

Abstract
Background
The horizontal transfer of expressed genes from Bacteria into Ciliates which live in close contact with each other in the rumen (the foregut of ruminants) was studied using ciliate Expressed Sequence Tags (ESTs). More than 4000 ESTs were sequenced from representatives of the two major groups of rumen Cilates: the order Entodiniomorphida (Entodinium simplex, Entodinium caudatum, Eudiplodinium maggii, Metadinium medium, Diploplastron affine, Polyplastron multivesiculatum and Epidinium ecaudatum) and the order Vestibuliferida, previously called Holotricha (Isotricha prostoma, Isotricha intestinalis and Dasytricha ruminantium).
Results
A comparison of the sequences with the completely sequenced genomes of Eukaryotes and Prokaryotes, followed by large-scale construction and analysis of phylogenies, identified 148 ciliate genes that specifically cluster with genes from the Bacteria and Archaea. The phylogenetic clustering with bacterial genes, coupled with the absence of close relatives of these genes in the Ciliate Tetrahymena thermophila, indicates that they have been acquired via Horizontal Gene Transfer (HGT) after the colonization of the gut by the rumen Ciliates.
Conclusion
Among the HGT candidates, we found an over-representation (>75%) of genes involved in metabolism, specifically in the catabolism of complex carbohydrates, a rich food source in the rumen. We propose that the acquisition of these genes has greatly facilitated the Ciliates' colonization of the rumen providing evidence for the role of HGT in the adaptation to new niches.
Background
Horizontal Gene Transfer (HGT) implicates the transfer of genetic material between species. Genes acquired by this process can provide novel functions to the recipient organism, particularly when that organism is naive for functions associated with the newly acquired gene(s). Therefore, HGT has the potential to play an important role in the exploitation of new niches. HGT has been inferred in many biological processes including the emergence and spread of virulence-factors, resistance to antibiotics, and the long-term maintenance of organelles [1]. Thus far, HGT on a large-scale has mainly been described from organelles to the nucleus transfer [2], and between different species of Bacteria and Archaea [3,4], and on a smaller scale from Eukaryotes to Bacteria [5]. The least well-documented form of large-scale HGT deals with the uptake of DNA into eukaryotic cells. Individual examples include transfer from Bacteria to Fungi [6] or Ciliates [7] in the rumen. The transfer of 16 bacterial genes to Nematodes [8] and of 96 such genes to Entamoeba histolytica [9] are the only examples where HGT from Bacteria to Eukaryotes has been investigated on a large-scale.
Here we investigate HGT from the Bacteria to rumen Ciliates – a monophyletic but rather diverse group of unicellular Eukaryotes. These organisms co-exist in the rumen under conditions that have been shown to allow HGT in vitro [10,11]. Ciliates form an extremely diverse taxonomic group of protozoa with an enormous diversity of known species. They are the most complex single cell Eukaryotes, some having genomes with more than 30,000 genes [12]. They are abundant in almost every aqueous environment, from ocean waters to small ponds and even pockets of soil water; and they can grow as symbionts, commensals or parasites in pelagic, benthic, sapropelic or intestinal ecosystems. One of the intestinal environments in which Ciliates have been described is the rumen, a highly specialized foregut differentiation in herbivorous mammals like cattle, sheep and goats. In these animals the rumen is the primary site for digestion of plant material consumed as a food source. Digestion is performed by a numerous and diverse microbiota including Bacteria, anaerobic Fungi, and Ciliates. The resulting fermentation products, such as short-chain fatty acids, but also the microbial biomass substantially contribute to the nutrition of the host. There is a certain degree of genomic plasticity within this environment, with some evidence for HGT between organisms [6] and one bacterial species being naturally transformable [11].
Given the close contact between Ciliates and Bacteria in the rumen, this environment promises optimal conditions to study HGT from Bacteria to Eukaryotes, in particular, because Ciliates engulf and digest Bacteria [13]. In the process of breakdown and digestion of the Bacteria, some of the bacterial DNA may be taken up by the Ciliates and incorporated into their genomes.
