

Abstract
The basic helix-loop-helix transcription factor Nex1/Math-2 belongs to the NeuroD subfamily, which plays a critical role during neuronal differentiation and maintenance of the differentiated state. Previously, we demonstrated that Nex1 is a key regulatory component of the nerve growth factor (NGF) pathway. Further supporting this hypothesis, this study shows that Nex1 has survival-inducing properties similar to NGF, as Nex1-overexpressing PC12 cells survive in the absence of trophic factors. We dissected the molecular mechanism by which Nex1 confers neuroprotection upon serum removal and found that constitutive expression of Nex1 maintained the expression of specific G1 phase cyclin-dependent kinase inhibitors and concomitantly induced a dynamic expression profile of key anti-apoptotic regulators. This study provides the first evidence of the underlying mechanism by which a member of the NeuroD-subfamily promotes an active anti-apoptotic program essential to the survival of neurons. Our results suggest that the survival program may be viewed as an integral component of the intrinsic programming of the differ entiated state.
Keywords: basic helix-loop-helix transcription factor, Bcl2-related protein bcl-w, Bcl2-related protein bcl-xL, cyclin-dependent kinase inhibitors, survivin, X-linked inhibitor of apoptosis
The basic helix-loop-helix (bHLH) transcription factors are implicated in the regulation of cell fate decision and differentiation in a wide variety of cell types. In the mammalian CNS, the neurogenic bHLH proteins have been classified into two groups based on sequence homology, temporal expression pattern, and gain and loss of functions (reviewed by Bertrand et al. 2002). The determination factors or proneural genes, which include MASH-1, MATH-1, MATH-5 and the neurogenins, are transiently expressed in neuronal progenitor cells at the early stages of CNS development, and play critical roles in determining various cell lineages. In contrast, the differentiation factors, which include the members of the NeuroD family, NeuroD, NeuroD2, Nex1/MATH-2 and MATH-3, are downstream regulatory genes that are partly under the transcriptional control of proneural genes. They display broadly overlapping patterns of expression during embryonic and postnatal development, resulting in functional compensation in knockout models. This is exemplified by Nex1-null mice, which do not display any discernable phenotype (Schwab et al. 1998). In addition to their differentiation-inducing functions, they are also believed to promote survival of specific neuronal lineages as suggested by the phenotypes of NeuroD and NeuroD2 knockout mice. Several studies on NeuroD-null mice reveal massive cell death of specific progenitor neurons, such as cerebellar granule cells, granule cells of the dentate gyrus and the inner ear sensory neurons (Miyata et al. 1999; Liu et al. 2000; Kim et al. 2001; Pennesi et al. 2003), whereas NeuroD2-null mice show decreased survival of mature cerebellar granule cells (Olson et al. 2001). Finally, double knockout Nex1/NeuroD mice display increased cell death of immature granule neurons of the dentate gyrus accompanied by a significant reduction in the overall hippocampal size (Schwab et al. 2000).
The molecular mechanism by which NeuroD members promote neuronal survival remains unresolved. It has been hypothesized that cell death, as visualized by terminal deoxynucleotidyl transferase αUTP nick-end labeling (TUNEL) assay, is most likely the result of failure in proper cell cycle withdrawal and differentiation (Miyata et al. 1999; Pennesi et al. 2003). However, a link with distinct neurotrophin pathways suggests a lead to a potential survival-promoting mechanism, based on the following observations. NeuroD-null mice fail to express the neurotrophin TrkB and TrkC receptors in the cochlear—vestibular ganglion (Kim et al. 2001), which is supported by a more recent study showing that NeuroD directly regulates the expression of the neurotrophin TrkB receptor gene at the transcriptional level (Liu et al. 2004). In addition, NeuroD2-null mice show altered expression of brain-derived neurotrophic factor (BDNF) and specific members of the ras/mitogen-activated protein kinase (MAPK) cascade (Olson et al. 2001). These observations are in accordance with our previous findings showing that Nex1 is as an important effector of the nerve growth factor (NGF) pathway in the PC12 cell system and that Nex1 also has the ability to promote neurite outgrowth and regeneration in the absence of NGF (Uittenbogaard and Chiaramello 2002). Thus, collectively, these observations lead us to ask whether the NeuroD members may be key transcriptional regulators of the neurotrophin-induced survival pathway.
To address this fundamental question, we explored whether Nex1 regulates the expression of specific anti-apoptotic regulators to promote survival of immature neurons using our previously established Nex1-overexpressing PC12 cell system. The rat PC12 pheochromocytoma cell line is a well established system with which to study apoptotic death and mechanisms of neuronal survival (Greene and Tischler 1976). As an experimental cell death paradigm, we used serum deprivation of PC12-Nex1 cells because naïve PC12 cells undergo apoptosis when cultured in serum-free medium, unless rescued by NGF treatment (Greene 1978; Batistatou and Greene 1991; Rukenstein et al. 1991). A major advantage of this approach is that we could investigate under restrictive conditions whether Nex1 regulates an intrinsic survival-promoting transcriptional program independently of any extrinsic differentiation signaling.
In this study, we provide the first direct evidence of the neuroprotective properties of Nex1 as well as the molecular mechanism by which Nex1 promotes long-term neuronal survival upon withdrawal of trophic factors. We found that serum-deprived PC12-Nex1 cells retain their neuronal phenotype and attain a non-dividing phenotype through the expression of key G1 phase cyclin-dependent kinase (CDK) inhibitors. Furthermore, we found that in the absence of an apoptotic stimulus Nex1 regulates the expression of the Bcl-w and X-linked inhibitor of apoptosis (XIAP) anti-apoptotic regulators. Most importantly, following serum deprivation, Nex1 promotes a dynamic anti-apoptotic response, acting at two distinct regulatory levels of the mitochondrial pathway, independently of the phosphatidylinositol 3-kinase (PI3K)/Akt intracellular signaling pathway. These results indicate that the Nex1-mediated intrinsic programming of the differentiated state includes the regulation of an anti-apoptotic program. Thus, our results suggest that the NeuroD family may play an active role in the regulation of the comprehensive differentiation/survival program during development and maintenance of the differentiated state.
Materials and methods
Cell culture
Rat pheochromocytoma PC12 cells (ATCC, Manassas, VA, USA), control PC12-bsd cells (previously referred to as PC12-OOF), Nex1/MATH-2-overexpressing PC12-Nex1 cells (clones A, B and C), and PC12-Nex1-mut1 cells (Uittenbogaard and Chiaramello 2002) were grown on collagen I-coated plates (Becton Dickinson Labware, San Jose, CA, USA) in F-12K modified medium (Kaighn's modification) supplemented with 2.5% fetal bovine serum and 15% horse serum (Gibco, Rockville, MD, USA; Invitrogen, Carlsbad, CA, USA), as recommended by ATCC. For cell survival assays involving trophic factor deprivation, cells were plated at 70% confluency and then washed the following day with serum-free medium. The cells were cultured for specific periods of time as indicated. When appropriate, the serum-free medium was changed three times a week; cells were no longer split during long-term serum deprivation as they stopped proliferating.
Quantification of cell death
Cell death was determined by the trypan blue exclusion assay, which detects cell membrane integrity. Values represent the mean ± SEM (n ≥ 9) of three independent experiments, and the trypan blue exclusion test results are expressed as percentage survival. Cell cytotoxicity was estimated in parallel by measuring the activity of extracellular lactate dehydrogenase (LDH), which is released into the cell culture supernatant upon damage of the plasma membrane. Experiments were performed in 24-well coated plates (100 000 cells/well). After serum deprivation, cells were removed and centrifuged at 800 g for 5 min; aliquots of supernatant were collected and stored at 4°C and assayed in triplicate following the manufacturer's instructions (Roche Molecular Biochemicals, Nutley, NJ, USA). Values represent the mean ± SEM (n ≥ 9) of three independent experiments and the results are expressed as percentage cytotoxicity.
Genomic DNA isolation and DNA fragmentation assay
Genomic DNA was isolated from the control PC12-bsd cells and PC12-Nex1 cells at different time points during serum deprivation using the suicide track™ DNA ladder isolation kit (Oncogene Research Products, Boston, MA, USA), according to the manufacturer's recommendations. DNA ladder fragments were separated by electrophoresis on a 1.5% agarose gel along with DNA molecular weight markers, and stained with ethidium bromide.
