

Abstract
Transcription co-activators and histone acetyltransferases, p300 and cyclic AMP responsive element-binding protein-binding protein (CBP), participate in hypoxic activation of hypoxia-inducible genes. Here, we show that exposure of PC12 and cells to 1–10% oxygen results in hyperphosphorylation of p300/CBP. This response is fast, long lasting and specific for hypoxia, but not for hypoxia-mimicking agents such as desferioxamine or Co2+ ions. It is also cell-type specific and occurs in pheochromocytoma PC12 cells and the carotid body of rats but not in hepatoblastoma cells. The p300 hyperphosphorylation specifically depends on the release of intracellular calcium from inositol 1,4,5-triphosphate (IP3)-sensitive stores. However, it is not inhibited by pharmacological inhibitors of any of the kinases traditionally known to be directly or indirectly calcium regulated. On the other hand, p300 hyperphosphorylation is inhibited by several different inhibitors of the glucose metabolic pathway from generation of NADH by glyceraldehyde 3-phosphate dehydrogenase, through the transfer of NADH through the glycerol phosphate shuttle to ubiquinone and complex III of the mitochondrial respiratory chain. Inhibition of IP3-sensitive calcium stores decreases generation of ATP, and this inhibition is significantly stronger in hypoxia than in normoxia. We propose that the NADH glycerol phosphate shuttle participates in generating a pool of ATP that serves either as a co-factor or a modulator of the kinases involved in the phosphorylation of p300/CBP during hypoxia.
Keywords: calcium, carotid body respiratory chain, hypoxia-inducible factor, intermittent hypoxia, transcription co-activators
Abbreviations used: 2-APB, 2-aminoethoxydiphenylborate; BAPTAAM, bis-(o-aminiphenoxy)-ethane-N,N,N1,N1-tetraacetoxy-methylester; [Ca2+]i, cytosolic free Ca2+ concentration; CaM, calmodulin; CH, cysteine–histidine-rich zinc finger; CREB, cyclic AMP responsive element-binding protein; CBP, CREB-binding protein; DF, desferioxamine; DMEM, Dulbecco’s modified Eagle’s medium; DMSO, dimethylsulfoxide; DPI, diphenyliodonium; ERK, extracellular signal regulated kinase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; G3PDH, glycerol-3-phosphate dehydrogenase; HIF, hypoxia-inducible factor; IP3, inositol 1,4,5-triphosphate; mTOR, mammalian target of rapamycin; P13K, phosphatidylinositol 3 kinase; PAGE, polyacrylamide gel electrophoresis; PKA, protein kinase A; PKC, protein kinase C; RyR, ryanodine; SDS, sodium dodecyl sulfate
Cyclic AMP responsive element-binding protein (CREB)-binding protein (CBP) and the adenovirus E1A-associated 300-kDa protein (p300) are homologous transcriptional co-activators that create a physical bridge between various transcription factors and the basal transcriptional machinery and RNA polymerase holoenzyme (for review see Goodman and Smolik 2000; Vo and Goodman 2001). The simultaneous interaction with multiple transcription factors contributes to transcriptional synergy. They also have histone acetyltransferase activity, which links transcription to chromatin remodeling. Both proteins have three cysteine–histidine-rich zinc finger (CH) domains, each of which interacts with different transcription factors. They are present at limiting concentrations, and their interaction with one group of specific regulators inhibits their co-activation with another group of regulators. Thus, p300 and CBP serve as integrators of multiple signal transduction pathways within the nucleus.
Both co-activators are of crucial importance for regulating the hypoxia-inducible genes. The CH1 domain of p300/CBP binds hypoxia-inducible factor (HIF), and the co-activators are necessary for the hypoxic activation of transcription from the hypoxia-responsive elements (Kallio et al. 1998; Ema et al. 1999) in genes such as vascular endothelial growth factor (Arany et al. 1996) and the glycolytic enzyme, lactate dehydrogenase (Ebert and Bunn 1998). HIF–p300/CBP interactions require abrogation of hydroxylation of a conserved asparagine within the C-terminal transactivation domain of HIF-α (Lando et al. 2002a, 2002b). The HIF–p300/CBP interaction is blocked by a hypoxia-inducible p35srj protein, which also interacts with the CH1 domain of co-activators (Bhattacharya et al. 1999).
p300 and CBP are phosphoproteins (Yaciuk and Moran 1991), but relatively little is understood about the kinases and phosphatases regulating their phosphorylation or about the role of phosphorylation in their function. Phosphorylation of different sites appears to modulate interactions of p300/CBP with different transcription factors. Phosphorylation of p300/CBP by cyclin E/Cdk2 negatively regulates the transactivating function of co-activators (Perkins et al. 1997) but increases their histone acetyltransferase activity (Ait-Si-Ali et al. 1998). Both p300 and CBP have consensus protein kinase A (PKA) sites adjacent to their CH3 domains (Chrivia et al. 1993; Xu et al. 1998). Phosphorylation of CBP by PKA near the CH3 domain appears to be necessary for the co-activation of transcription by phosphorylated CREB and Pit-1, a Pou family transcription factor. PKA-responsive elements were also proposed to exist within the N-terminal transactivation domain of CBP, where they enhance CREB-mediated transcription in PC12, but not in F9 or Cos-7 cells (Swope et al. 1996). More recently, phosphorylation of p300 on serine 89 by PKA was shown to dramatically decrease its in vitro and in vivo interactions with several nuclear receptors (e.g. retinoic acid receptor) but did not affect its interaction with transcription factors such as p53, GATA4 or E1A (Yang et al. 2001). A protein kinase C (PKC) site on a conserved serine 89 was also identified, and this phosphorylation appears to inhibit p300 transcriptional activity (Yuan and Gambee 2000). p300/CBP phosphorylation is also induced by differentiation of F9 cells by retinoic acid (Kitabayashi et al. 1995) and by mitogen-activated protein kinase during nerve growth factor treatment of PC12 cells (Liu et al. 1999). Calcium/calmodulin (CaM) kinase IV phosphorylates CBP, but not p300, and this phosphorylation is necessary for the full CREB/CBP-mediated transcriptional activation (Chawla et al. 1998). Most recently it has been demonstrated that high concentrations of transcription factors, such as C/EBPb, c-jun and c-Ets, binding to the CH3 domain of p300 trigger massive hyperphosphorylation of p300 (Schwartz et al. 2003). Although it is not currently known which specific kinases are involved in this process, mutational analysis of p300 phosphorylation sites reveals that the kinase may be proline directed (Schwartz et al. 2003).