Thus far, there have been reports of HGT to the rumen Ciliates of a xylanase [7], a cellulase [14] and a glutamate dehydrogenase [15]. Whether these are incidental occurrences or whether there is indeed evidence for large-scale HGT from Bacteria to Ciliates within this environment remained to be discovered. Here we have undertaken random cDNA sequencing of rumen dwelling Ciliates in order to identify expressed HGT candidates. As part of the EU-funded programs ERCULE (European Rumen ciliates CULturE collection), and CIMES (CIliates as Monitors for the Environmental Safety of GMOs) cDNA libraries were constructed from ten species of rumen Ciliates and were sequenced randomly (these ciliates were cultivated as mono-cultures in fistulated sheep). Thus we have obtained a large set of cDNAs (4768 sequences) from Ciliates belonging to the order Entodiniomorphida: i.e. Entodinium simplex, Entodinium caudatum, Eudiplodinium maggii, Metadinium medium, Diploplastron affine, Polyplastron multivesiculatum and Epidinium ecaudatum and from Isotricha prostoma, Isotricha intestinalis and Dasytricha ruminantium, which belong to the order Vestibuliferida. Here we examine these EST data for HGT from Bacteria. Using large-scale sequence comparisons and phylogenetic analyses we find 148 genes that are likely to have been horizontally transferred to the Ciliates. The majority of these genes are involved in the catabolism of complex carbohydrates and in the adaptation to an anaerobic environment. This supports the hypothesis that HGT plays an important role in the exploitation of new niches.
Results
Best Hit within the Bacteria
A total of 4768 sequences were generated. Following filtering of sequences (see methods section) we clustered the remaining 4324 ESTs to remove redundancy. Grouping sequences with >97% identity over a stretch of at least 100 nucleotides resulted in 377 clusters containing between two and 21 sequences, and 3186 single sequence clusters (More details in Table 1). In order to find potential cases of HGT, we first performed a Best Hit search (translated nucleotides versus protein) against the predicted proteomes of 148 completely sequenced genomes. Examining the longest sequence per cluster resulted in 2307 clusters out of 3563 (64.75%) with a Best Hit in one of the complete genomes. The species distribution of the Best Hits (Figure 1), ranging from Plasmodium falciparum with the most hits, followed by Arabidopsis thaliana and Danio rerio is in agreement with the phylogeny of the Eukaryotes proposed by Baldauf et al. [16]. According to that phylogeny the sub-class Ciliophora is most closely related to the Apicomplexa (P. falciparum), then to the Viridiplantae (A. thaliana) and then to the Opisthokonts (D. rerio, Homosapiens, Neurosporacrassa).
Table 1.
Species distribution of the ciliate dataset. Indicated are the total number of sequences per species and the number of clusters that were obtained after combining the sequences likely derived from a same gene. 377 clusters have a size varying between 2 and 21 sequences, the rest of the clusters are singletons.
| Organism | Number of sequences | Number of clusters |
number of clusters ≥2 sequences (size maximum of the cluster) |
number of clusters singletons |
| P. multivesiculatum | 715 | 502 | 115 (21) | 387 |
| Epid. ecaudatum | 595 | 530 | 46 (5) | 484 |
| Eud. maggii | 543 | 500 | 32 (4) | 468 |
| I. prostoma | 546 | 442 | 43 (12) | 399 |
| I. intestinalis | 84 | 81 | 2 (3) | 79 |
| Das. ruminantium | 591 | 421 | 61 (20) | 360 |
| Ent. caudatum | 1062 | 901 | 76 (18) | 825 |
| Ent. simplex | 27 | 27 | 0 | 27 |
| Dip. affine | 10 | 10 | 0 | 10 |
| M. medium | 151 | 149 | 2 (2) | 147 |
| TOTAL | 4324 | 3563 | 377 | 3186 |
Figure 1.