4-[3-(4-Iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate (WST-1) assay for metabolic activity
The metabolic activity of PC12-Nex1 cells (clones A, B and C) during serum deprivation was measured by the WST-1 assay (Roche Molecular Biochemicals) following the manufacturer's instructions. Cleavage of the tetrazolium salt WST-1 to formazan by mitochondrial dehydrogenases of viable PC12-Nex1 cells was measured using a microplate reader at 480 nm with the reference wavelength > 600 nm. Data represent mean ± SD of three independent experiments performed in triplicate.
Western blot analysis
Control PC12-bsd and PC12-Nex1 cells were lysed in M-Per mammalian protein extraction buffer (Pierce, Rockford, IL, USA) in the presence of a cocktail of protease inhibitors (Roche, Molecular Biochemicals), as described by Uittenbogaard and Chiaramello (2002). Cells extracts were spun at 12 000 g for 10 min, and protein concentration was determined by the Bradford assay (Bio-Rad, Hercules, CA, USA). Proteins (40 μg) were resolved on 10% NuPAGE Bis-Tris gels (Invitrogen) with either MES-sodium dodecyl sulfate (SDS) or MOPS-SDS running buffers, as recommended by the manufacturer, and transferred on to nitrocellulose. Nitrocellulose membranes were stained with Ponceau S (Sigma, St Louis, MO, USA) to confirm uniform transfer of proteins, and subsequently blocked using Superblock™ blocking buffer (Pierce) in phosphate-buffered saline containing 0.05% Tween-20. The membranes were probed with various primary antibodies described in Table 1 and corresponding secondary horseradish peroxidase-conjugated antibodies (Pierce). The antigen—antibody complex was detected using the Supersignal West Pico Chemiluminescent Substrate Kit (Pierce). When mentioned, blots were stripped using Restore™ western blot stripping buffer (Pierce) according to the manufacturer's recommendations, and re-probed with a monoclonal antibody against βIII tubulin to confirm equal protein loading.
Table 1.
Primary antibodies used in this study
| Antibody | Type | Source (Catalog no.) | Remarks |
|---|---|---|---|
| Nex1 | Rabbit pAb | Chiaramello laboratory | Uittenbogaard and Chiaramello (2002) |
| βIII tubulin | Mouse mAb | Covance (mms-435P) | |
| GAP-43 | Rabbit pAb | Chemicon (AB 5220) | |
| p21waf/cip | Mouse Ab | Santa-Cruz (sc-6246) | |
| p27KIP1 | Rabbit pAb | StressGen (KAP-CC02) | |
| p16INK4 | Rabbit pAb | SantaCruz (sc-1207) | |
| CDC47 | Mouse mAb | Neomarkers (MS-862) | |
| Bcl-xL | Rabbit pAb | Santa Cruz (sc-7195) | Specific to Bcl-xL only (amino acids 126-188) |
| Bcl-w | Rabbit pAb | Santa Cruz (sc-11422) | Recognizes (amino acids 55-193) |
| XIAP | Mouse mAb | StressGen (AAM-050) | |
| Survivin | Rabbit pAB | Chemicon (AB 3611) | |
| PTEN | Rabbit pAb | Upstate (07-016) | |
| p-PTEN | Rabbit pAb | Cell Signaling (9551) | Recognizes only p-PTEN at ser380 |
| Akt | Rabbit pAb | Cell Signaling (9272) | |
| p-Akt | Mouse mAb | Cell Signaling (4057) | Recognizes only p-Akt at ser473 |
| PARP | Rabbit pAb | Cell Signaling (9545) | Recognizes cleaved PARP (larger fragment only) |
mAb monoclonal antibody; pAb, polyclonal antibody; p-PTEN, phosphorylated form of PTEN; p-Akt, phosphorylated form of Akt.
Results
Nex1 prevents apoptosis of PC12-Nex1 cells upon withdrawal of trophic factor
In our previous study, we established a stable PC12-Nex1 cell line that constitutively overexpresses the bHLH differentiation factor Nex1/MATH-2 under the control of the cytomegalovirus promoter, as naïve PC12 cells do not express endogenous Nex1, as shown by RT-PCR and immunoblot analysis (Bartholoma and Nave 1994; Uittenbogaard and Chiaramello 2002). We showed that at least three distinct PC12-Nex1 clones A, B, and C displayed identical neuronal phenotype and gene expression pattern, thus excluding clonal variability in data interpretation (Uittenbogaard and Chiaramello 2002). In our original study, we simultaneously generated a control stable PC12 cell line, referred to as PC12-OOF, that did not express Nex1 because of an out-of-frame insertion of the Nex1 cDNA into pcDNA6/HisB, creating a stop codon right after the first Nex1 amino acid (Uittenbogaard and Chiaramello 2002). In this study, these control PC12-OOF cells are now referred as PC12-bsd cells in both the text and figures to reflect that they underwent the same selection process as the PC12-Nex1 cells. We also concomitantly generated the stable PC12-Nex1-mut1 cell line that expresses a dominant mutant-like form of Nex1 (Nex1-mut1), which was shown to be transcriptionally inactive owing to the deletion of the TAD1 activation domain (Uittenbogaard et al. 2003), and fails to display spontaneous neurite outgrowth and to differentiate upon exposure to NGF (Uittenbogaard and Chiaramello 2002).
In this study, we investigated whether Nex1 could rescue PC12 cells from undergoing apoptosis upon withdrawal of trophic factors. Control PC12-bsd, PC12-Nex1-mut1 and PC12-Nex1 cells (clones A, B and C) were cultured in serum-free medium for different periods of time after extensive washes and cell death was determined by trypan blue exclusion assay and LDH release assay. As expected, about 60% of both PC12-bsd and PC12-Nex1-mut1 cells died after 2 days of culture in serum-free medium, whereas the three PC12-Nex1 clones (A, B and C) displayed identical survival characteristics, with the majority (95%) of the cells remaining viable after 9 days of serum deprivation (Figs 1a and b). Furthermore, we failed to detect DNA fragmentation, a hallmark of apoptosis, in serum-deprived PC12-Nex1 cells Fig. 1c).
Fig. 1.

Overexpression of Nex1 promotes survival of PC12-Nex1 cells upon serum deprivation. (a) Prolonged survival of PC12-Nex1 cells (clones A, B and C) compared with control PC12-bsd and PC12-Nex1-mut1 cells after serum deprivation. Trypan blue exclusion test results are expressed as percentage survival. (b) Overexpression of Nex1 results in increased cell viability, as measured by LDH assay. Values represent the mean ± SEM (n = 9) of three independent experiments.(c) Overexpression of Nex1 prevents DNA fragmentation upon serum deprivation in PC12-Nex1 cells compared with control PC12-bsd cells. Apoptotic DNA was isolated from PC12-bsd and PC12-Nex1 cells at different time points during serum deprivation treatment, according to the manufacturer's recommendations. Equal amounts of DNA were run on a 1.5% agarose gel and stained with ethidium bromide. DNA markers were electrophoresed as a base pair reference. (d) PC12-Nex1 cells (clones A, B, and C) remain metabolically active after a prolonged 15-day serum deprivation treatment. The metabolic activity of PC12-Nex1 cells was measured by a colorimetric WST-1 assay. Data represent mean ± SD of three independent experiments performed in triplicate.
We then assessed the metabolic activity of the serum-deprived PC12-Nex1 cells at different time points using the WST-1 assay. WST-1, a water-soluble tetrazolium salt, is used to detect cellular reducing activities involving the pyridine nucleotide co-factors NADH and NADPH as well as the mitochondrial succinate tetrazolium reductase system, which are active only in viable cells. We observed fairly constant cleavage of WST-1 to formazan throughout the duration of the serum deprivation treatment with the three PC12-Nex1 clones, indicating that PC12-Nex1 cells remain metabolically active during prolonged Nex1-induced survival (Fig. 1d). Thus, constitutive expression of Nex1 rescues all three PC12-Nex1 clones from undergoing apoptosis upon withdrawal of trophic factors, suggesting that Nex1 has survival-inducing properties.