Here we report that exposure to hypoxia stimulates hyperphosphorylation of p300 and CBP in different cells, including oxygen-sensitive PC12 and rat carotid body cells. We also show that this hyperphosphorylation depends on the release of calcium from 1,4,5-triphosphate (IP3)-sensitive stores and on a metabolic pathway generating ATP in an IP3-sensitive manner.
Materials and methods
Cell cultures and reagents
Rat pheochromocytoma PC12, insulinoma INS-1 and RINm5F cell lines, and human embryonic kidney HEK293T and hepatoblastoma HepG2 cells, were grown in Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium with 10% fetal calf serum, 15 mm Hepes, pH 7.4, 100 U/mL penicillin and 100 μg/mL streptomycin. All cells were used 48 h after plating, at a final cell density of 1.5–2.5 × 105 cells/cm2. In some cases, the medium was replaced by fresh serum-free or, where indicated, calcium-free medium before the beginning of experiments. All drugs were added 15 min before cells were exposed to hypoxia. Hypoxic exposures were performed in a Hypoxic Workstation (Coy Laboratory Products Inc., Grasslake, MI, USA). Cells were collected and lysed in the hypoxic environment to prevent reoxygenation. All chemicals were purchased from Sigma (St Louis, MO, USA) or Calbiochem (La Jolla, CA, USA). The anti-p300 (C-20) and anti-CBP (C-20) used for immunoblotting were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-p300 MN11 antibody used for immunoprecipitations was from Pharmingen (Lexington, KY, USA) and the anti-HIF-1α antibody was from Novus Biologicals (Littleton, CO, USA).
Exposure of animals to hypoxia
Male Sprague–Dawley rats (175–200 g) were exposed to intermittent or sustained hypoxia for 1, 3, 7, 14 and 30 days in commercially designed chambers (Oxycycler model A44XO; BioSpherix, Red-field, NY, USA) as described in Hui et al. (2003). Control rats were exposed to room air. At least six rats were used for each time point, and series of experiments were repeated independently at least three times. The chambers were operated under a 12-h light–dark cycle. Gas was circulated around each chamber at 60 L/min. Oxygen concentration was continuously measured by an O2 analyzer and was changed throughout the 12 h of light time by a computerized system controlling the gas valve outlets, so that the moment-to-moment desired oxygen concentration of the chamber was programmed and adjusted automatically. The intermittent hypoxia profile consisted of alternating room air and 10% O2 every 90 s for the 12-h light period for up to 30 days, whereas the sustained hypoxia paradigm consisted of exposure to 10% O2 throughout. At the end of exposures, animals were anesthetized, killed by decapitation, and carotid bodies, superior cervical ganglia and adrenal glands were quickly removed and frozen in liquid nitrogen. All procedures involving animals were approved by the Institutional Animal Care and Use Committee of the University of Louisville and are consistent with guidelines provided by the National Institutes of Health (NIH).
Preparation of nuclear extracts, western blots and immunoprecipitation
These procedures were performed as described in Schnell et al. (2003) and Hui et al. (2003). For western blot analysis we used 100 μg of lysates from cell lines, 80 μg from carotid bodies and 150 μg from superior cervical ganglia. For all immunoprecipitation reactions, the protein G-bound agarose beads were pre-coated with bovine serum albumin and then pre-conjugated with the primary antibodies. Binding reactions were performed in buffer containing 250 mm NaCl, 20 mm Na2HPO4, pH 7.4, 0.1% Triton, standard protease and phosphatase inhibitors, and 200 μg PC12 lysate. Immunoprecipitated proteins were eluted by boiling in sodium dodecyl sulfate (SDS) sample buffer, resolved by SDS–polyacrylamide gel electrophoresis (PAGE) on 4–22% gradient gels, and detected by immunoblotting. Western blotting was performed as described by Schnell et al. (2003).
Measurement of intracellular calcium
Cytosolic free Ca2+ concentration ([Ca2+]i) was evaluated using the fluorescent Ca2+ indicator Fura-2, as described previously (Zhu et al. 1996). PC12 cells were incubated with DMEM/F12 medium containing 1 μm Fura-2 acetoxymethyl ester and pluronic F127 (0.01%) for 30 min at 25°C. The cells were then rinsed twice with the same medium without Fura-2 and maintained in either drug-free medium or medium containing vehicle [dimethylsulfoxide (DMSO) 1 : 1000 dilution], 200 μm 2-aminoethoxydiphenylborate (2-APB) or 10 μm ryanodine (RyR) for 30 min. Imaging experiments were then performed in Ca2+-free Ringer’s solution containing 155 mm NaCl, 4.5 mm KCl, 1 mm MgCl2, 10 mM Hepes, 10 mM glucose and 2 mm EGTA, pH 7.4 (Zhu et al. 1996). Fura-2 intensity was measured on a Cyt-Im2 Ca2+ imaging system (Intracellular Imaging, Cincinnati, OH, USA). Cells were imaged on a Nikon (Garden City, NY, USA) inverted epifluorescence microscope equipped with a 20 × superfluor objective, NA 0.75, and the dye was excited alternately with 340 and 380 nm light from a xenon arc lamp. The ratio of F340/F380 was used as an indicator of [Ca2+]i. The acute effect of progressively lower O2 tension (pO2) was studied by switching from a perfusion medium bubbled with air (21% O2, normoxia) to a medium equilibrated with 100% N2 (PO2 in the perfusion chamber approximately 20 mmHg, hypoxia) followed by a medium equilibrated with 100% N2 in presence of 1 mM sodium dithionate (Na2S2O4; an O2 chelator; pO2 in the perfusion chamber approximately 0 mmHg, pH 7.4, anoxia). Changes in [Ca2+]i were calculated by measuring plateau values and expressing them as the change in fluorescence ratio from basal levels (normoxia), determined for each recording.