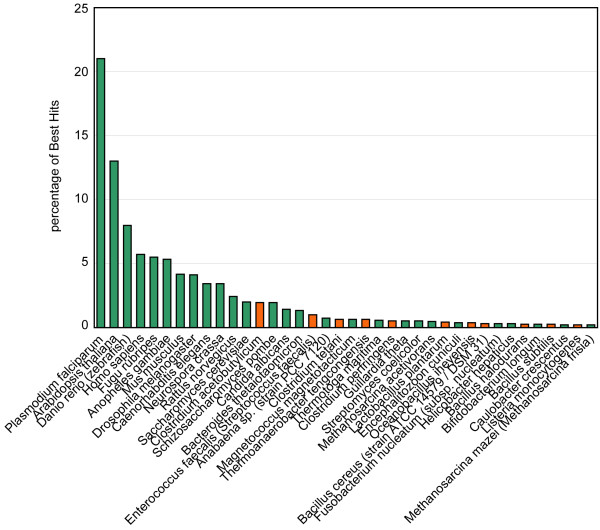
Distribution of Best Hits over the proteomes. Only proteomes with a number of Best Hits ≥ 0.2% are displayed. The 12 most represented proteomes are eukaryotic, the remaining set contains a large fraction of Firmicutes proteomes (orange).
Of the 38 Best Hit proteomes shown in Figure 1, the top 12 proteomes are eukaryotic. Nevertheless, we also found a substantial number of ESTs with a bacterial Best Hit. The Bacterium with the most Best Hits is Clostridiumacetobutylicum, a Firmicute which has previously been isolated from bovine rumen fluid [17]. A total of 11 different Firmicutes were identified with Best Hits, plus nine "other" Bacteria and two Archaea species.
Firmicutes, as with other "intestinal" Bacteria are likely HGT donors because they live in close contact with the studied Ciliates in the gastrointestinal tract of the ruminants [18]. According to Edwards et al., low G+C Gram positive Bacteria represent 54% of the rumen bacterial ecosystem, followed by the Cytophaga-Flexibacter-Bacteroides group (40%) [19]. Nelson et al. (2003) also show that Gram negative Bacteria were poorly represented in the gastrointestinal tract of wild herbivores [20].
A Best Hit approach can only provide an indication of the relationship between sequences in different organisms, and it does not always reflect the closest neighbour [21]. Therefore, we used a phylogenetic approach to further analyse those ciliate sequences which have a Best Hit in the bacterial genomes.
Furthermore we show details of the complete SWX comparison against the 148 proteomes in Additional file 2. Among 292 sequences with a Best Hit in Bacteria, 138 (47%) only hit Bacteria and this number raise to 151 (52%) that hit both Bacteria and Archaea.
Phylogenetic analysis
Of the 362 sequences that have a bacterial sequence as Best Hit, 224 had enough homologs (minimally three) to construct phylogenetic trees (see Methods). In 133 of these 224 trees, the ciliate sequence clusters within the Bacteria (in these trees the second-smallest partition of the tree that contains the ciliate sequence and otherwise only contains Bacterial sequences, see Methods). Further examination of these 133 trees shows that in 34 trees the ciliate sequence clusters within Firmicutes, in nine trees within Proteobacteria, in three trees within Actinobacteria, in three trees within Bacteroidetes and in one tree within Spirochetes. In the remaining 83 trees the ciliate sequence clustered within a taxonomically more varied set of Bacteria. We also considered 13 ciliate sequences that clustered between the Bacteria and Archaea as HGT candidates, as well as two that clustered within the Archaea. Thus a total of 148 sequences were studied in more detail. We included all trees that showed evidence of HGT, irrespective of their statistical support, because we are interested in an estimate of the amount of HGT. The dominance of one functional class among the HGT candidates (see below) indicates the robustness of our results.
No bias of codon usage was detected, indicating complete adaptation to the codon usage of the Ciliate host and confirming that the HGT candidates are not contaminations (data not shown).
Over-representation of genes involved in anaerobic metabolism among HGT candidates
Out of 3563 clusters in our database, 2280 were assigned to at least one KOG or COG. Among the HGT candidates there is an over representation of genes involved in metabolism: while in the complete EST dataset the functions involved in Cellular process and signalling (47.0%) are prevalent, most of the HGT candidates are involved in Metabolism (75.4%) (See Figure 2).(Note that this number is an underestimate as it does not include 15 of the 30 sequences which do not belong to a KOG/COG – among which are eight xylanases, two cellulases, three pectate lyases, one uridine kinase and one α-glucosidase). Comparing the numbers of ESTs per cluster we found no indication that horizontally transferred genes are higher expressed than non-transferred ones (data not shown). 125 sequences out of the 148 HGT candidates encode enzymes (~84%), more than 35% of them are glycosyl hydrolases (GH), enzymes that are involved in the degradation of carbohydrates from the plant cell wall.