Serum-deprived PC12-Nex1 cells retain their neuronal characteristics and a constant expression of Nex1 protein
Both the results presented in Fig. 1 and those described in our previous study (Uittenbogaard and Chiaramello 2002) illustrate the fact that the three PC12-Nex1 clones display identical neuronal phenotype, gene expression pattern and survival characteristics. We therefore conducted our following analysis using the PC12-Nex1 clone B, which was also used in expression profiling studies of Nex1-MATH-2-mediated neuritogenesis (Uittenbogaard and Chiaramello 2004). Figure 2(a) illustrates that the PC12-Nex1 cells retained their differentiated morphology accompanied by substantial neurite outgrowth after 21 days of serum deprivation. However, they also displayed a flatter and wider cell body than the PC12-Nex1 cells grown in serum. Furthermore, these cells stopped growing and survived well beyond a month of serum deprivation, retained their known NGF responsiveness, and regained their growth properties in the presence of serum (data not shown). We initially examined whether Nex1 protein is expressed at constant levels during serum deprivation, by using PC12-Nex1 cell extracts isolated at successive times after serum deprivation for immunoblot analysis using our polyclonal anti-Nex1 antibody. Figure 2 shows that the expression of Nex1 protein remained constant and at high levels during the whole period of serum deprivation.
Fig. 2.
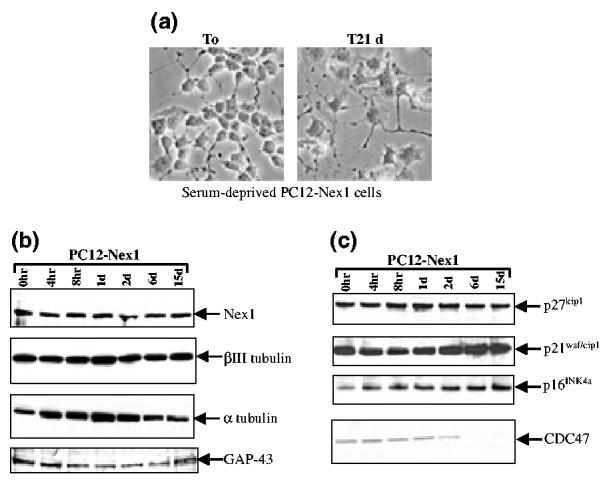
Expression profile of neuronal markers and cell cycle regulators in serum-deprived PC12-Nex1 cells. (a) Phase-contrast micrographs of serum-deprived PC12-Nex1 cells reveal a maintained neuronal phenotype accompanied by a flattened and wide cell body and significant neurite outgrowth. Serum-grown PC12-Nex1 cells, τ = 0 days; serum-deprived PC12-Nex1 cells, τ = 21 days. (b) Nex1 expression and that of specific neuronal markers remain constant during serum deprivation of PC12-Nex1 cells. PC12-Nex1 cells extracts isolated at successive times after serum deprivation as indicated above the panel were subjected to immunoblot analyses using appropriate antibodies described in Table 1 and corresponding secondary antibodies. The antigen—antibody complexes were detected by chemiluminescence. The membrane was stripped with Restore™ western blot stripping buffer and re-probed with the anti-βIII tubulin, anti-α tubulin, and anti-GAP-43 antibodies separately. (c) Constitutive expression of Nex1 results in sustained expression of specific CDK inhibitors upon serum deprivation. Immunoblot analyses using PC12-Nex1 cell extracts were performed as described above with antibodies described in Table 1. The membrane was stripped with Restore™ stripping buffer and re-probed with the anti-p27cip1, anti-p16INK4a and CDC47 antibodies separately. Data shown are representative of at least three independent experiments.
Because overexpression of Nex1 results in increased expression of neuronal markers such as growth-associated protein-43 (GAP-43) and βIII tubulin in the absence of NGF (Uittenbogaard and Chiaramello 2002), we examined their expression profile in serum-deprived PC12-Nex1 cells. We extended our analysis to the expression of α tubulin, a major cytoskeletal component of growing axons. Immunoblot analysis revealed reasonably constant levels of expression of both βIII tubulin and GAP-43 proteins throughout the duration of serum deprivation, whereas we detected a transient and modestly increased expression of α tubulin within 24 h of serum removal followed by a decline to original levels (Fig. 2b). Together, these results are in agreement with the sustained neuronal morphology observed in serum-deprived PC12-Nex1 cells, and suggest that Nex1 may link neuronal survival to the differentiation process.
Expression of CDKI is maintained in serum-deprived PC12-Nex1 cells
Our previous studies have demonstrated that overexpression of Nex1 induces the expression of the CDK inhibitor p21WAP/crp1 in the absence of NGF, resulting in a doubling time of PC12-Nex1 cells twice as long as that of PC12 cells (Uittenbogaard and Chiaramello 2002). More recently, we found high expression levels of two additional CDK inhibitors, p27kip1 and p16INK4, upon overexpression of Nex1 in PC12-Nex1 cells (Uittenbogaard and Chiaramello 2004 that overexpression of the CDKI p21WAF/cip, p27kip1 and p16INK4 proteins, and treatment with pharmacological CDK inhibitors and G1/S blockers, protect serum-deprived naïve PC12 cells from apoptosis (Farinelli and Greene 1996; Park et al. 1997b), we investigated whether the constitutive expression of Nex1 could maintain the expression of these CDK inhibitors in serum-deprived PC12-Nex1 cells. As shown in Fig. 2(c), PC12-Nex1 cells exhibited substantial levels of expression of both p21cip1 and p27kip1 proteins before withdrawal of trophic factors. Such high levels were sustained throughout the serum deprivation process in PC12-Nex1 cells. In contrast, we observed an increased expression of the p16INK4a protein just after 4 h of withdrawal of trophic factors, which remained constant for the duration of the treatment.
To investigate the time at which serum-deprived PC12-Nex1 cells become quiescent, we monitored the expression of the CDC47 protein, a member of the minichromosome maintenance (MCM) family, whose expression is repressed in quiescent cells, such as serum-starved fibroblasts and post-mitotic neurons (Fujita et al. 1996; Klein et al. 2002). We found that the expression of the CDC47 protein had already decreased significantly after 2 days of serum deprivation and declined to undetectable levels after 6 days of treatment (Fig. 2c). These results suggest that specific CDK inhibitor proteins from the CIP/KIP and INK4 families are key components of the Nex1-mediated transcriptional network promoting neuronal survival, and that removal of growth-promoting factors allows the activities of the CDK inhibitors to take full effect in inducing cell cycle arrest.
Nex1 induces the expression of the anti-apoptotic Bcl2 members Bcl-XL and Bcl-w upon serum deprivation
Next, we investigated whether Nex1 could promote survival of serum-deprived PC12-Nex1 cells by modulating the expression of known anti-apoptotic regulators. We initially focused our attention on the multidomain members of the Bcl2 family, Bcl-XL and Bcl-w, as they are known to regulate the survival of NGF-dependent sensory neurons (Middleton et al. 2001). Furthermore, the expression of the Bcl-XL isoform is NGF dependent in PC12 cells, and is not sustained upon NGF withdrawal (Rong et al. 1999). In this study, we did not examine the expression of the Bcl2 protein because the Bcl2 gene is not expressed in naïve PC12 cells, and is expressed only at very low levels in NGF-treated PC12 cells, which do not appear to be sufficient to promote neuronal survival (Batistatou et al. 1993; Rong et al. 1999). For the Bcl-XL immunoblot analysis, we used a polyclonal antibody raised against the 63 amino acids present only in the Bcl-XL isoform, to avoid cross-reaction with Bcl-XS (Table 1).
Figure 3(a) shows that PC12-Nex1 and control PC12-bsd cells expressed very low levels of Bcl-XL in the presence of serum. However, only the serum-deprived PC12-Nex1 cells displayed a dramatic increase in Bcl-XL expression after 2 days without serum, followed by a sustained expression up to 6 days after serum deprivation, before declining to residual levels after 15 days of treatment (Fig. 3a). Interestingly, we detected an additional major band migrating around 22 kDa that most probably corresponds to the Bcl-XΔ™ isoform, based on its apparent molecular weight and the presence of the 63-amino acid domain containing the BH1 and BH2 conserved domains critical for anti-apoptotic properties (Fig. 3a). This Bcl-xΔ™ splice variant was originally discovered in murine pre-B cell lines and found to rescue an interleukin-3-dependent cell line from apoptosis following growth factor withdrawal (Fang et al. 1994). Furthermore, this study revealed that high levels of Bcl-xΔ™ expression were also detected in adult brain. In serum-deprived PC12-Nex1 cells, its expression preceded that of Bcl-xL, as significant levels were detected by 8 h after withdrawal of trophic factors. Its expression peaked at day 2 and remained detectable even after 15 days of serum deprivation, a time at which Bcl-xL expression returned to negligible levels (Fig. 3a). The membrane was re-probed with the anti-βIII tubulin antibody to confirm equal protein loading (Fig. 3a).