Results
Hypoxia induces hyperphosphorylation of p300 and CBP
Exposure of PC12 cells to hypoxia resulted in a fast and reversible shift in mobility of p300 and CBP as assessed by SDS–PAGE (Fig. 1a). This response started at fairly mild levels of 10% O2 (Fig. 1b) and was specific for hypoxia, but not for the hypoxia-mimicking agents desferioxamine mesylate or CoCl2 (Fig. 1c). A similar shift in p300 mobility was measured in extracts from O2-sensitive tissues physiologically involved in respiratory and cardiovascular response to hypoxic stress, such as the carotid bodies from animals exposed to either sustained or intermittent hypoxia (Fig. 1d). This shift was also present, but weaker, in extracts obtained from superior cervical ganglia; the shift was stronger in ganglia obtained from animals exposed to intermittent hypoxia (Fig. 1d). A shift in p300 or CBP migration was also measured in rat pancreatic β cell lines (data not shown) and a delayed shift was present in HEK293T cells (Fig. 1e). In contrast, we failed to measure any visible shift in extracts from hepatoblastoma HepG2 cells (data not shown). Thus, although the effects of hypoxia on p300 and CBP are fairly general, they show also some cell-type specificity.
Fig. 1.
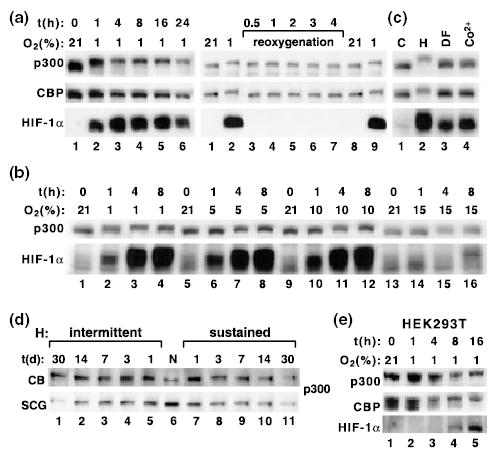
Cell specific effects of hypoxia on mobility of p300 and CBP. Hypoxia induces a specific shift in the mobility of p300 and CBP in rat PC12 cells (a–c), rat carotid bodies (CB) and superior cervical ganglia (SCG) (d), and in human HEK293T cells (e). All results are shown as western blots. (a) Analysis of p300, CBP and HIF1α in nuclear extracts from PC12 cells exposed to 1% O2 for indicated periods of time (left). Reoxygenation of cells after 4 h of hypoxia abolished the shift in p300 mobility (right). (b) Effects of different levels of hypoxia on the occurrence of the shift in p300 mobility. (c) The response was specific for hypoxia but not for treatment of cells with desferioxamine (DF; 100 μm) or Co2+ (100 μm). (d) Analysis of p300 mobility in rats exposed to sustained or intermittent hypoxia for indicated numbers of days. N, normoxia. (e) Effects of hypoxia on p300, CBP and HIF-1α in HEK293T cells.
The shift in p300 mobility resulted from hyperphosphorylation because it was abolished by treating the immunoprecipitated p300 reactions with λ protein phosphatase (Fig. 2a, lanes 5, 6, 9 and 10). The effects of phosphatase were prevented by addition of an inhibitor, sodium fluoride (lanes 7 and 8). Treatment of normoxic PC12 cells with okadaic acid, a serine–threonine phosphatase inhibitor, also led to a major shift in p300 mobility similar to that observed during hypoxia (Fig. 2b). This indicates that hyperphosphosrylation of p300 can indeed lead to a robust change in its mobility on SDS–PAGE.
Fig. 2.
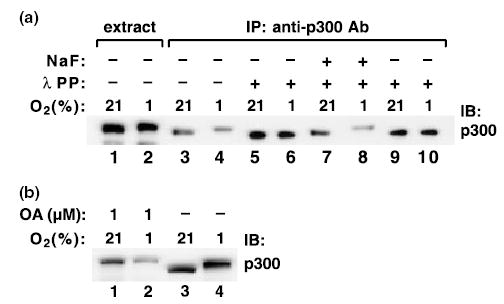
Shift in p300 mobility results from hyperphosphorylation. (a) p300 was immunoprecipitated (IP) with the MN11 antibody and the immunoprecipitates were treated with lambda protein phosphatase (λ PP) in the absence or presence of NaF (10 mm). (b) PC12 cells were treated with 1 μm okadaic acid (OA) for 1 h, and the extracts were analyzed for p300. IB, immunoblotting.