Figure 2.
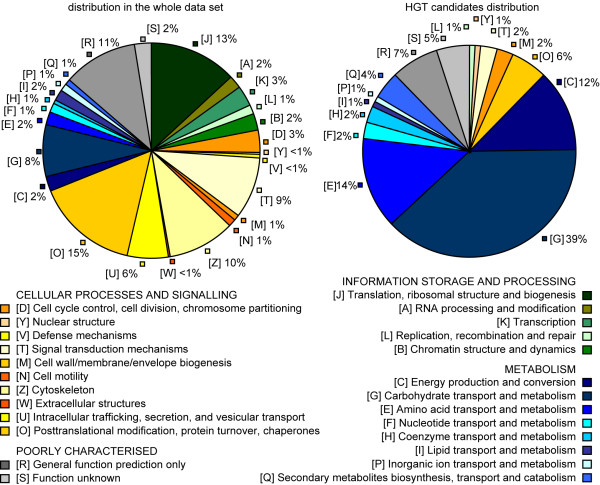
Distribution of functional classes in HGT and in the complete set of sequences. There is a clear over-representation of the metabolic KOG/COGs within the HGT candidates. green – the KOG/COG corresponding to the "information storage and processing"; yellow-orange – "cellular processes and signalling"; blue – "metabolism"; grey – "poorly characterised".
More specifically, most of the horizontally transferred enzymes are involved in the catabolism rather than in the anabolism. A description of these 148 candidates can be found in Additional file 2. Notably, we find a spectrum of enzymes in the Entodiniomorphids that is very different from that we find in the Vestibuliferids. The Vestibuliferids expressed some enolases, fructokinases, glucokinases that we did not find in the Entodiniomorphids. In contrast, the Entodiniomorphids expressed enzymes such as cellobiose phosphorylases, cellulases, xylanases, pectate lyase, aspartate-ammonia lyase and nitroreductase that were not found in the Vestibuliferid sequences. Moreover the only GHs that were found in the Vestibuliferids (lysozyme, β-hexoaminidase, 6-phospho β-glucosidase) are not fibrolytic enzymes. This matches the observation of Williams and Coleman (1992) that Vestibuliferids do not ingest fibres. These Ciliates depend on the uptake of starch and Bacteria.
Glycosyl hydrolases (GH) and other enzymes involved in the degradation of complex carbohydrates
The HGT candidates are dominated by the presence of one class of enzymes, namely the glycosyl hydrolases. These are a widespread group of enzymes that hydrolyse the glycosidic bonds between carbohydrates, or between a carbohydrate and a non-carbohydrate moiety. Below, we described these enzymes, and more specifically the xylanases and cellulases.
We found twelve xylanases within the 148 HGT candidates. All of them are restricted to the Entodiniomorphids [P. multivesiculatum (2), Epi. ecaudatum (3), Eud. maggii (2), Dip. affine (1) and M. medium (4)]. Five of them are homologous to three genes of C. acetobutylicum (possible xylan degradation enzymes Q97TI1, Q97TI3 and Q97TI6) that are located in a single operon indicating that there might have been only a single transfer event involving a piece of DNA containing the whole operon. In this case, we may expect that the other homologues of the xylanase genes that were located on the same operon in C. acetobutylicum but that were not present in our set, are present in M. medium, P. multivesiculatum and E. maggii genomes. On the other hand, without complete genome data, we cannot exclude gene loss either. Table 2 describes domains present in these xylanases genes found in Ciliates, and the domains found in the other glycosyl hydrolases.
Table 2.
