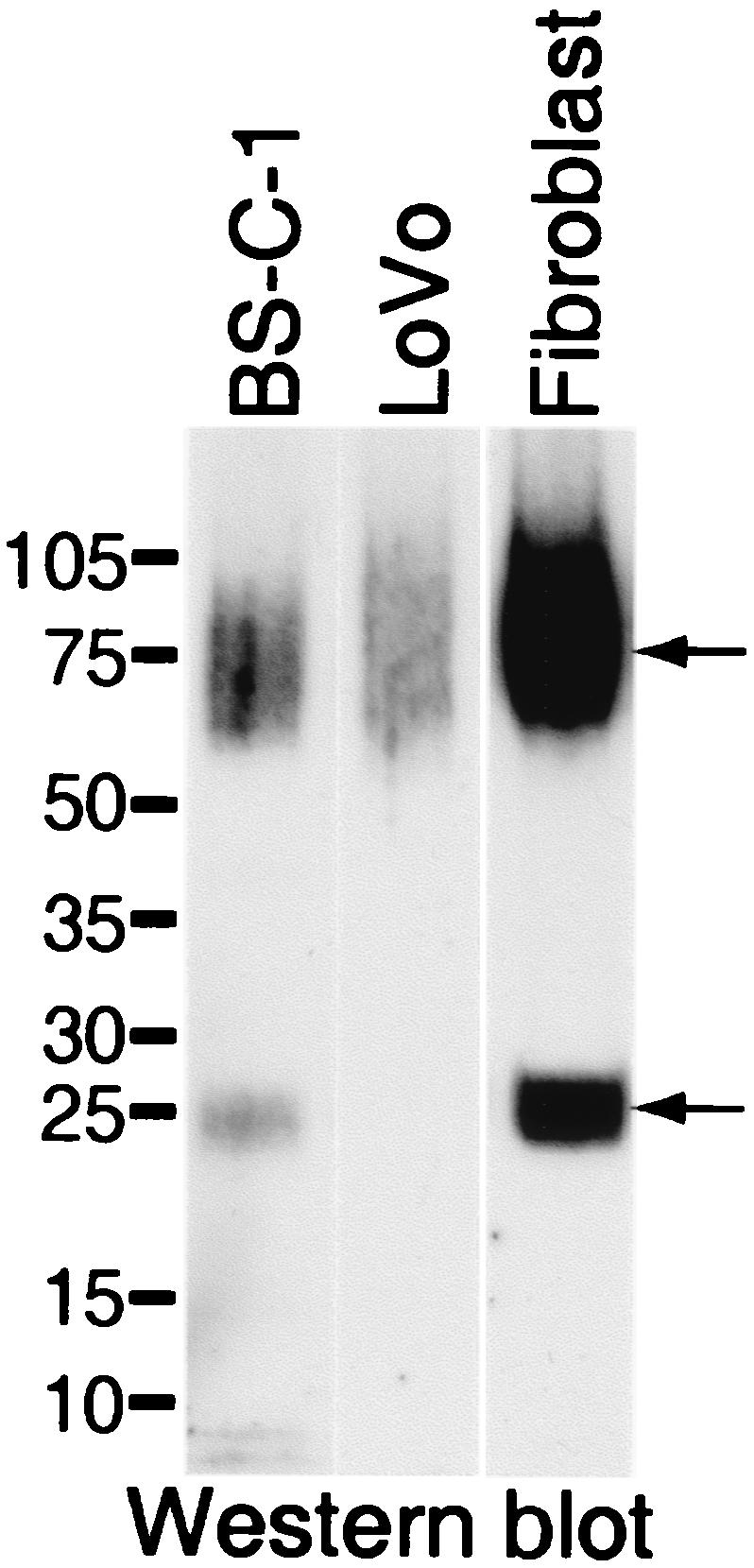
Expression of recombinant MC54L proteins in furin-competent and -deficient cells. BS-C-1 cells, furin-deficient LoVo cells, or human primary foreskin fibroblasts were infected with a recombinant vaccinia virus encoding MC54L protein with a C-terminal six-histidine tag. Secreted recombinant MC54L proteins were purified by metal affinity chromatography, subjected to SDS-PAGE, and detected by chemiluminescence after Western blotting with a MAb to the polyhistidine tag. The positions of molecular size markers (sizes are in kilodaltons) are shown on the left. The arrows point to the full-length (top) and C-terminal fragment (bottom) forms of the MC54L protein.