Hyperphosphorylation of p300 depends on calcium release from IP3-sensitive stores
Calcium is a documented intermediary in hypoxic adaptation in excitable PC12 and carotid body cells (Zhu et al. 1996; Lopez-Barneo et al. 2001). Pretreatment of PC12 cells with chelators of intracellular calcium, BAPTA-AM or EGTA-AM, abolished hypoxia-induced hyperphosphorylation of p300 (Fig. 3a) . In contrast, chelation of extracellular calcium with EGTA in calcium free-medium had no effect (Fig. 3b). Furthermore, two inhibitors of Ca2+ release from IP3-sensitive stores, 2-APB and xestospongin C, inhibited hyperphosphorylation (Fig. 3c), whereas inhibitors of the mitochondrial (RU360) and RyR-sensitive calcium pools did not (Fig. 3c). These results are consistent with our observation that in PC12 cells hypoxia released calcium from 2-APB-sensitive but not from RyR-sensitive stores (Fig. 3d). Treatment of PC12 cells with the calcium ionophore ionomycin during normoxia did not result in p300 hyperphosphorylation (not shown), an indication that increased intracellular calcium concentration by itself was insufficient to evoke hyperphosphorylation.
Fig. 3.
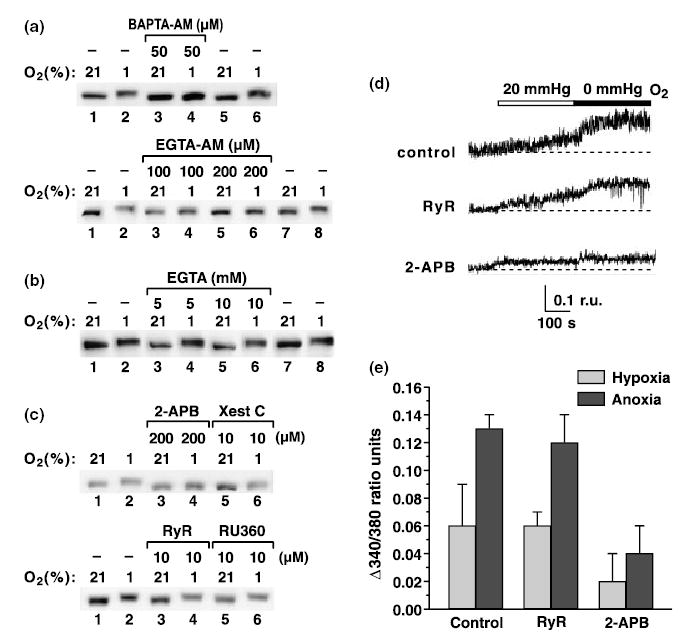
Hypoxia-induced p300 hyperphosphorylation depends on release of intracellular calcium from IP3-sensitive stores. PC12 cells were treated with the indicated concentrations of (a) BAPTA-AM and EGTA-AM, (b) EGTA, (c) 2-APB and Xestospongin C (Xest C) (inhibitors of IP3 receptors), RyR or RU360 (inhibitor of mitochondrial calcium release) and exposed to 1% hypoxia for 3 h. p300 was analyzed by western blotting. (d) Representative measurements of [Ca2+]i increase evoked by hypoxia (20 mmHg) and anoxia (0 mmHg) in PC12 cells pretreated with DMSO (1 : 1000; control), RyR (10 μm) and 2-APB (200 μm). Basal levels of [Ca2+]i (determined before application of hypoxia) were not significantly different in control PC12 cells (181 cells), 2-APB (74 cells) and RyR-treated cells (67 cells). (e) Averaged rise in [Ca2+]i during hypoxia and anoxia in control, 2-APB- and RyR-treated cells. Values are mean ± SD of 2–3 independent experiments in each case, from a total of 61 control cells, 74 2-APB-treated cells and 67 RyR-treated cells. r.u., Ratio units.
Pharmacological inhibitors of kinases known to be regulated directly or indirectly by calcium CaM kinase II (KN93), PKA (H89), PKC (Gö6976), ERK (PD98059), PI3K (wortmannin, LY294002) and mTOR (rapamycin) in various doses and combinations failed to affect the hypoxia-induced p300 hyperphosphorylation (Fig. 4). These data indicate that calcium regulates p300 phosphorylation either by activating an unknown kinase, by inhibiting a phosphatase, or both.
Fig. 4.

Hypoxia-induced hyperphosphorylation of p300 is independent of traditional directly or indirectly calcium-regulated kinases. Western blot of p300 from cells exposed to normoxia and hypoxia in response to treatment with the following inhibitors of directly or indirectly calcium-regulated kinase pathways: KN93, calcium CaM kinase II; H89, PKA; Gö6976, classical PKC; PD98059, extracellular signal-regulated kinases; wortmannin and LY257002, PI3K; rapamycin, mTOR.
Hyperphosphorylation of p300 requires activation of a specific glucose metabolic pathway
Calcium in physiological concentrations, in particular the pool released from the IP3-sensitive stores, increases intramitochondrial calcium and oxidative phosphorylation (Rizzuto et al. 1993; Bootman et al. 2002). Calcium is an allosteric activator of four mitochondrial dehydrogenases (Nichols and Denton 1995), including mitochondrial glycerol-3-phosphate dehydrogenase (G3PDH). G3PDH participates in the transfer of NADH generated by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) during glycolysis from the cytosol to mitochondria. NADH cannot penetrate the mitochondrial membrane, but it is transferred by glycerol phosphate and malate–aspartate shuttles. G3PDH on the cytosolic side utilizes NADH to reduce dihydroacetone to membrane-permeable glycerol-3-phosphate. G3PDH on the outer surface of the inner mitochondrial membrane is a flavoprotein that uses FAD to oxidize glycerol-3-phosphate back to dihydroacetone, and generates FADH2, which transfers reducing equivalents on the respiratory chain to ubiquinone, and then complex III within the respiratory electron transfer chain, omitting complex I or II. G3PDH contains an EF-hand, a calcium-binding motif on its cytosolic site, and is known to be stimulated by calcium (Nichols and Denton 1995). Western blot analysis of G3PDH protein expression in PC12 cells confirmed the presence of a single band migrating around 80 kDa (data not shown). Thus, we hypothesized that calcium released from IP3-sensitive stores during hypoxia in PC12 cells regulates transfer of reducing equivalents from glycolysis, through the glycerol phosphate shuttle to complex III of the mitochondrial respiratory chain, maintaining formation of ATP during hypoxia, and that such a pool of ATP may be necessary for p300 hyperphosphorylation.