Enzymes involved in the degradation of complex carbohydrates that appear to have been acquired by HGT.
| Organism | Accession number | Annotation | Domains |
| Das. ruminantium | AM051680 | 6-phospho-beta-glucosidase | glycosyl hydrolase family 1 |
| P. multivesiculatum | AM055068 | Aldose 1-epimerase precursor | aldose 1 epimerase |
| Epi. ecaudatum | AM053198 | alpha glucanotransferase | 4-alpha-glucanotransferase |
| P. multivesiculatum | AM055071 | alpha-glucosidase | no |
| P. multivesiculatum | AM055169 | Alpha-xylosidase | Glyco_hydro_31 |
| Iso. prostoma | AM054399 | Beta-hexosaminidase | glycosyl hydrolase family 3, N terminal domain |
| Epi. ecaudatum | AM053419 | carbohydrate esterase | rhamnogalaturan acetyltransferase |
| Epi. ecaudatum | AM053258 | Cellobiose phosphorylase | CBM_X |
| Epi. ecaudatum | AM053258 | Cellobiose phosphorylase | Glyco_transf_36 |
| Eud. maggii | AM053819 | Cellobiose phosphorylase | CBM_X |
| Eud. maggii | AM053819 | Cellobiose phosphorylase | Glyco_transf_36 |
| Eud. maggii | AM053820 | Cellobiose phosphorylase | no |
| P. multivesiculatum | AM055116 | Cellobiose phosphorylase | no |
| Epi. ecaudatum | AM053290 | cellulase | Glyco_hydro_5 |
| Epi. ecaudatum | AM053291 | cellulase | Glyco_hydro_5 |
| Epi. ecaudatum | AM053292 | cellulase | Glyco_hydro_5 |
| Epi. ecaudatum | AM053294 | cellulase | Glyco_hydro_5 |
| Epi. ecaudatum | AM053295 | cellulase | Glyco_hydro_5 |
| Epi. ecaudatum | AM053297 | cellulase | Glyco_hydro_5 |
| Epi. ecaudatum | AM053298 | cellulase | Glyco_hydro_9 |
| P. multivesiculatum | AM055058 | cellulase | endoglucacanase E like |
| P. multivesiculatum | AM055119 | cellulase | Glyco_hydro_5 |
| Epi. ecaudatum | AM053266 | Chitinase D precursor | Glyco_hydro_18 |
| Dip. affine | AM052171 | Endo-1,4-beta-xylanase | Glyco_hydro_10 |
| Epi. ecaudatum | AM053241 | Endo-1,4-beta-xylanase | Glyco_hydro_10 |
| M. medium | AM055031 | Endo-1,4-beta-xylanase precursor | Glyco_hydro_11 |
| M. medium | AM055032 | Endo-1,4-beta-xylanase precursor | Glyco_hydro_11 |
| Epi. ecaudatum | AM053745 | Endo-1,4-beta-xylanase T | Glyco_hydro_11 |
| Das. ruminantium | AM051785 | Fructokinase | ROK |
| Das. ruminantium | AM051789 | Fructokinase | ROK |
| Ent. caudatum | AM052478 | galactoside O-acetyltransferase | Trimeric LpxA like enzyme family |
| Epi. ecaudatum | AM053257 | glycosidase | NLPC_P60 |
| P. multivesiculatum | AM055193 | glycosidase | chitinase_glyco_hydro_19 |
| Iso. prostoma | AM054501 | Lysozyme | Phage_lysozyme |
| Epi. ecaudatum | AM053368 | Pectate lyase | no |
| Epi. ecaudatum | AM053374 | Pectate lyase | no |
| P. multivesiculatum | AM055221 | Pectate lyase | no |
| M. medium | AM054934 | pectin degradation protein | no |
| P. multivesiculatum | AM055224 | Polygalacturonase | Glyco_hydro_28 |
| Epi. ecaudatum | AM053746 | xylanase | Glyco_hydro_10 |
| Eud. maggii | AM053864 | xylanase | cellulose binding domain: CBD_IV |
| Eud. maggii | AM054010 | xylanase | Glyco_hydro_10 |
| M. medium | AM055027 | xylanase | alpha-L arabinanase |
| M. medium | AM055030 | xylanase | cellulose binding domain: CBD_II |
| P. multivesiculatum | AM055577 | xylanase | glycol_hydro_30 |
| P. multivesiculatum | AM055581 | xylanase | cellulose binding domain: CBD_IV alpha-L arabinanase |