Fig. 3.
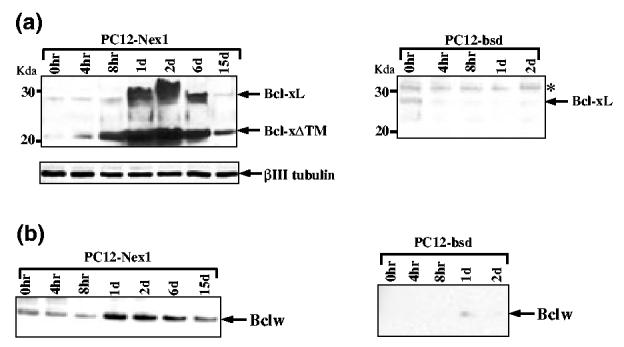
Expression profile of the anti-apoptotic Bcl2 members upon withdrawal of trophic factors. (a) Constitutive expression of Nex1 induces expression of Bcl-XL and Bcl-XΔ™ proteins in serum-deprived PC12-Nex1 cells. PC12-Nex1 and PC12-bsd cells were grown in the absence of serum for periods of time indicated above each lane. Whole-cell extracts were prepared and equal amounts of total proteins (40 μg) were electrophoresed and subjected to immunoblot analysis with a polyclonal antibody raised against the 63-amino acid domain specific to the splice variant Bcl-XL to avoid cross-reaction with Bcl-XS. Equal loading was verified by staining the membrane with Ponceau S after transfer and by probing with the β-III tubulin antibody. Molecular weight markers are indicated on the left side of each figure. The asterisk indicates a non-specific anti-Bcl-XL immunoreactive polypeptide migrating more slowly than the Bcl-XL protein, and serves as an internal control for equal loading. (b) Constitutive expression of Nex1 results in increased expression of Bcl-w upon serum deprivation. Immunoblot analyses were performed as described above after stripping the membrane before probing with an anti-Bcl-w polyclonal antibody that does not cross-react with other Bcl2 proteins. Data shown are representative of at least three independent experiments.
Finally, we observed an increased expression of the Bcl-w protein, although this was more modest than that of Bcl-xL, in serum-deprived PC12-Nex1 cells, which was sustained even after 15 days of serum removal (Fig. 3b). In contrast, the control PC12-bsd cells failed to express the Bcl-w protein upon withdrawal of trophic factors (Fig. 3b). Interestingly, PC12-Nex1 cells grown in serum expressed substantial levels of the Bcl-w protein, suggesting that Nex1 might directly modulate the expression of the Bcl-w gene. Thus, Nex1 may promote neuronal survival of PC12-Nex1 cells by inducing the expression of a combination of key anti-apoptotic regulators, such as Bcl-w, Bcl-XL and its splice variant Bcl-XΔ™, upon serum deprivation. Furthermore, these factors displayed a distinct temporal pattern of expression, suggesting that they may play different roles at specific stages of the survival process.
Nex1 stimulates the expression of XIAP and survivin in serum-deprived PC12-Nex1 cells
Previous studies have reported that upon serum deprivation activation of both caspase-2 and caspase-3 is observed in apoptotic cell death of naïve PC12, resulting in cleavage of apoptotic substrates, such as poly (ADP-ribose) polymerase (PARP) (Haviv et al. 1998; Stefanis et al. 1998). We therefore examined whether Nex1 could exert its anti-apoptotic effects by inducing the expression of members of the inhibitor of apoptosis (IAP) family, which are known to interfere with caspase activity (Deveraux and Reed 1999). We turned our attention to XIAP, which behaves as the most potent inhibitor of caspase-3 activity (Deveraux et al. 1997; Roy et al. 1997), and to survivin, which may target caspase-2 activity (Beltrami et al. (2004). Immunoblotting analysis revealed that both IAP regulators were expressed in serum-deprived PC12-Nex1 cells, albeit with distinct temporal patterns (Figs 4a and b). We observed a steady level of XIAP expression throughout serum deprivation treatment of PC12-Nex1 cells (Fig. 4a). In contrast, survivin expression was triggered after 8 h of serum deprivation, and peaked by 24 h before gradually declining to lower but still substantial levels after 15 days of treatment (Fig. 4b). Serum-deprived PC12-bsd cells expressed only negligible levels of survivin throughout the treatment.
Fig. 4.
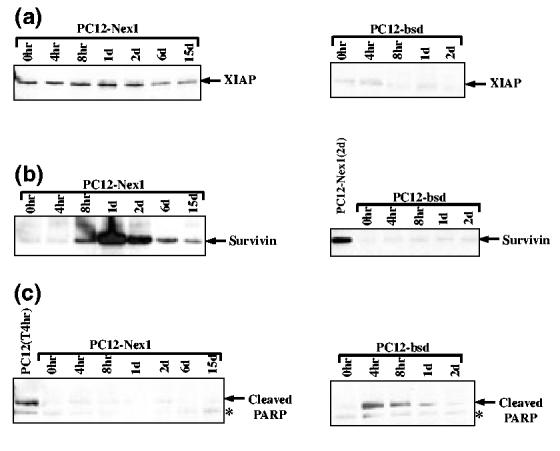
Overexpression of Nex1 induces the expression of members of the IAP family. (a) XIAP remains expressed in serum-deprived PC12-Nex1 cells. PC12-Nex1 and PC12-bsd cell extracts were prepared at different times during serum deprivation as indicated above each panel and subjected to immunoblot analysis using an anti-XIAP antibody. The antigen—antibody complexes were detected by chemiluminescence. (b) Survivin expression is only triggered upon serum deprivation of PC12-Nex1 cells. PC12-Nex1 and PC12-bsd cell extracts were prepared as described above and survivin expression was examined by western blotting. The antigen—antibody complexes were detected by chemiluminescence. (c) Serum deprivation of PC12-Nex1 cells does not induce PARP cleavage. PC12-Nex1 and PC12-bsd cell extracts were prepared and subjected to immunoblot analysis using a polyclonal antibody that only recognizes the large cleaved fragment of the rat PARP protein. The asterisk indicates a non-specific anti-PARP immunoreactive polypeptide migrating slightly lower than the large cleaved PARP fragment.
As a measure of a lack of endogenous caspase activity, we monitored PARP cleavage in serum-deprived PC12-Nex1 cells and control PC12-bsd cells. As shown in Fig. 4(c), we observed PARP cleavage only in serum-deprived PC12-bsd cells, with a similar time course to that previously reported (Stefanis et al. 1996; Francçois et al. 2001). Thus, our results suggest that both IAP regulators may function as key players in the Nex1-mediated anti-apoptotic cascade leading to long-term survival of serum-deprived PC12-Nex1 cells.
Activation of Akt is not required for the sustained survival effects of Nex1
It was previously suggested that the PI3K signaling pathway might be one of the pathways participating in growth factor-induced survival of PC12 cells grown in serum-containing medium (Yao and Cooper 1995). We therefore examined whether the PI3K-Akt intracellular signaling cascade is involved in the Nex1-mediated survival of serum-deprived PC12-Nex1 cells. PI3K generates phosphatidylinositol 3,4,5-triphosphate, which in turn activates Akt resulting in the phosphorylation of the Ser-473 residue (Alessi et al. 1996). This activated form of Akt influences the activity of Bad, caspase-9, CAMP-response element binding protein (CREB), nuclear factor (NF)-κB and forkhead transcription factors to promote neuronal survival (Datta et al. 1999). To ascertain the status of Akt at distinct phases of the Nex1-mediated survival cascade, we made use of a monoclonal antibody that specifically recognizes the Akt protein phosphorylated at Ser-473 (Table 1). PC12-Nex1 cells grown in the presence of serum failed to express detectable levels of phosphorylated Akt, and withdrawal of trophic factors resulted in a negligible modulation of active Akt expression (Fig. 5a). In contrast, total levels of Akt protein remained raised and reasonably constant throughout serum deprivation, indicating that the majority of Akt protein present in serum-deprived PC12-Nex1 cells was unphosphorylated (Fig. 5a). Furthermore, we did not detect the formation of an Akt cleavage product with an apparent molecular weight of 49 kDa in serum-deprived PC12-Nex1 cells, reflecting a lack of caspase-3 activity, as caspase-3 has the ability to cleave signaling molecules promoting cell survival such as Akt (Bachelder et al. 1999; Francçois and Grimes 1999). These results are in agreement with the PARP cleavage data shown in Fig. 4(c).