First, we determined that glucose was necessary for p300 hyperphosphorylation. Exposure of cells to hypoxia in glucose-free medium or in the presence of 2-deoxyglucose, an inhibitor of an early step of glycolysis, not only prevented the hyperphosphorylation but actually decreased the mobility of p300 on SDS–PAGE, indicating dephosphorylation below normoxic levels (Fig. 5a). A similar effect was observed with a low concentration of iodoacetate, an inhibitor of GAPDH, an enzyme that catalyzes the initial step in glycolysis that generates NADH (Fig. 5a). All of these treatments prevented hypoxic accumulation of the HIF-1α transcription factor as well. Next, we treated cells with hydroxybutarate, pyruvate or α-ketoglutarate in the presence of 2-deoxyglucose. These substrates enter the Krebs cycle and contribute NADH at the level of complex I, and so can activate the mitochondrial respiratory chain independently of glycolysis. However, activation of the Krebs cycle by these substrates did not prevent inhibition of p300 phosphorylation (Fig. 5b), indicating that glycolysis is necessary for p300 hyperphosphorylation. Because NAD is the product of cytosolic G3PDH activity, we anticipated that by increasing the concentration of NAD we would inhibit the activity of the enzyme and, as a result, hyperphosphorylation of p300. Indeed, treatment with NAD, but not NADP, NADH or NADPH, in increasing concentrations inhibited p300 hyperphosphorylation (Fig. 5c). Finally, we found that treatment of PC12 cells with palmitoyl CoA, a competitive inhibitor of G3PDH (Edgar and Bell 1979), abolished hypoxic hyperphosphorylation of p300 (Fig. 5d). Hyperphosphorylation of p300 was also abolished by treatment of cells with diphenyliodonium (DPI) (Chakraborty and Massey 2002), a specific inhibitor of flavoproteins (G3PDH on the mitochondrial site is a flavoprotein) in a dose-dependent manner (Fig. 5e). To differentiate among other potentially relevant flavoproteins that might be also blocked by DPI, such as mitochondrial complex II and NADPH oxidases, cells were treated with numerous inhibitors specific for complex II (malonate, methyl malonate, 3-nitropropionic acid, tenoyltrifluoroacetate) or for NADPH oxidase (phenylarsine and neopterine). However, these inhibitors failed to affect p300 hyperphosphorylation (data not shown). Interestingly, although DPI inhibited accumulation of HIF-1α, neither NAD nor palmitoyl CoA had any effect on HIF-1α accumulation. Inhibition of the malate–aspartate shuttle with a specific inhibitor, amino-oxyacetate, did not affect p300 phosphorylation (data not shown). Consistent with the idea that reducing equivalents are transported from G3PDH on to ubiquinone and then complex III, inhibition of complex III with both antimycin A and myxothiazol inhibited p300 hyperphosphorylation (Fig. 5f). In contrast, inhibition of complex I with rotenone (Fig. 5f) or complex II (date not shown) had no effect. Thus, we concluded that Ca2+ released from IP3-sensitive stores during hypoxia is necessary for synthesis of a specific pool of ATP that participates in the phosphorylation of p300. Inhibition of IP3-sensitive pools with 2-APB decreased the amount of ATP generated during both normoxia and hypoxia, but the effect was significantly stronger during hypoxia (Fig. 5g). These data indicate that during hypoxia the IP3-sensitive pool of calcium results in production of ATP specifically through the glycerol phosphate shuttle.
Fig. 5.
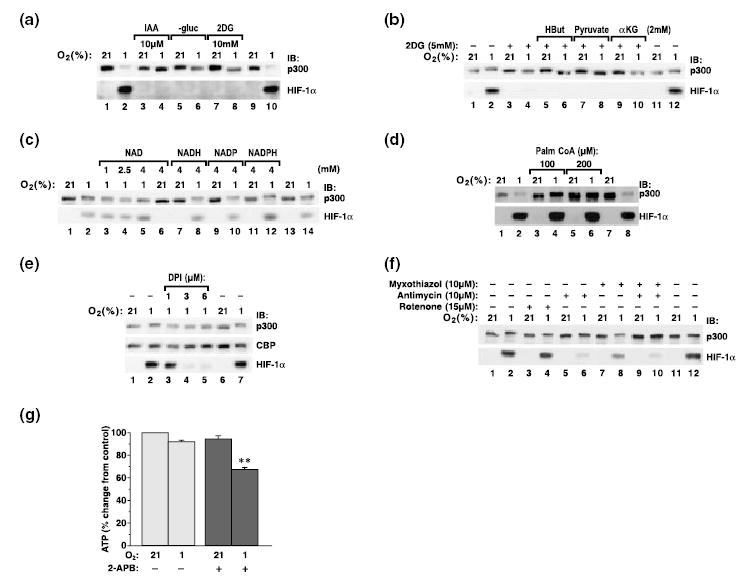
Hypoxic hyperphosphorylation of p300 is regulated by glycerol-3-phosphate metabolic pathway. Hyperphosphorylation of p300 was analyzed by western blotting. Hyperphosphorylation of p300 was abolished by inhibitors of glycolysis: iodoacetate (IAA), deoxyglucose (2DG) and glucose-free medium (–gluc) (a). This effect was not prevented by supplementing of substrates for the Krebs cycle, such as hydroxybutyrate (HBut), pyruvate, α-ketoglutarate (αKG) (b). Hyperphosphorylation was abolished by treatment of cells with increasing concentrations of NAD, but not NADH, NADP or NADPH (c). It was also abolished by palmitoyl CoA, a competitive inhibitor of G3PDH (d); DPI, an inhibitor of flavoptroteins (e); and by inhibition of mitochondrial complex III (f). Inhibition of IP3 stores with 2-APB reduced the amount of ATP produced in hypoxic cells (g).