The set of HGT candidates also contains nine cellulases, which were only found in the Entodiniomorphids (Epi. ecaudatum (7) and P. multivesiculatum (2)). Seven of them belong to GH family 5 and only contain a cellulase domain. AM053298 belongs to GH family 9 and contains a glycol_hydro_9 domain. AM055058 contains an endoglucanase E like domain. Some cellulolytic Bacteria and rumen Fungi have cellulosome, which is a multi-enzyme complex for the degradation of plant cell wall polysaccharides [22]. This cellulosome consists of a scaffoldin protein that binds several glycosyl hydrolases via specialized intermodular "cohesin-dockerin" interactions. Finding such protein could reinforce the prokaryotic origin of the cellulases. However, we found no evidence for the presence of a scaffoldin and any cohesin domain was detected in the ciliate sequences. This could be explained by the fact that the occurrence of cellulases was restricted to those species that are able to ingest plant fibres. Therefore, it is likely that these cellulases do not require cohesin domains as is the case in bacterial or fungal cellulases, which are excreted into the environment. Our results on expressed genes are consistent with previous work on enzyme activities. We found cellulases in Epidinium confirming previous work using recombinant DNA technology [14], in which an Epidinium cellulase was shown to be functional. Furthermore, the role of P. multivesiculatum in cellulose digestion [23] is consistent with the presence of a cellulase in our Polyplastron sequences. Conversely, the absence of cellulase genes among the ESTs of Entodinium, is consistent with in vitro studies of Entodinium in which no cellulase activity was detected [24,25].
Beside the xylanases and cellulases mentioned above, we also found other enzymes involved in the degradation of the complex carbohydrates: an α-glucanotransferase, a β-hexosaminidase, a 6-phospho-β-glucosidase, three pectate lyases, a pectin degradation protein, four cellobiose phosphorylases, two glycosidases, an α-xylosidase, an α-glucosidase, a chitinase, a lysozyme, a polygalacturonase, a carbohydrate esterase, a sorbitol dehydrogenase and a galactoside O-acetyltransferase, two fructokinases, an aldose epimerase precusrsor.
Adaptation to an anaerobic environment
Among the other HGT candidates are an Fe-hydrogenase and a malic enzyme, which are of interest because of their link with hydrogenosomes, membrane-bound organelles present in a number of anaerobic organisms such as the ciliate Nyctotherus ovalis [26], the chytrid fungi Neocallimastix and Piromyces, and the Parabasalian flagellate Trichomonas vaginalis. Most hydrogenases are found in such membrane-bound organelles, but there are exceptions for example in Giardia and Entamoeba where hydrogenases are located in the cytoplasm [27,28]. Similarly, also malic enzymes are found in various compartments of the eukaryotic cell: in Trichomonas vaginalis, a cytosolic malic enzyme of bacterial origin was found as well as a hydrogenosomal homologue [29]. Until now hydrogenosomes of rumen ciliates have not been studied in more detail, and it remains unclear as to whether all rumen Ciliates possess hydrogenosomes [30].
Another enzyme, Pyrophosphate-fructose 6-phosphate 1-phosphotransferase (EC: 2.7.1.90) can also be linked to adaptation to an anaerobic environment. This enzyme catalyzes the phosphorylation of D-fructose 6-phosphate to D-fructose 1,6 biphosphate, using PPi rather than the standard 6-phosphofructokinase (EC: 2.7.1.11) which uses ATP. PPi dependent phosphofructokinase typically occurs, besides in plants, among anaerobic unicellular species, and has been suggested to be an adaptation to a situation where the glycolysis is the sole source of ATP [31-33].