Additional experiments were carried out with the specific furin inhibitor dec-RVKR-cmk (21). Predominantly full-length MC54L protein was purified by metal affinity chromatography from transfected 293T cells when the furin inhibitor was present, whereas only smaller fragments were detected in the absence of the inhibitor, as found by Coomassie blue staining or Western blotting with a MAb to the polyhistidine tag (Fig. 3A). The furin inhibitor also increased the amount of full-length MC54L made in BS-C-1 cells by the recombinant vaccinia virus expression vector (Fig. 3B). There was also a concomitant decrease in the amount of smaller protein that was purified along with full-length MC54L (Fig. 3B). In some gels, the smaller protein, which stained with the polyhistidine MAb, appeared as a doublet (Fig. 3B), probably because of cleavage at one or the other of the overlapping furin cleavage sites between residues 158 and 162 of MC54L (Fig. 1).
FIG. 3.

Expression of recombinant MC54L protein in the presence (+) or absence (−) of a furin inhibitor. (A) 293T cells were transiently transfected with a plasmid encoding MC54L protein with a C-terminal six-histidine tag. (B) BS-C-1 cells were infected with a recombinant vaccinia virus encoding MC54L protein with a C-terminal six-histidine tag. Transfected or infected cells were incubated in medium with (+) or without (−) 50 μM dec-RVKR-cmk. Secreted recombinant MC54L proteins were purified by metal affinity chromatography, and the eluted fractions indicated by number were subjected to SDS-PAGE. The proteins were detected by Coomassie blue staining (left) or by chemiluminescence after Western blotting with a MAb to the polyhistidine tag (right). The arrows point to the full-length (top) and C-terminal fragment (bottom) forms of the MC54L protein. The values on the left indicate the mobilities and masses in kilodaltons of marker proteins.
In vitro cleavage of MC54L and localization of the furin cleavage site.
In the above-described experiments, the putative N-terminal cleavage product containing the IL-18BD was lost during the metal affinity purification step because it lacked the six-histidine tag. To directly demonstrate both products of furin cleavage, partially purified full-length MC54L was subjected to in vitro digestion with recombinant furin. The digestion resulted in major products of approximately 46 and 25 kDa (Fig. 4) but only the full-length uncleaved protein and the 25-kDa product reacted with the polyhistidine MAb (data not shown), indicating that the 46-kDa band represented the N-terminal fragment. These apparent masses are higher than those predicted on the basis of the amino acid sequence because of N-glycosylation (24). The specificity of furin cleavage was demonstrated by the complete inhibition produced by the furin inhibitor dec-RVKR-cmk (Fig. 4).
FIG. 4.

In vitro cleavage of MC54L with recombinant furin. MC54L proteins that were full length or had an internal deletion of Δ(142-173) or Δ(140-235) were expressed individually in BS-C-1 cells by recombinant vaccinia viruses and purified by metal affinity chromatography. Recombinant MC54L proteins were incubated with (+) or without (−) recombinant furin and with (+) or without (−) dec-RVKR-cmk and then resolved by SDS-PAGE and detected by Coomassie staining. The values on the left indicate the mobilities and masses in kilodaltons of marker proteins.
The MC54L proteins with deletions Δ(140-235) and Δ(142-173) lack the five arginines comprising the predicted cleavage site (Fig. 1). As shown in Fig. 4, these proteins were completely resistant to furin digestion. In addition, when the latter proteins were expressed in 293T cells by a nonviral expression vector, only the uncleaved forms, which bound IL-18 with high affinity, were detected (22).
The full-length MC54L protein binds to glycosaminoglycans with high affinity via the C-terminal tail.