Fig. 5.
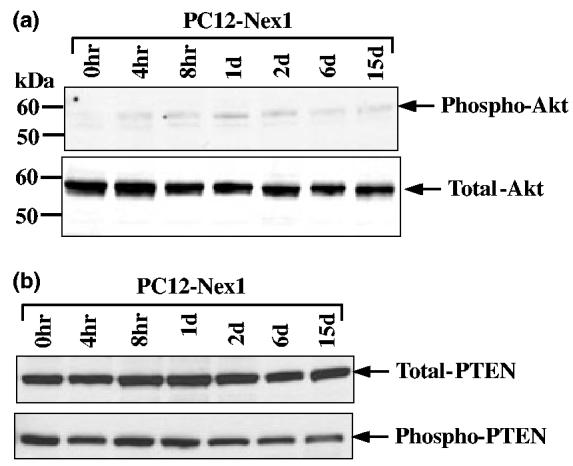
Nex1 promotes long-term survival of serum-deprived Pc12-Nex1 cells, independently of the Akt pathway. (a) Serum deprivation of PC12-Nex1 cells does not lead to phosphorylation of Akt at Ser-437. PC12-Nex1 cells were deprived of serum for the times indicated above the panel and equal amounts of total protein were subjected to immunoblot analysis. Amounts of total and phosphorylated Akt were detected by using anti-Akt and anti-phosphorylated Akt (Ser-437) antibodies respectively. The antigen—antibody complexes were detected by chemiluminescence. The membrane was stripped before probing with the anti-Akt antibody. Molecular weight markers are indicated on the left side of each figure. Data shown are representative of at least three independent experiments. (b) The majority of PTEN protein exists in an inactive phosphorylated form in serum-deprived PC12-Nex1 cells. PC12-Nex1 cells were deprived of serum for times indicated above the panel and whole-cell extracts were analyzed by immunoblotting. Amounts of total and phosphorylated PTEN were detected by using anti-PTEN and anti-phosphorylated PTEN (Ser-380) antibodies respectively. The antigen—antibody complexes were detected by chemiluminescence. The membrane was stripped before probing with the anti-phosphorylated PTEN antibody. Data shown are representative of at least three independent experiments.
We next explored whether the lack of activated Akt was a consequence of the negative regulation by phosphate and tensin homolog (PTEN), a lipid phosphatase that dephosphorylates phosphatidylinositol 3,4,5-triphosphate and antagonizes the PI3K pathway to prevent activation of Akt (Vasquez and Sellers 2000). Moreover, PTEN antagonistic activities are negatively regulated by casein kinase 2, which phosphorylates the PTEN tail and induces an inactive ‘open’ conformation to prevent its membrane localization (Vasquez et al. 2000, 2001). Immunoblot analyses showed that the total amount of PTEN protein remained constant throughout the serum deprivation treatment and that the PTEN protein existed predominantly in a phosphorylated (Ser-380) and inactive state before and after withdrawal of trophic factors, except at day 15 when a slight decline in phosphorylated PTEN was observed (Fig. 5b). Collectively these results indicate that the lack of Akt activation observed in serum-deprived PC12-Nex1 cells is not a reflection of the negative regulation by PTEN, but is rather due to a lack of PI3K signaling. Taken together, these findings reveal that activation of Akt is not required for the sustained survival effects of Nex1 upon withdrawal of trophic factors.
Discussion
Many studies have demonstrated that neurotrophic factors are critical to neuronal differentiation and survival. How they each contribute to the specificity and survival of various neuronal lineages is under intense scrutiny, as the elucidation of their molecular mechanisms will most likely lead to therapeutic strategies. In our previous study, we showed that in PC12 cells Nex1 is an important effector of the NGF-mediated differentiation pathway (Uittenbogaard and Chiaramello 2002). In the present report, we extend our study by addressing the question of whether Nex1 can substitute for the NGF effect in promoting neuronal survival. Numerous studies on neuronal survival have shown that growth factors are key modulators of the apoptotic/anti-apoptotic equilibrium. Such regulation has been shown to occur at all levels controlling gene expression and protein activity, from transcription to post-translational modifications. At the transcriptional level, critical studies have shown that neuronal death during vertebrate development occurs through a lack of trophic support, and leads to the activation of the core apoptotic machinery in a transcription-dependent mechanism (Oppenheim 1991). Examination of the transcriptional mechanism regulating either the pro-apoptotic or anti-apoptotic pathways in different cellular systems has shown a rather conserved use of transcription factors. These include the activation of c-Jun, p53, Forkhead-FOXO and E2F by the pro-apoptotic pathway, and the activation of CREB and NF-κB by the anti-apoptotic pathway. Overall, all these transcription factors behave as immediate regulators of gene expression through the mediation of a conserved apoptotic/anti-apoptotic signaling network, which necessitates a rapid response (Finkbeiner 2000; Mattson and Camandola 2001).
In contrast, this report provides evidence that the transcription-dependent anti-apoptotic program is regulated at an additional level, which involves the intrinsic programming of neuronal specificity. First, we demonstrate that constitutive expression of Nex1 prevents cell death of PC12-Nex1 cells upon trophic factor withdrawal, whereas naïve control PC12 cells undergo rapid apoptosis. Second, serum-deprived PC12-Nex1 cells remain metabolically active and retain their neuronal phenotype throughout the entire duration of starvation. Third, Nex1 induces the expression of several G1 phase CDK inhibitors, such as p21Cip1, p27kip1 and p16INK4a in the presence of serum, and thereby conditions the PC12-Nex1 cells to adequately undergo cell cycle arrest upon withdrawal of trophic factors. Fourth, Nex1 triggers the expression of two distinct anti-apoptotic regulators, Bcl-w and XIAP, in the absence of any apoptotic stimulus. Finally, upon serum withdrawal, Nex1 induces an Akt-independent dynamic and comprehensive anti-apoptotic response that acts at different levels in the mitochondrial pathway, as discussed below.
The anti-apoptotic Bcl2-related proteins Bcl-XL and Bcl-w display distinct kinetics of expression suggesting that they are differentially regulated upon withdrawal of trophic factors. The fact that the Bcl-w protein is steadily expressed in the presence and absence of serum raises the possibility of a direct Nex1-mediated regulation of the Bcl-w gene. In contrast, the Nex1-mediated modulation of the expression of both the Bcl-XL protein and its splice variant Bcl-XΔ™ appears to be indirect upon serum removal. It is noteworthy that the expression levels of both anti-apoptotic isoforms undergo a dramatic fluctuation from barely detectable to striking levels within 24 h of serum removal. However, only the Bcl-XΔ™ protein remains expressed during long-term survival, suggesting that Bcl-XL activity is no longer necessary at this late phase of the survival process. Previous studies on the regulation of Bcl-X gene expression revealed a complex and cellular context-dependent modulation mediated by the control of several transcription factors, such as the Pit-Oct-Unc (POU) transcription factor Brn-3a, p53 and NF-κB (Glasgow et al. 2000; Sugars et al. 2001). Thus, it is plausible that the complex transcriptional network regulated by Nex1 may interface with these transcription factors, and ultimately affect the expression and splicing pattern of the Bcl-X gene. The expression of the transmembrane-deleted splice variant Bcl-XΔ™ was originally discovered in activated T and B lymphocytes. It was shown to be particularly abundant in the mouse brain, diffusely expressed in the cytosol, and to possess anti-apoptotic properties (Fang et al. 1994). Therefore, the concomitant expression of both splice variants most likely provides an enhanced survival phenotype by recruiting the anti-apoptotic machinery in two separate intracellular pools, one membrane bound and the other cytosolic, with complementary functions.
The dynamic and distinct expression profile of the anti-apoptotic Bcl-2-related proteins suggests that Nex1-mediated survival requires an appropriate combination of key regulators, an optimal expression level, and a specific temporal pattern of expression throughout the survival process. In support of this concept is the in vivo analysis of the regulatory effect of the Bcl-XL and Bcl-w proteins on the survival of nodose and trigeminal sensory neurons. This study revealed that their effectiveness in promoting neuronal survival is age dependent and correlates with their abundance and pattern of expression during brain development, Bcl-X being predominantly expressed in differentiating embryonic neurons and Bcl-w expression persisting in mature neurons (Hamneér et al. 1999; Middleton et al. 2001). This may partly explain why overexpression of either Bcl2 or Bcl-XL alone does not promote long-term survival of PC12 cells (Batistatou et al. 1993; Rong et al. 1999).