In the HGT candidates we also found some enzymes involved in nitrogen metabolism: four nitroreductases, two aspartate-ammonia ligases (EC: 6.3.1.1), an aspartate-ammonia lyase (4.3.1.1) and a NAD specific glutamate dehydrogenase (EC: 1.4.1.3). These last three enzymes catalyse the reversible reactions between ammonia and the respective amino-acids: L-Asparagine, L-Aspartate and L-Glutamate. In the rumen, the proteins are degraded into amino-acids, which are then rapidly deaminated to form ammonia. Ammonia can then be excreted via urine and faeces or, alternatively assimilated by ruminal Bacteria [34] or by the Ciliates themselves. Indeed a glutamate dehydrogenase (GDH) probably acquired through HGT, has already been described in Entodinium caudatum. This GDH was cloned and expressed in E. coli, where it expressed a high affinity for ammonia and α-ketoglutarate and a low affinity for glutamate, suggesting that the enzyme may be involved in the assimilation of ammonia by Ent. caudatum in the rumen [15].
Discussion
Detecting HGT by a phylogenetic approach after selecting candidates by Best Hit
A number of methods have been proposed to detect horizontal gene transfer, varying from Best Hit approaches [35] to phylogenetic methods [36] and methods that examine aberrant codon usage [37]. Here we have combined a fast screening "Best Hit" method for the detection of potential candidates followed by two levels of phylogenetic analyses. First, we used the Best Hit method to restrict the data set of potential candidates. Then, the fast Neighbor-Joining method highlighted the sequences that clustered within the Bacteria. In the final step we used a Maximum Likelihood approach which is the more time consuming, but also a more reliable phylogenetic method. Such a multi-step approach appears well suited for the detection of likely candidates when doing Maximum Likelihood analyses for all genes would be too time consuming. The large number of phylogenetic trees that had to be examined prompted us to use an automated method for their analysis: the examination of the species composition of the second smallest partition, allowing us to pinpoint the Firmicutes as the largest group of potential donors of genes.
Our large-scale analysis allowed us nevertheless to identify the few genes previously shown from small scale analyses, to have arisen through HGT (cellulase [14], xylanase [7,15]), thereby confirming the usefulness and validity of our method.
HGT frequency
HGT seems to be an important process in the evolution of rumen Ciliates. In total 4.1% of the ESTs available for these ciliates appear to have been acquired by HGT. This number is at least one order of magnitude higher than the level of HGT found in (plant-pathogenic) nematodes (16 in the whole genome, i.e. 16 in roughly 19,000 genes, less than 0.1%) [8] and about four times the 1% of potential HGT's found in (the parasite) Entamoeba histolytica [9], although these differences might be biased due to the usage of EST data instead of DNA based gene predictions. However, even under the (unrealistic) assumption that the 148 candidates genes for HGT in rumen ciliates described here represent the vast majority of all HGT genes in the genome of a "standard" rumen ciliate with more than 30,000 genes, the incidence of HGT would still be about 0.5%.
The low number of transfers in nematodes (and multicellular eukaryotes in general) might be due to the fact the transferred genes must be incorporated into the germ line to be inherited. This hampers, for principal reasons, HGT into the genome of multicellular organisms. In contrast, Entamoeba histolytica, which exhibits a genome-wide HGT incidence of about 1%, is a unicellular eukaryote without any germ-line soma differentiation. Ciliates are in an intermediate position between unicellular organisms and multicellular organisms, being unicellular organisms, but possessing a differentiated germ-line (the micronucleus), and a somatic nucleus (the macronucleus), which accounts for all transcriptional activities during the vegetative growth of the cell. Therefore, the high incidence of evolutionary HGT might be surprising in the first instance. However, the unusually high incidence of HGT in ciliates can be explained by (i) their bacteriovory, which eventually leads to the release of bacterial DNA into the cytoplasm, and (ii) their nuclear dimorphism, which allows amplification of foreign DNA in the macronucleus [38]. Furthermore, Ciliates possess a sophisticated machinery of enzymes facilitating extended genome reorganizations obviously allowing the assembly of DNA into novel genetic environments [39].
The level of HGT in Ciliates seems more comparable to that observed in Bacteria (~10%) [37]. Most of the studies about HGT within Bacteria are based on the nucleotide composition as it has been postulated that transferred genes retain the nucleotide composition of the donor for some time [40]. This is however a relatively poor indicator[41], particularly in the case of genes which have been incorporated long ago.