Approximately half of the amino acids from residue 190 to the C terminus of MC54L are basic (Fig. 1), suggesting that this region might bind negatively charged biomolecules such as glycosaminoglycans. Full-length MC54L bound to heparin-agarose very tightly, as the binding was prevented only by salt concentrations of >0.55 M (Fig. 5A). The binding was specific, as it was inhibited by excess free heparin (Fig. 5A) and no binding between MC54L and control protein A-agarose was observed (data not shown). The heparin binding site was localized to the C terminus of MC54L, as the MC54LΔ(140-235) protein failed to bind to heparin-agarose whereas the MC54LΔ(142-173) protein bound to heparin-agarose like full-length MC54L (Fig. 5A). As furin cleavage products of MC54L, in addition to full-length MC54L, are released from infected cells, their abilities to bind to heparin were also tested. The furin digestion products were produced by in vitro cleavage of purified full-length MC54L and incubated with heparin-agarose. As predicted, the C-terminal furin cleavage products of MC54L were able to bind to heparin-agarose while the N-terminal furin cleavage product failed to bind to heparin (Fig. 5B).
FIG. 5.

Heparin binding properties of full-length and mutated forms of MC54L. MC54L proteins that were full length or lacked amino acids 142 to 173 or 140 to 235 were expressed individually in BS-C-1 cells by recombinant vaccinia viruses and purified by metal affinity chromatography. (A) Except for the control lanes, recombinant MC54L proteins were incubated with 30 μl of heparin-agarose in the presence of various concentrations of NaCl or heparin (indicated below the lanes). Proteins that bound to heparin-agarose were eluted after extensive washing and detected by Western blotting with a MAb against the polyhistidine tag. In the control lanes, approximately half of the material that was applied to the heparin-agarose was directly run on the gel and analyzed by Western blotting. (B) Full-length MC54L protein was incubated with (+) or without (−) recombinant furin, incubated with 30 μl of heparin-agarose in the presence of 0.35 M NaCl, and analyzed by SDS-PAGE and Coomassie blue staining. Control lanes have the same meaning as in panel A. The arrows point to the full-length (top) and C-terminal fragment (bottom) forms of the MC54L protein. The values on the left indicate the mobilities and masses in kilodaltons of marker proteins.
The binding affinity of MC54L for heparin was measured by surface plasmon resonance assay with a BIAcore apparatus. The artificial proteoglycan albumin-heparin and control albumin were immobilized on two different flow cells of a BIAcore sensor chip. Various concentrations of full-length MC54L were then injected over the chip, and the sensorgrams were globally fitted to a one-to-one binding model (Fig. 6). As a positive control, a known heparin binding protein, the heparin binding epidermal growth factor-like growth factor (HB-EGF), was also analyzed. The kinetic and affinity constants that were obtained from four repeat experiments are summarized in Table 1. Full-length MC54L and HB-EGF bound to heparin-albumin with Kds of 0.52 and 12 nM, respectively, indicating that the viral protein had the greater affinity. Deletion of residues 142 to 173 of MC54L did not significantly affect heparin binding (Table 1).
FIG. 6.

Kinetic analyses of the binding of MC54L proteins to immobilized heparin. Heparin-albumin-biotin and the control albumin-biotin were immobilized in two different flow cells of a BIAcore streptavidin sensor chip (SA chip). MC54L proteins that were expressed in the presence of the furin inhibitor were purified by metal affinity chromatography. MC54L proteins at concentrations of 0.02, 0.05, 0.09, 0.19, and 0.38 nM were injected at a flow rate of 50 μl/min. The colored and black lines are the actual responses in resonance units (RU) and globally fitted curves, respectively. The residual responses represent deviations of the actual responses from the fitted curves. The root mean square deviation was 2.41.
TABLE 1.
Kinetic and affinity constantsa
| Protein | Kon, 106/ms | Koff, 10−3/s | Kd, nM |
|---|---|---|---|
| Full-length MC54L | 8.8 ± 0.1 | 4.6 ± 0.3 | 0.52 ± 0.03 |
| MC54LΔ (142-173) | 15 ± 5 | 5.4 ± 0.1 | 0.4 ± 0.2 |
| HB-EGF | 0.14 ± 0.02 | 1.66 ± 0.09 | 12 ± 2 |
The kinetic and affinity constants shown are from four independent experiments similar to that shown in Fig. 6.