Another aspect of the regulatory impact of the anti-apoptotic Bcl-2-related proteins their involvement in maintaining mitochondrial integrity to prevent cytochrome c leakage into the cytosol and facilitate mitochondrial exchange of metabolites and ATP/ADP. A study of the constitutive expression of Bcl-XL in non-transformed pro-B cells FL5.12 has shown that it promotes cell survival by modulating intrinsic bioenergetics pathways, which allow the cells to be less dependent on extracellular nutrients, and to overcome reduced glycolytic activity upon growth factor deprivation (Plas et al. 2001). Taken together, these results suggest that upon serum removal Nex1 is able to coordinate survival by positively affecting the metabolic activity of mitochondria, thereby promoting long-term survival. Such a physiological adaptation may be necessary during neuronal development when neurons need to survive before making contact with their target cells, and during the period of synaptogenesis that requires a high concentration of mitochondria to generate enough energy. Likewise, it may also be necessary for the survival of axotomized neurons as well as a prerequisite for CNS regeneration (Goldberg and Barres 2000).
Our results also indicate that Nex1 suppresses serum deprivation-induced apoptosis by modulating the expression of XIAP and survivin, two members of the IAP family, known to interfere with the activities of specific caspases (Deveraux and Reed 1999). XIAP has been shown to behave as the most potent inhibitor of both the initiator caspase-9 and the effector caspase-3 and caspase-7 (Deveraux et al. 1997; Roy et al. 1997). Of interest is the observation that XIAP is expressed in PC12-Nex1 cells in the absence of an apoptotic stimulus, suggesting a possible direct regulation of the XIAP gene by Nex1. Furthermore, XIAP expression remains constant throughout the entire serum deprivation treatment, which may be a consequence of a combined transcriptional and translational regulation. It has been demonstrated that under cellular stress, such as serum starvation, the expression of XIAP is controlled at the translation initiation via a potent internal ribosome entry site element located in the 5′ untranslated region of the XIAP gene (Holcik et al. 1999). In contrast, little is known about the transcriptional regulation of the XIAP gene. However, an additional IAP protein is most probably involved in this regulation, because XIAP does not inhibit the activity of caspase-2, and serum-deprived PC12 cells undergo apoptosis in a caspase-2 dependent manner (Troy et al. 1997; Haviv et al. 1998; Stefanis et al. 1998). A likely candidate is survivin, which confers cytoprotection by regulating the upstream events of the mitochondrial-dependent pathway, and is believed to target caspase-2 (Beltrami et al. 2004). In our experimental paradigm, survivin expression is triggered within hours of serum deprivation and remains at substantial levels after 15 days of treatment. This dynamic expression pattern implies that survivin expression can be modulated independently of cell cycle progression. However, this stands in contrast with the fact that its expression is cell cycle-regulated at the G2/M phase (Li et al. 1998). Normally, its expression is undetectable in differentiated neurons, except when they are transformed, such as in neuroblastoma cells associated with an unfavorable prognosis (Adida et al. 1998). In the G1 phase of cycling cells, survivin expression is down-regulated at the transcriptional level via the cell cycle dependent element (CDE)/cell cycle homology region (CHR) G1-repressor elements (Li and Altieri 1999). This suggests that in quiescent serum-deprived PC12-Nex1 cells the survivin gene is regulated by a different transcriptional mechanism in which the G1 repressor is inactive, thus allowing distinct positive regulators to stimulate survivin promoter activity. It is also possible that the increased expression of the survivin protein may partly result from a post-translational regulation affecting its half-life. In cycling cells, survivin is short-lived with a half-life of 30 min owing to ubiquitination and degradation by the ubiquitin-protea-some pathway (Zhao et al. 2000). Thus, our study provides the first evidence that survivin is a regulatory component of the neuronal survival pathway.
Another important facet of the apoptotic/anti-apoptotic network is the link to the cell cycle. Several studies on PC12 cells and sympathetic neurons have shown that after loss of trophic support inadequate cell cycle withdrawal or inappropriate cell cycle re-entry leads to apoptosis, suggesting a key participation by the cyclin-CDK machinery (Rukenstein et al. 1991; Farinelli and Greene 1996; Park et al. 1997b). Pharmacologically active G1 blockers delay apoptosis of serum-deprived naïve PC12 cells (Farinelli and Greene 1996; Park et al. 1997b). Furthermore, the CDK inhibitors olomoucine and flavopiridol block cell death of neuronal PC12 cells as well as sympathetic neurons deprived of trophic support (Park et al. 1997a). In accordance with these findings are several additional lines of evidence showing that overexpression of specific CDK inhibitors leads to increased survival. Overexpression of the CDK inhibitor p16INK4a protects differentiated neuroblastoma cells from cell death caused by trophic factor deprivation (Kranenburg et al.1996). Similar neuroprotective effects are observed upon overexpression of p21cip1 and p27kip1 in NGF-deprived sympathetic neurons (Park et al. 1997a). Our study reveals that overexpression of Nex1 results in expression of the p16INK4a, p21cip1 and p27kip1 proteins in serum-grown PC12-Nex1 cells, and consequently conditions the cells to adequately undergo cell cycle arrest upon withdrawal of trophic factors. Moreover, the fact that the expression of the tumor suppressor p16INK4a protein is increased and sustained upon serum removal is in accordance with a recent report demonstrating its critical role in G1 phase growth arrest (Ohtani et al. 2001). Finally, several studies have demonstrated that members of the NeuroD family promote cell cycle exit during the differentiation program, by direct regulation of the p21cip1 and p27kip1 genes (Farah et al. 2000; Uittenbogaard and Chiaramello 2002; Liu et al. 2004). However, none of these studies has yet reported their roles in regulating neuronal survival through the anti-apoptotic pathway. Our study therefore strengthens the hypothesis that the anti-apoptotic/apoptotic network is tightly linked to the cell cycle.
Of interest are the complementary studies demonstrating that the Bcl-2-related proteins Bcl2 and Bcl-xL also influence cell cycle arrest in the absence of trophic factors by a mechanism that remains unclear. Overexpression of Bcl-2 in hemopoietic cells not only leads to survival upon growth factor deprivation, but also hastens cell cycle arrest in the G0 phase (Vaux et al. 1988; Nunez et al. 1990; Vairo et al.1996). One line of study actually suggests that Bcl-2 has a cell cycle inhibitory function involving the phosphorylation of the conserved Tyr-28 which is separate from its survival functions; however, this finding remains controversial (Huang et al. 1997; Janumyan et al. 2003). Additional evidence has linked the overexpression of Bcl-2 with the up-regulation of two essential cell cycle arrest mediators, the pRB relative p130 and p27kip1, leading to the speculation that Bcl-2 may be producing repressive p130-E2F4 complexes to inhibit the G1-S phase transition (Vairo et al. 2000). Thus, it is plausible that high concentrations of Bcl-xL and Bcl-xΔ™ combined with their specific temporal expression during serum deprivation of PC12-Nex1 cells may be needed to drive the cells in the G1 phase of the cell cycle. Modulation of the cell cycle has also been extended to XIAP, which induces cell cycle arrest of vascular endothelial cells through the activation of NF-κB transcription factor and induction of the CDK inhibitor proteins p21cip1 and p27kip1 (Levkau et al.2001). Finally, whether survivin mediates cell cycle arrest remains to be further investigated.
In conclusion, we present compelling evidence that Nex1 mediates long-term neuronal survival by orchestrating several distinct but interconnected programs, such as neuronal differentiation, cell cycle withdrawal and the anti-apoptotic pathway. This report provides the first evidence that a NeuroD member promotes survival by modulating dynamic expression of key anti-apoptotic proteins. More importantly, our results combined with those of knockout studies imply that the anti-apoptotic program needs to be activated in a transcription-dependent manner during the establishment of the differentiation process, and sustained during maintenance of the differentiated state. Finally, our results highlight the existence of an additional regulatory layer contributing to the neuronal survival pathway, suggesting that the survival program can be viewed as an integral component of the intrinsic programming of differentiation. Together, our results may have profound implications regarding the design of novel therapeutic strategies for neurodegenerative disorders and CNS regeneration.
Acknowledgements
We thank Dr John Reed for critically reviewing the manuscript and for his helpful suggestions. We also thank Dr Robyn Rufner for her technical help at the Center for Microscopic Image Analysis. This work was supported in part by National Institutes of Health grant R01-NS41391 to AC and by P30HD40677.