Discussion
Here we have identified a novel reversible hyperphosphorylation of two transcription co-activators, p300 and CBP. This hyperphosphorylation shows some tissue specificity; it is particularly pronounced in O2-sensitive PC12 cells and carotid body cells. It is completely absent, however, in heptoblastoma HepG2 cells. Hypoxic hyperphosphorylation of p300 most likely requires the activity of kinases, but possibly also inhibition of a phosphatase activity (Taylor et al. 2000). So far, we have not been able to identify the specific kinase/phosphatase involved in this phosphorylation. It is possible that the activity of such a kinase/phosphatase can be regulated by calcium, but it is also possible that the primary effect of calcium is through the metabolic pathway and generation of ATP, whereas the activity of the putative kinase/phosphatase is regulated by another aspect of hypoxic signaling, such as a more reducing environment owing to decreased generation of reactive oxygen species (H2O2) during hypoxia, which we have consistently measured in PC12 cells (Kroll and Czyzyk-Krzeska 1998; A. Zakrzewska and M. F. Czyzyk-Krzeska, unpublished observations). Our observations are in line with the previously reported evidence that hypoxia stimulates phosphorylation of a p300/CBP partner, CREB, on serine 133 (Beitner-Johnson and Millhorn 1998). In that case, the authors were also unable to identify a kinase/phosphatase involved in the pathway. Thus, perhaps a novel hypoxia signaling pathway participates in both situations. Interestingly, it also appears that recruitment of p300 by transcription factors such as C/EBPβ, Ets and c-Jun induces a major hyperphosphorylation of p300 that results in a change in its mobility (Schwartz et al. 2003). These experiments were, however, performed in quail and chicken fibroblasts, so their relevance to mammalian systems is not clear.
Importantly, we have determined that p300 hyperphosphorylation requires release of calcium from IP3-sensitive stores, and a pool of ATP generated in the pathway involving the first step of glycolysis catalyzed by GAPDH, cytosolic–mitochondrial glycerol phosphate shuttle and mitochondrial complex III. We have demonstrated that inhibition of this pathway at several pertinent steps (Fig. 6) prevents hyperphosphorylation of p300. This finding is interesting because very little is known about pools of ATP used in different intracellular phosphorylation events. The notion of ATP as a regulator of kinase/phosphatase activity is particularly interesting in view of the recent report that a kinase important in regulation of translation, the mammalian target of rapamycin, mTOR, acts as an ATP sensor, and ATP regulates mTOR activity towards its downstream targets, affecting translation (Dennis et al. 2001). Our observation is also in line with published evidence demonstrating that only a pool of ATP generated by the transport of reducing equivalents from glucose-induced glycolysis through glycerol phosphate–malate shuttles inhibits the activity of ATP-sensitive potassium channels, leading to depolarization of the β-cell plasma membrane and insulin secretion (Eto et al. 1999a, 1999b; Tan et al. 2002).
Fig. 6.
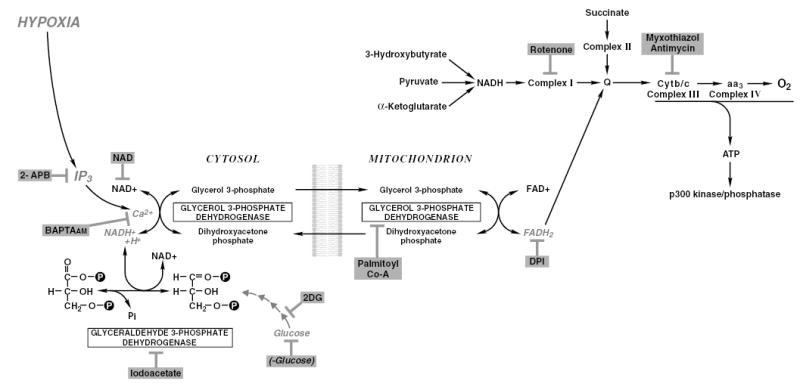
Schematic representation of the metabolic pathway participating in p300 hyperphosphorylation during hypoxia.