The ability of MC54L to bind other glycosaminoglycans was measured in competition experiments. In the experiment depicted in Fig. 7, about 500 resonance units of MC54L bound to immobilized albumin-heparin in the absence of a competing glycosaminoglycan. In the presence of increasing concentrations of heparin, heparan sulfate, or chondroitin sulfate A, B, or C, a decreasing amount of MC54L was bound to the immobilized heparin-albumin (Fig. 7). The concentrations of heparan and chondroitin sulfates needed to reduce binding were higher than those of heparin, indicating weaker affinities for MC54L (Fig. 7). The relative affinities of MC54L for glycosaminoglycans correlated with the densities of their negative charges.
FIG. 7.

Competitive inhibition of MC54L protein to heparin by glycosaminoglycans. Heparin-albumin-biotin was immobilized on a BIAcore SA sensor chip as described in the legend to Fig. 6. MC54L protein (2 nM) was injected alone or with increasing concentrations of the indicated competing glycosaminoglycans. The responses, in resonance units (RU), after 10 min of injection were plotted as a function of the concentration of the competing glycosaminoglycan.
Full-length MC54L was also shown to bind to IL-18 and heparin simultaneously. Full-length MC54L was first injected over a BIAcore sensor chip coated with albumin-heparin. After the injection but before MC54L completely dissociated from the immobilized heparin, recombinant murine IL-18 was injected over the sensor chip. The binding of IL-18 to those MC54L proteins that were still attached to the immobilized heparin resulted in a significant increase in the signal (Fig. 8). The signal then decreased as the IL-18-MC54L complex and MC54L dissociated from the heparin. IL-18 did not associate with the surface of a control albumin-heparin chip or the surface of a control albumin-heparin chip containing HB-EGF bound to albumin-heparin (data not shown).
FIG. 8.

Simultaneous binding of MC54L to heparin and IL-18. A BIAcore sensor chip that was coated with heparin-albumin-biotin was injected first with MC54L proteins and then with murine IL-18. The response, in resonance units (RU), is shown.
Full-length MC54L binds to cells via the C-terminal tail.
As glycosaminoglycans are components of proteoglycans on the cell surface and in the extracellular matrix, MC54L was also tested for the ability to bind to cells. To minimize sticking to the culture plates, the wells were blocked successively with 10% serum and 1% BSA or with 1% BSA alone (data not shown). The cells were incubated with recombinant MC54L proteins in medium containing 10% fetal calf serum at 4°C for 2 h. Recombinant MC54L proteins that were attached to the cells were detected by Western blotting with an antibody against the six-histidine tag. A sharp background band that migrates similarly to that of full-length MC54L was observed in the wild-type and mutant CHO cell lines, but it was not observed in BS-C-1 cells, suggesting that it represents a cellular protein in hamster cells that cross-reacted with antibodies in the Western blot assay. While MC54LΔ(140-235) failed to bind to cells, full-length MC54L or MC54LΔ(142-173) did bind to BS-C-1 cells, CHO cells (Fig. 9), and primary human fibroblasts (data not shown). Full-length MC54L and MC54LΔ(142-173) also bound to PgsA-745 (Fig. 9), a CHO cell line deficient in glycosaminoglycans because of a genetic defect in their biosynthetic pathway (7), suggesting an interaction of the C-terminal tail of MC54L with additional negatively charged surface molecules such as polysaccharides.
FIG. 9.

Binding of MC54L to cells. Empty wells or wells containing confluent CHO, PgsA-745, or BS-C-1 cells in a 48-well plate were preincubated at 4°C with medium containing 10% fetal calf serum for 1 h and containing 1% BSA for another hour and then incubated with recombinant MC54L proteins in medium containing 10% fetal calf serum for 2 h at 4°C. After removal of the medium, the cell monolayers or empty wells were washed extensively and resuspended in SDS-gel loading buffer. The proteins that associated with cells were detected in a Western blot with a MAb against the polyhistidine tag.