Abbreviations used:
- Bcl2
B-cell leukemia/lymphoma 2
- BDNF
brain-deprived neurotrophic factor
- bHLH
basic helix-loop-helix
- CDE
cell cycle dependent element
- CDK
cyclin-dependent kinase
- CHR
cell cycle homology region
- CREB
c-amp response element binding protein
- GAP-43
growth-associated protein-43
- IAP
inhibitor of apoptosis
- LDH
lactate dehydrogenase
- MAPK
mitogen-activated protein kinase
- MCM
minichromosome maintenance; NF-κB
- NF-κB
nuclear factor kappa B
- NGF
nerve growth factor
- PARP
poly (ADP-ribose) polymerase
- PI3K
phosphatidylinositol 3-kinase
- POU
Pit-Oct-Unc
- PTEN
phosphate and tensin homolog
- SDS
sodium dodecyl sulfate
- TUNEL
terminal deoxynucleotidyl transferase αUTP nick-end labeling
- XIAP
X-linked inhibitor of apoptosis
- WST-1
4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate
References
- Adida C, Berrebi D, Peuchmaur M, Reyes-Mugica M, Altieri DC. Anti-apoptosis gene, survivin, and prognosis of neuroblastoma. Lancet. 1998;351:882–883. doi: 10.1016/S0140-6736(05)70294-4. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Bachelder RE, Ribick MJ, Marchetti A, Falcioni R, Soddu S, Davis KR, Mercurio AM. p53 inhibits α6β4 integrin survival signaling by promoting the caspase 3-dependent cleavage of AKT/PKB. J. Cell Biol. 1999;147:1063–1072. doi: 10.1083/jcb.147.5.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholoma A, Nave K-A. Nex1: a novel brain-specific helix-loop-helix protein with autoregulation and sustained expression in mature cortical neurons. Mech. Dev. 1994;48:217–228. doi: 10.1016/0925-4773(94)90061-2. [DOI] [PubMed] [Google Scholar]
- Batistatou A, Merry DE, Korsmeyer SJ, Greene LA. Bcl-2 affects survival but not neuronal differentiation of PC12 cells. J. Neurosci. 1993;13:4422–4428. doi: 10.1523/JNEUROSCI.13-10-04422.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrami E, Plescia J, Wilkinson JC, Duckett CS, Altieri DC. Acute ablation of survivin uncovers p53-dependent mitotic checkpoint functions and control of mitochondrial apoptosis. J. Biol. Chem. 2004;279:2077–2084. doi: 10.1074/jbc.M309479200. [DOI] [PubMed] [Google Scholar]
- Bertrand N, Castro DS, Guillemot F. Proneural genes and the specification of neural cell types. Nat. Rev. Neurosci. 2002;3:517–530. doi: 10.1038/nrn874. [DOI] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Deveraux QL, Reed JC. IAP family proteins - suppressors of apoptosis. Genes Dev. 1999;13:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- Fang W, Rivard JJ, Mueller DI, Behrens TW. Cloning and molecular characterization of mouse bcl-x in B and T lymphocytes. J. Immunol. 1994;153:4388–4398. [PubMed] [Google Scholar]
- Farah MH, Olson JM, Sucic HB, Hume RI, Tapscott SJ, Turner DL. Generation of neurons by transient expression of neural bHLH proteins in mammalian cells. Development. 2000;127:693–702. doi: 10.1242/dev.127.4.693. [DOI] [PubMed] [Google Scholar]
- Farinelli SE, Greene LA. Cell cycle blockers mimosine, ciclopirox, and deferoxamine prevent the death of PC12 cells and postmitotic sympathetic neurons after removal of trophic factor. J. Neurosci. 1996;16:1150–1162. doi: 10.1523/JNEUROSCI.16-03-01150.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S. CREB couples neurotrophin signals to survival messages. Neuron. 2000;25:11–14. doi: 10.1016/s0896-6273(00)80866-1. [DOI] [PubMed] [Google Scholar]
- Francçois F, Grimes ML. Phosphorylation-dependent Akt cleavage in neural cell in vitro reconstitution of apoptosis. J. Neurochem. 1999;73:1773–1776. doi: 10.1046/j.1471-4159.1999.731773.x. [DOI] [PubMed] [Google Scholar]
- Francçois F, Godinho MJ, Dragunow M, Grimes ML. A population of PC12 cells that is initiating apoptosis can be rescued by nerve growth factor. Mol. Cell. Neurosci. 2001;18:347–362. doi: 10.1006/mcne.2001.1035. [DOI] [PubMed] [Google Scholar]
- Fujita M, Kiyono T, Hayashi Y, Ishibashi M. hCDC47, a human member of the MCM family. J. Biol. Chem. 1996;271:4349–4354. doi: 10.1074/jbc.271.8.4349. [DOI] [PubMed] [Google Scholar]
- Glasgow JN, Wood T, Perez-Polo JR. Identification and characterization of nuclear factor κB binding sites in the murine bcl-x promoter. J. Neurochem. 2000;75:1377–1389. doi: 10.1046/j.1471-4159.2000.0751377.x. [DOI] [PubMed] [Google Scholar]
- Goldberg JL, Barres BA. The relationship between neuronal survival and regeneration. Annu. Rev. Neurosci. 2000;23:579–612. doi: 10.1146/annurev.neuro.23.1.579. [DOI] [PubMed] [Google Scholar]
- Greene LA. Nerve growth factor prevents the death and stimulates neuronal differentiation of clonal PC12 pheochromocytoma in serum-free medium. J. Cell Biol. 1978;78:747–755. doi: 10.1083/jcb.78.3.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl Acad. Sci. USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamneér S, Skogloösa Y, Lindholm D. Differential expression of bcl-w and Bcl-X messenger RNA in the developing and adult rat nervous system. Neuroscience. 1999;91:673–684. doi: 10.1016/s0306-4522(98)00642-3. [DOI] [PubMed] [Google Scholar]
- Haviv R, Lindenboim L, Yuan J, Stein R. Need for caspases-2 in apoptosis of growth-factor-deprived PC12 cells. J. Neurosci. Res. 1998;52:491–497. doi: 10.1002/(SICI)1097-4547(19980601)52:5<491::AID-JNR1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Holcik M, Lefebre CA, Yeh C, Chow T, Korneluk RG. A new internal-ribosome-entry-site motif potentiates XIAP-mediated cytoprotection. Nat. Cell. Biol. 1999;1:190–192. doi: 10.1038/11109. [DOI] [PubMed] [Google Scholar]
- Huang DCS, O'Reilly LA, Strasser A, Cory S. The anti-apoptosis function of Bcl-2 can be genetically separated from its inhibitory effect on cell cycle entry. EMBO J. 1997;16:4628–4638. doi: 10.1093/emboj/16.15.4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janumyan YM, Sansam CG, Chattopadhyay A, Cheng N, Soucie EL, Penn LZ, Andrews D, Knudson MC, Yang E. Bcl-XL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. EMBO J. 2003;22:5459–5470. doi: 10.1093/emboj/cdg533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W-Y, Fritzsch B, Seris A, Bakel LA, Huang EJ, Reichardt LF, Barth DS, Lee JE. NeuroD-null mice are deaf due to a severe loss of the inner ear sensory neurons during development. Development. 2001;128:417–426. doi: 10.1242/dev.128.3.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein JA, Longo-Guess CM, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL. The harlequin mouse mutation down-regulates apoptosis-inducing factor. Nature. 2002;419:367–374. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- Kranenburg O, van der Eb AJ, Zantena A. Cyclin D1 is an essential mediator of apoptotic neuronal cell death. EMBO J. 1996;15:46–54. [PMC free article] [PubMed] [Google Scholar]
- Levkau B, Garton KL, Ferri N, Kloke K, Nofer J-R, Baba HA, Raines EW, Breithard G. xIAP induces cell-cycle arrest and activates nuclear factor-κB. Circ. Res. 2001;88:282–290. doi: 10.1161/01.res.88.3.282. [DOI] [PubMed] [Google Scholar]
- Li F, Altieri DC. Transcriptional analysis of human survivin gene expression. Biochem. J. 1999;344:305–311. doi: 10.1042/0264-6021:3440305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, Altieri DC. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580–584. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- Liu M, Pereira FA, Price SD, Chu M-J, Shope C, Himes D, Eatock RA, Brownell WE, Lysakowski A, Tsai M-J. Essential role of Beta2/NeuroD1 in development of the vestibular and auditory systems. Genes Dev. 2000;14:2839–2854. doi: 10.1101/gad.840500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Encinas M, Comella JX, Aldea M, Gallego C. Basic helix-loop-helix proteins bind to TrkB and p21Cip1 promoters linking differentiation and cell cycle arrest in neuroblastoma cells. Mol. Cell. Biol. 2004;24:2662–2672. doi: 10.1128/MCB.24.7.2662-2672.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Camandola S. NF-kappaB in neuronal plasticity and neurodegenerative disorders. J. Clin. Invest. 2001;107:247–254. doi: 10.1172/JCI11916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton G, Wyatt S, Ninkina N, Davies AM. Reciprocal developmental changes in the roles of Bcl-w and Bcl-XL in regulating sensory neuron survival. Development. 2001;128:447–457. doi: 10.1242/dev.128.3.447. [DOI] [PubMed] [Google Scholar]