Currently the function of p300 hyperphosphorylation is not well understood. We have, however, determined that this phosphorylation does not lead to any form of p300 ubiquitylation or degradation, and that the approximate half-life of p300 protein is between 8 and 10 h both in normoxia and hypoxia (data not shown), which is in the range described by others (Poizat et al. 2000). We also determined that hyperphosphorylation did not affect histone acetyltransferase activity of p300 in an in vitro assay (data not shown), and was without effect on p53 polyubiquitylation (Grossman et al. 2003). The most likely possibility is that hyperphosphorylated p300 and CBP interact with certain transcription factors, such as HIFs, stimulating their activity during hypoxia. This possibility is potentially consistent with the data reported by Yang et al. 2001, who showed that the metabolic situation of the cell may affect p300 interactions with different transcription factors. It is also supported by a correlation between hyperphosphorylation of p300/CBP and the accumulation of HIF-1α. With the exception of palmitoyl CoA and NAD, all other compounds, including calcium chelators (M. F. Czyzyk-Krzeska, unpublished data), that prevented p300 hyperphosphorylation also prevented accumulation of HIF-1α. Similarly, in HEK293 cells late hyperphosphorylation of p300/CBP correlated with a fairly late accumulation of HIF-1α. Our suggestion of a role of complex III in p300 hyperphosphorylation also agrees with the role this complex plays in stabilization of HIF-1α protein as proposed by Chandel et al. (2000). These authors showed, however, that reactive oxygen species rather than ATP mediate the effects of complex III inhibition. Our further direct investigations of the function of p300 hyperphosphorylation have been restricted because we have been unable to express, even transiently, p300 mutants in PC12 cells and because p300 hyperphosphorylation appears to be sensitive to the method of transfection, which most probably indicates that some membrane-associated events are necessary. An interesting result is that hypoxic accumulation of HIF-1α requires glucose. This is a novel observation, and it is not clear at the present time whether glucose plays a role in HIF-1α stabilization and/or translation.
Acknowledgments
This work was supported by NIH grants HL58687 and HL66312, and American Cancer Society Research Scholar Grant GMC-101430 to MFC-K, and start-up funds from the Genome Research Institute. DG was supported by NIH grant HL69932. We thank Glenn Doermann for preparation of the figures.
References
- Ait-Si-Ali S, Ramirez S, Barre FX, et al. Histone acetyltransferase activity of CBP is controlled by cycle-dependent kinases and oncoprotein E1A. Nature. 1998;396:184–186. doi: 10.1038/24190. [DOI] [PubMed] [Google Scholar]
- Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, Bunn HF, Livingstone DM. An essential role for p300/CBP in cellular response to hypoxia. Proc Natl Acad Sci USA. 1996;93:12 969–12 973. doi: 10.1073/pnas.93.23.12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beitner-Johnson D, Millhorn DE. Hypoxia induces phosphorylation of the cyclic AMP response element-binding protein by a novel signaling mechanism. J Biol Chem. 1998;273:19 834–19 839. doi: 10.1074/jbc.273.31.19834. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Michels CL, Leung MK, Arany ZP, Kuang AL, Livingstone DM. Functional role of p35srj, a novel p300/cbp binding protein during transactivation by HIF-1. Genes Dev. 1999;13:64–75. doi: 10.1101/gad.13.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bootman MD, Berridge MJ, Roderick HL. Activating calcium release through inositol 1,4,5-trisphosphate receptors without inositol 1,4,5-trisphosphate. Proc Natl Acad Sci USA. 2002;99:7320–7322. doi: 10.1073/pnas.132254299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Massey V. Reaction of reduced flavins and flavoproteins with diphenyliodonium chloride. J Biol Chem. 2002;277:41 507–41 516. doi: 10.1074/jbc.M205432200. [DOI] [PubMed] [Google Scholar]
- Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1a during hypoxia. J Biol Chem. 2000;275:25 130–25 138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- Chawla S, Hardingham GF, Quinn DR, Bading H. CBP: a signal regulated transcriptional coactivator controlled by nuclear calcium and CaM kinase IV. Science. 1998;281:1505–1509. doi: 10.1126/science.281.5382.1505. [DOI] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma S, Thomas G. Mammalian TOR: a homeostatic ATP sensor. Science. 2001;294:1102–1105. doi: 10.1126/science.1063518. [DOI] [PubMed] [Google Scholar]
- Ebert B, Bunn HF. Regulation of transcription by hypoxia requires a multiprotein complex that induces hypoxia-inducible factor 1, an adjacent transcription factor, and p300/CREB binding protein. Mol Cell Biol. 1998;18:4089–4096. doi: 10.1128/mcb.18.7.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar JR, Bell RM. Biosynthesis Escherichia coli of sn-glycerol 3-phosphate, a precursor of phospholipids: palmitoyl-CoA inhibition of the biosynthetic sn-glycerol-3-phosphate dehydrogenase. J Biol Chem. 1979;254:1016–1021. [PubMed] [Google Scholar]
- Ema M, Hirota K, Mimura J, Abe H, Yodoi J, Sogawa K, Poellinger L, Fuji-Kuriyama Y. Molecular mechanisms of transcription activation by HLF and HIF1a in response to hypoxia: their stabilization and redox signal-induced interaction with CBP/p300. EMBO J. 1999;18:1905–1914. doi: 10.1093/emboj/18.7.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto K, Suga S, Wakui M, et al. NADH shuttle system regulates K+ ATP channel-dependent pathway and steps distal to cytosolic Ca2+ concentration elevation in glucose-induced insulin secretion. J Biol Chem. 1999a;274:25 386–25 392. doi: 10.1074/jbc.274.36.25386. [DOI] [PubMed] [Google Scholar]