DISCUSSION
The IL-18BPs of poxviruses share 20 to 40% amino acid sequence identity with the IL-18BPs of mammals, suggesting that the viruses may have acquired these genes from their hosts and then retained and modified them by natural selection. The pirating of IL-18BPs by poxviruses and their use as decoy receptors is consistent with the critical role of IL-18 in defense against virus infections and provides a mechanism for evasion of the immune system. The MCV IL-18BP MC54L is unique in that it contains a long C-terminal tail. We found that the tail confers high-affinity glycosaminoglycan and cell binding properties on the MC54L protein. Some insight regarding potential advantages of this appendage can be gained by examining other biological processes in which glycosaminoglycan interactions play a role, such as the sequestration and concentration of cell signaling factors, virus attachment, and cell adhesion. Basic fibroblast growth factor (bFGF) is one of the best-studied glycosaminoglycan binding proteins (reviewed in reference 15). The binding of bFGF with glycosaminoglyans increases the affinity of bFGF for the FGF receptor and protects bFGF from circulating proteases. The interaction with glycosaminoglycans on the cell surface and in the extracellular matrix also creates a local reservoir of bFGF, contributing to the strict spatial regulation of FGF signaling that was shown to be important in limb development (5). Similarly, the binding of MC54L with glycosaminoglycans may increase its IL-18 binding ability, prolong the half-life of MC54L, and concentrate MC54L in the immediate vicinity of MCV-infected cells, where protection against IL-18 activity may be most needed. Since MC54L can bind to IL-18 and glycosaminoglycans simultaneously, multiple MC54L proteins could cluster by binding to the same glycosaminoglycan, providing increased avidity for IL-18. The same region of MC54L that facilitates its glycosaminoglycan binding was also important for binding to cells, presumably via the same mechanism. However, MC54L bound to cells that were deficient in glycosaminoglycan synthesis, suggesting additional interactions with other polyanions on the cell surface, such as polysaccharides.
The apparent Kd for the binding between MC54L and heparin-albumin is remarkably low. Although this apparent Kd indicated that MC54L binds heparin with very high affinity and with fast on and fast off kinetics, its absolute value needs to be compared to the affinity constants of other heparin binding proteins with caution for the following reasons. First, the reliability of the affinity constant obtained with BIAcore is affected by the purity of the analyte and the correct measurement of the concentration of the analyte (in this case, MC54L). We estimated that full-length MC54L was 80% of the total protein by comparing the intensities of the Coomassie blue-stained bands (see Fig. 3B, fraction 3). If some of the contaminating proteins bind to heparin, then the affinity constant for MC54L may be exaggerated. Furthermore, because MC54L is heavily glycosylated and migrates as a diffused band on SDS-PAGE, the mass or concentration of MC54L that was measured with molecular weight standards by SDS-PAGE or with BSA as the standard in the Bradford assay may not be accurate and may result in an underestimate of the molar concentration of MC54L. For instance, if the MC54L molar concentration were underestimated 10-fold, then the affinity constant would decrease almost 10-fold. However, even under these circumstances, the Kd of MC54L would be low compared to those of other heparin binding proteins. In addition, the affinity constants were obtained after fitting the BIAcore data to a one-to-one binding model that assumes that one molecule of MC54L binds to a homogeneous binding site on heparin-albumin. It is possible that the binding between MC54L and heparin-albumin is more complex than the simple one-to-one binding model and may affect the affinity constant.
Several other poxvirus-encoded immune modulators bind to cell surface glycosaminoglycans. These include the vaccinia virus complement control protein (18) and the myxoma virus CC-chemokine inhibitor M-T1 (16). Such findings have changed the view that secreted poxvirus immune modulators necessarily function as soluble factors and suggest that the cell surface and extracellular matrix are important targeting sites. Our present studies with MC54L provide additional support for this view.
We found that full-length MC54L is partially processed by cellular furin into N- and C-terminal fragments. The N-terminal fragment of MC54L contained the IL-18BD but did not bind glycosaminoglycans or cells, making it similar to IL-18BPs encoded by other poxviruses and their mammalian hosts. Compared to full-length MC54L, the N-terminal fragment of MC54L and other IL-18BPs may diffuse to sites distant from infected cells and therefore extend the effective activity range. The cleavage site on MC54L was mapped to a 32-amino-acid segment that contains five consecutive arginines that form two overlapping furin consensus sites. Despite substantial sequence variation at the C-terminal tail of MC54L, the five consecutive arginines are conserved in all of the strains of MCV that have been analyzed (19, 22). The amount of MC54L that is cleaved during infection with MCV is unknown, as the virus does not productively infect either cultured cells or experimental animals. By using vaccinia virus as a surrogate poxvirus expression vector, we found some variation in different cell types. Less than 50% of MC54L was cleaved in monkey BS-C-1 and primary human fibroblast cells, while substantially more MC54L was cleaved in human 293T cells, probably reflecting different levels of furin.