- Miyata T, Maeda T, Lee JE. NeuroD is required for differentiation of the granule cells of the cerebellum and hippocampus. Genes Dev. 1999;13:1647–1652. doi: 10.1101/gad.13.13.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez G, London L, Hockenberry D, Alexander M, McKearn JP, Korsmeyer SJ. Deregulated Bcl-2 gene expression selectively prolongs survival of growth factor-deprived hemopoietic cell lines. J. Immunol. 1990;144:3602–3610. [PubMed] [Google Scholar]
- Ohtani N, Zebedee Z, Huot TJG, Stinson JA, Sugimoto M, Oshashi Y, Sharrocks AD, Peters G, Hara E. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature. 2001;409:1067–1070. doi: 10.1038/35059131. [DOI] [PubMed] [Google Scholar]
- Olson JM, Asakura A, Snider L, Hawkes R, Strand A, Stoeck J, Hallahan A, Pritchard J, Tapscott SJ. NeuroD2 is necessary for development and survival of central nervous system neurons. Dev. Biol. 2001;234:174–187. doi: 10.1006/dbio.2001.0245. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Annu. Rev. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Park DS, Levine B, Ferrari G, Greene LA. Cyclin dependent kinase inhibitors and dominant negative cyclin dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J. Neurosci. 1997a;17:8975–8983. doi: 10.1523/JNEUROSCI.17-23-08975.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DS, Morris E, Greene LA, Geller HM. G1/S cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis. J. Neurosci. 1997b;17:1256–1270. doi: 10.1523/JNEUROSCI.17-04-01256.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennesi ME, Cho J-H, Yang Z, Wu SH, Zhang J, Wu SM, Tsai M-J. Beta/NeuroD1 null mice: a new model for transcription factor-dependent photoreceptor degeneration. J. Neurosci. 2003;23:453–461. doi: 10.1523/JNEUROSCI.23-02-00453.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plas DR, Talapatra S, Edinger AL, Rathmell JC, Thompson CB. Akt and Bcl-XL promote growth factor-independent survival through distinct effects on mitochondrial physiology. J. Biol. Chem. 2001;276:12041–12048. doi: 10.1074/jbc.M010551200. [DOI] [PubMed] [Google Scholar]
- Rong P, Bennie AM, Epa WR, Barret GL. Nerve growth factor determines survival and death of PC12 cells by regulation of the bcl-x, bax, and caspases-3 genes. J. Neurochem. 1999;72:2294–2300. doi: 10.1046/j.1471-4159.1999.0722294.x. [DOI] [PubMed] [Google Scholar]
- Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J. 1997;16:6914–6925. doi: 10.1093/emboj/16.23.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rukenstein A, Rydel RE, Greene LA. Multiple agents rescue PC12 cells from serum-free death by translation-and transcription independent mechanisms. J. Neurosci. 1991;11:2552–2563. doi: 10.1523/JNEUROSCI.11-08-02552.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab MH, Druffel-Augustin S, Gass P, Jung M, Klugmann M, Bartholomae A, Rossner MJ, Nave K-A. Neuronal basic helix-loop-helix proteins (Nex1, NeuroD, NDRF): spatio-temporal expression and targeted disruption of the Nex gene in transgenic mice. J. Neurosci. 1998;18:1408–1418. doi: 10.1523/JNEUROSCI.18-04-01408.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab MH, Bartholomae A, Heimrich B, Feldmeyer D, Druffel-Augustin S, Goebbels S, Naya FJ, Zhao S, Frotscher M, Tsai M-J, Nave K-A. Neuronal basic helix-loop-helix proteins (Nex and Beta2/NeuroD) regulate terminal granule cell differentiation in the hippocampus. J. Neurosci. 2000;20:3714–3724. doi: 10.1523/JNEUROSCI.20-10-03714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanis L, Park S, Yan C-YI, Farinelli SE, Troy CM, Shelanski ML, Greene LA. Induction of CPP-32-like activity in PC12 cells by withdrawal of trophic factor. J. Biol. Chem. 1996;271:30 663–30 671. doi: 10.1074/jbc.271.48.30663. [DOI] [PubMed] [Google Scholar]
- Stefanis L, Troy CM, Qi H, Shelanski ML, Greene LA. Capsase-2 (Nedd-2) processing and death of trophic factor-deprived PC12 cells and sympathetic neurons occur independently of caspases-3 (CPP32)-like activity. J. Neurosci. 1998;18:9204–9215. doi: 10.1523/JNEUROSCI.18-22-09204.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugars K, 1 Budhram-Mahadeo V, Pacham G, Latchman DS. A minimal Bcl-x promoter is activated by Brn-3a and repressed by p53. Nucl. Acids Res. 2001;29:4530–4540. doi: 10.1093/nar/29.22.4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy CM, Stefanis L, Greene LA, Shelanski ML. Nedd2 is required for apoptosis after trophic factor withdrawal, but not superoxide dismutase (SOD1) downregulation, in sympathetic neurons and PC12 cells. J. Neurosci. 1997;17:1911–1918. doi: 10.1523/JNEUROSCI.17-06-01911.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uittenbogaard M, Chiaramello A. Constitutive overexpression of the basic helix-loop-helix Nex1/MATH-2 transcription factor promotes neuronal differentiation of PC12 cells and neurite regeneration. J. Neurosci. Res. 2002;67:235–245. doi: 10.1002/jnr.10119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uittenbogaard M, Chiaramello A. Expression profiling upon Nex1/MATH-2-mediated neuritogenesis in PC12 cells and its implication in regeneration. J. Neurochem. 2004;91:1332–1343. doi: 10.1111/j.1471-4159.2004.02814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uittenbogaard M, Martinhu DL, Chiaramello A. The basic helix-loop-helix differentiation-factor Nex1/MATH-2 functions as a key activator of the GAP-43 gene. J. Neurochem. 2003;84:673–683. doi: 10.1046/j.1471-4159.2003.01572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vairo G, Innes KM, Adams JM. Bcl-2 has a cell cycle inhibitory function separable from its enhancement of cell survival. Oncogene. 1996;13:1511–1519. [PubMed] [Google Scholar]
- Vairo G, Soos TJ, Upton TM, Zalvide J, deCaprio JA, Ewen ME, Koff A, Adams JM. Bcl-2 retards cell cycle entry through p27kip1, pRB relative p130, and altered E2F regulation. Mol. Cell. Biol. 2000;20:5745–4753. doi: 10.1128/mcb.20.13.4745-4753.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez F, Sellers WR. The PTEN tumor suppressor protein: an antagonist of phosphoinositide 3-kinase signaling. Biochem. Biophys. Acta. 2000;1470:M21–M35. doi: 10.1016/s0304-419x(99)00032-3. [DOI] [PubMed] [Google Scholar]
- Vasquez F, Grossman SR, Takahashia Y, Rokas MV, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J. Biol. Chem. 2001;276:48 627–48 630. doi: 10.1074/jbc.C100556200. [DOI] [PubMed] [Google Scholar]
- Vasquez F, Ramaswamy S, Nakamura N, Seller WR. Phosphorylation of PTEN tail regulates protein stability and function. Mol. Cell. Biol. 2000;20:5010–5018. doi: 10.1128/mcb.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- Yao R, Cooper GM. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science. 1995;267:2003–2006. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- Zhao J, Tenev T, Martins LM, Downward J, Lemoine NR. The ubiquitin-proteasome pathway regulates survivin degradation in a cell cycle-dependent manner. J. Cell Sci. 2000;113:4363–4371. doi: 10.1242/jcs.113.23.4363. [DOI] [PubMed] [Google Scholar]