- Eto K, Tsubamot Y, Terauchi Y, et al. Role of NADH system in glucose-induced activation of mitochondrial metabolism and insulin secretion. Science. 1999b;283:981–985. doi: 10.1126/science.283.5404.981. [DOI] [PubMed] [Google Scholar]
- Goodman RH, Smolik S. CBP/p300 in cell growth, transformation and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- Grossman SR, Deato ME, Grignone C, Chan HM, Kung AL, Tagami H, Nakatani Y, Livingston DM. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science. 2003;300:342–344. doi: 10.1126/science.1080386. [DOI] [PubMed] [Google Scholar]
- Hui AS, Striet JB, Gudelsky G, et al. Regulation of catecholamines by sustained and intermittent hypoxia in neuroendocrine cells and sympathetic neurons. Hypertension. 2003;42:1130–1136. doi: 10.1161/01.HYP.0000101691.12358.26. [DOI] [PubMed] [Google Scholar]
- Kallio PJ, Okamoto K, O’Brien S, Carrero P, Makino Y, Tanaka H, Poellinger L. Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia indcuible factor-1a. EMBO J. 1998;17:6573–6586. doi: 10.1093/emboj/17.22.6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitabayashi I, Eckner R, Arany Z, Chiu R, Gachelin G, Livinstone DM, Yokoyama KK. Phosphorylation of the adenovirus E1A associated 300 kDa protein in response to retinoic acid and E1A during the differentiation of F9 cells. EMBO J. 1995;14:3496–3509. doi: 10.1002/j.1460-2075.1995.tb07356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroll SL, Czyzyk-Krzeska MF. Role of H2O2 and heme- containing O2 sensors in hypoxic regulation of tyrosine hydroxylase gene expression. Am J Physiol. 1998;274:C167–C174. doi: 10.1152/ajpcell.1998.274.1.C167. [DOI] [PubMed] [Google Scholar]
- Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002a;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Aspargine hydroxylation of HIF transactivation domain: a hypoxic switch. Science. 2002b;295:858–861. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- Liu YZ, Thomas NSB, Latchman DS. CBP associates with the p42/p44 MAPK enzymes and is phosphorylated following NGF treatment. Neuroreport. 1999;10:1239–1243. doi: 10.1097/00001756-199904260-00016. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Pardal R, Ortega-Saenz P. Cellular mechanism of oxygen sensing. Annu Rev Physiol. 2001;63:259–287. doi: 10.1146/annurev.physiol.63.1.259. [DOI] [PubMed] [Google Scholar]
- Nichols BJ, Denton RM. Towards the molecular basis for the regulation of mitochondrial dehydrogenases by calcium ions. Mol Cell Biochem. 1995;149/150:203–212. doi: 10.1007/BF01076578. [DOI] [PubMed] [Google Scholar]
- Perkins ND, Felzien LK, Betts JC, Leung K, Beach DH, Nabel GJ. Regulation of NF-kappaB by cyclin-dependent kinases associated with the p300 coactivator. Science. 1997;275:523–527. doi: 10.1126/science.275.5299.523. [DOI] [PubMed] [Google Scholar]
- Poizat C, Sartorelli V, Chung G, Kloner RA, Kedes L. Proteasome-mediated degradation of the coactivator p300 impairs cardiac transcription. Mol Cell Biol. 2000;20:8643–8654. doi: 10.1128/mcb.20.23.8643-8654.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262:744–747. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- Schnell PO, Ignacak ML, Bauer AL, Striet JB, Paulding WR, Czyzyk-Krzeska MF. Regulation of tyrosine hydroxylase promoter activity by the von Hippel–Lindau tumor suppressor protein and hypoxia-inducible trasncritption factors. J Neurochem. 2003;85:483–491. doi: 10.1046/j.1471-4159.2003.01696.x. [DOI] [PubMed] [Google Scholar]
- Schwartz C, Beck K, Mink S, Schmolke M, Budde B, Wenning D, Klempnauer KH. Recruitment of p300 by C/EBP-β triggers phosphorylation of p300 and modulates coactivator activity. EMBO J. 2003;22:882–892. doi: 10.1093/emboj/cdg076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swope DL, Mueller CL, Chriva JC. CREB-binding protein activates transcription through multiple domain. J Biol Chem. 1996;271:28 138–28 145. doi: 10.1074/jbc.271.45.28138. [DOI] [PubMed] [Google Scholar]
- Tan C, Tuch BE, Tu J, Brown SA. Role of NADH shuttles in glucose-induced insulin secretion from fetal βcells. Diabetes. 2002;51:2989–2996. doi: 10.2337/diabetes.51.10.2989. [DOI] [PubMed] [Google Scholar]
- Taylor CT, Furuta GT, Synnestvedt K, Colgan SP. Phosphorylation-dependent targeting of cAMP response element binding protein to the ubiquitin/proteasome pathway in hypoxia. Proc Natl Acad Sci USA. 2000;97:12 091–12 096. doi: 10.1073/pnas.220211797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. J Biol Chem. 2001;276:13505–13508. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- Xu L, Lavinsky RM, Dasen JS, et al. Signal-specific co-activator domain requirements for Pit-1 activation. Nature. 1998;395:301–306. doi: 10.1038/26270. [DOI] [PubMed] [Google Scholar]
- Yaciuk P, Moran E. Analysis of specific polyclonal antiserum indicates that the E1a associated 300 kDa product is a stable nuclear phosphoprotein that undergoes cell cycle specific modification. Mol Cell Biol. 1991;11:5389–5397. doi: 10.1128/mcb.11.11.5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Hong YH, Shen XQ, Frankowski C, Camp HS, Leff T. Regulation of transcription by AMP-activated protien kinase: phosphorylation of p300 blocks its interaction with nuclear receptors. J Biol Chem. 2001;276:38 341–38 344. doi: 10.1074/jbc.C100316200. [DOI] [PubMed] [Google Scholar]
- Yuan LW, Gambee JE. Phosphorylation of p300 at serine 89 by protein kinase C. J Biol Chem. 2000;275:40 946–40 951. doi: 10.1074/jbc.M007832200. [DOI] [PubMed] [Google Scholar]
- Zhu WH, Conforti L, Czyzyk-Krzeska MF, Millhorn DE. Membrane depolarization and dopamine secretion in PC12 cells during hypoxia are regulated by an O2-sensitive K+ current. Am J Physiol. 1996;271:C658–C665. doi: 10.1152/ajpcell.1996.271.2.C658. [DOI] [PubMed] [Google Scholar]