It is not unusual for proteins that are produced in the secretory pathway of eukaryotic cells to undergo activation by endoproteolytic cleavage. Cellular proteins such as growth factors, receptors, and serum components are among the substrates for furin. The envelope proteins of viruses are also frequently processed by endoproteases. For example, the susceptibility of influenza virus envelope proteins to cleavage is an important factor in viral pathogenicity (reviewed in reference 26). However, it is uncommon for a viral protein other than a surface glycoprotein to be processed in this way.
MCV frequently persists in hosts for many months, far longer than other poxviruses. Undoubtedly, many factors contribute to persistence, including cell tropism and types of immune defense modulators, that vary greatly among poxviruses of different genera. The properties of individual immune modulators, such as the IL-18BPs, may also contribute to the varied virus-host interactions. The presence of a furin cleavage site separating the IL-18BD and cell binding domain appears to be an efficient way for MCV to produce two proteins with complementary properties from a single open reading frame: a full-length IL-18BP that acts locally and a truncated version that can diffuse to distant sites.
Acknowledgments
We thank Alison McBride of the National Institutes of Health for the gift of human foreskin fibroblasts.
REFERENCES
- 1.Born, T. L., L. A. Morrison, D. J. Esteban, T. VandenBos, L. G. Thebeau, N. H. Chen, M. K. Spriggs, J. E. Sims, and R. M. L. Buller. 2000. A poxvirus protein that binds to and inactivates IL-18, and inhibits NK cell response. J. Immunol. 164:3246-3254. [DOI] [PubMed] [Google Scholar]
- 2.Buller, R. M., S. Chakrabarti, J. A. Cooper, D. R. Twardzik, and B. Moss. 1988. Deletion of the vaccinia virus growth factor gene reduces virus virulence. J. Virol. 62:866-877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calderara, S., Y. Xiang, and B. Moss. 2001. Orthopoxvirus IL-18 binding proteins: affinities and antagonist activities. Virology 279:22-26. [DOI] [PubMed] [Google Scholar]
- 4.Carroll, M. W., and B. Moss. 1995. E. coli β-glucuronidase (GUS) as a marker for recombinant vaccinia viruses. BioTechniques 19:352-355. [PubMed] [Google Scholar]
- 5.Cohn, M. J., J. C. Izpisua-Belmonte, H. Abud, J. K. Heath, and C. Tickle. 1995. Fibroblast growth factors induce additional limb development from the flank of chick embryos. Cell 80:739-746. [DOI] [PubMed] [Google Scholar]
- 6.Dinarello, C. A. 1999. IL-18: A TH1-inducing, proinflammatory cytokine and new member of the IL-1 family. J. Allergy Clin. Immunol. 103:11-24. [DOI] [PubMed] [Google Scholar]
- 7.Esko, J. D., T. E. Stewart, and W. H. Taylor. 1985. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc. Natl. Acad. Sci. USA 82:3197-3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esposito, L. J., and F. Fenner. 2001. Poxviruses, p. 2885-2921. In D. M. Knipe and P. M. Howley (ed.), Fields virology, vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 9.Fenner, F., D. A. Henderson, I. Arita, Z. Jezek, and I. D. Ladnyi. 1988. Smallpox and its eradication, first ed. World Health Organization, Geneva, Switzerland.
- 10.Gottlieb, S. L., and P. L. Myskowski. 1994. Molluscum contagiosum. Int. J. Dermatol. 33:453-461. [DOI] [PubMed] [Google Scholar]
- 11.Mackett, M., G. L. Smith, and B. Moss. 1982. Vaccinia virus: a selectable eukaryotic cloning and expression vector. Proc. Natl. Acad. Sci. USA 79:7415-7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molloy, S. S., P. A. Bresnahan, S. H. Leppla, K. R. Klimpel, and G. Thomas. 1992. Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J. Biol. Chem. 267:16396-16402. [PubMed] [Google Scholar]
- 13.Nakanishi, K., T. Yoshimoto, H. Tsutsui, and H. Okamura. 2001. Interleukin-18 regulates both Th1 and Th2 responses. Annu. Rev. Immunol. 19:423-474. [DOI] [PubMed] [Google Scholar]
- 14.Novick, D., S.-H. Kim, G. Fantuzzi, L. L. Reznikov, C. A. Dinarello, and M. Rubinstein. 1999. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity 10:127-136. [DOI] [PubMed] [Google Scholar]
- 15.Powers, C. J., S. W. McLeskey, and A. Wellstein. 2000. Fibroblast growth factors, their receptors and signaling. Endocr. Relat. Cancer 7:165-197. [DOI] [PubMed] [Google Scholar]
- 16.Seet, B. T., J. Barrett, J. Robichaud, B. Shilton, R. Singh, and G. McFadden. 2001. Glycosaminoglycan binding properties of the myxoma virus CC-chemokine inhibitor, M-T1. J. Biol. Chem. 276:30504-30513. [DOI] [PubMed] [Google Scholar]
- 17.Senkevich, T. G., J. J. Bugert, J. R. Sisler, E. V. Koonin, G. Darai, and B. Moss. 1996. Genome sequence of a human tumorigenic poxvirus: prediction of specific host response-evasion genes. Science 273:813-816. [DOI] [PubMed] [Google Scholar]
- 18.Smith, S. A., N. P. Mullin, J. Parkinson, S. N. Shchelkunov, A. V. Totmenin, V. N. Loparev, R. Srisatjaluk, D. N. Reynolds, K. L. Keeling, D. E. Justus, P. N. Barlow, and G. J. Kotwal. 2000. Conserved surface-exposed K/R-X-K/R motifs and net positive charge on poxvirus complement control proteins serve as putative heparin binding sites and contribute to inhibition of molecular interactions with human endothelial cells: a novel mechanism for evasion of host defense. J. Virol. 74:5659-5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith, V. P., N. A. Bryant, and A. Alcami. 2000. Ectromelia, vaccinia and cowpox viruses encode secreted interleukin-18-binding proteins. J. Gen. Virol. 81:1223-1230. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi, S., K. Kasai, K. Hatsuzawa, N. Kitamura, Y. Misumi, Y. Ikehara, K. Murakami, and K. Nakayama. 1993. A mutation of furin causes the lack of precursor-processing activity in human colon carcinoma LoVo cells. Biochem. Biophys. Res. Commun. 195:1019-1026. [DOI] [PubMed] [Google Scholar]
- 21.Vey, M., W. Schafer, B. Reis, R. Ohuchi, W. Britt, W. Garten, H. D. Klenk, and K. Radsak. 1995. Proteolytic processing of human cytomegalovirus glycoprotein B (gpUL55) is mediated by the human endoprotease furin. Virology 206:746-749. [DOI] [PubMed] [Google Scholar]
- 22.Xiang, Y., and B. Moss. 2001. Correspondence of the functional epitopes of poxvirus and human interleukin-18-binding proteins. J. Virol. 75:9947-9954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiang, Y., and B. Moss. 2001. Determination of the functional epitopes of human interleukin-18-binding protein by site-directed mutagenesis. J. Biol. Chem. 276:17380-17386. [DOI] [PubMed] [Google Scholar]
- 24.Xiang, Y., and B. Moss. 1999. Identification of human and mouse homologs of the MC51L-53L-54L family of secreted glycoproteins encoded by the molluscum contagiosum poxvirus. Virology 257:297-302. [DOI] [PubMed] [Google Scholar]
- 25.Xiang, Y., and B. Moss. 1999. IL-18 binding and inhibition of interferon gamma induction by human poxvirus-encoded proteins. Proc. Natl. Acad. Sci. USA 96:11537-11542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zambon, M. C. 2001. The pathogenesis of influenza in humans. Rev. Med. Virol. 11:227-241. [DOI] [PubMed] [Google Scholar]