Adaptive value of HGT in exploitation of new niches
The vast majority of the transferred genes that we found in this study are involved in the degradation of plant cell wall derived carbohydrates, indicating an adaptation to the carbohydrate-rich environment in which these Ciliates live.
Interestingly, also the few other examples of HGT from Bacteria to Eukaryotes tend to involve complex carbohydrate degradation enzymes. Previously documented examples include an endo-1,4-mannanase (a glycosyl hydrolase) in Piromyces [42], a β-1,4-endoglucanase (cellulase) [8,43], an endoglucanase and probably other glycoside hydrolases in Orpinomyces, a rumen fungus [6] and pectinases in the Nematodes [8]. Here we have shown that carbohydrate degrading enzymes also dominate in a large set of horizontally transferred genes. In contrast, in the analysis of horizontal gene transfer between e.g. Archaea and Bacteria, the transferred functions are not specifically involved in the breakdown of complex carbohydrates although there is a dominance of "operational" proteins over "informational" proteins [44]. The HGT candidates we found are mainly encoding proteins involved in the degradation of complex plant cell wall derived carbohydrates reinforcing the interpretation that genes were only retained that provide an evolutionary advantage for the host in the particular ecological niche. Thus, we were able to substantiate our postulate that the lifestyle of ciliates in the gastro-intestinal tract of animals and their unique genome organisation, i.e. the presence of a macronucleus with highly processed, gene-dense chromosomes, together with a germ-line nucleus that tolerates large genome rearrangements [39], would provide favourable circumstances for the acquisition of foreign DNA. The observed high rate of HGT makes rumen Ciliates ideal bio-monitors for identification of potential gene transfer events in the gastro-intestinal tract of farm animals. The frequency of the historical transfers observed here approaches the levels of bacterial HGTs. As Ciliates ingest Bacteria, it also supports the "You are what you eat" hypothesis, since the sources for the historical HGT were clearly Bacteria which are related to Bacteria which are abundant in the rumen and gut of herbivorous mammals.
Conclusion
We have screened a large set of ESTs from Rumen Ciliates, Eukaryotes that live in the foregut of the ruminant, by combining two methods: Best Hit and phylogenetic analyses. The amount of HGT (4.1%) is larger than what has ever been described in Eukaryotes. The results are consistent in the sense that the Horizontal Transfers are dominated by genes that internally encode enzymes involved into a few, specialized metabolic processes. Rumen Ciliate species live an anaerobic enviroment that is characterized by the availability of a rich source of plant polymers. Since Ciliates per se (c.f. the Tetrahymena genome) seem to possess only a limited repertoire of enzymes allowing the catabolism of such carbohydrates, a substantial fraction of the genes involved in the breakdown of carbohydrates in the rumen Ciliates appears to be acquired from Firmicutes and other Bacteria, which are well represented in the rumen.
Thus the horizontally transferred genes identified in this research may have greatly facilitated the Ciliates' colonization of their rumen habitat.
Methods
Construction of a cDNA library and sequencing
cDNA libraries were constructed from the Ciliates Ent. simplex, Ent. caudatum, Eud. maggii, M. medium, Dip. affine, P. multivesiculatum and Epi. ecaudatum (all Entodiniomorphids) and the Vestibuliferids I. prostoma, I. intestinalis and Das. ruminantium as described previously [45] and cloned into λ-Zap (Stratagene) following the manufacturer's instructions. Randomly picked clones were selected from P. multivesiculatum (721), Epid. ecaudatum (681), Eud. maggii (638), Ent. caudatum (1207), I. prostoma (638), and Das. ruminantium (595) for sequencing. In addition 96 clones were selected randomly from each of the other libraries for sequencing. The ciliates were maintained as well-defined monocultures in fistulated sheep.
DNA sequencing was performed on sequencers purchased from either PE Applied Biosystems, Beckman Coultard or Shimazu (PE Applied Biosystems ABI 373A and 377, a Genetic Analyzer – CEQ 8000 by Beckman).
ESTs sequences
4768 ESTs (Expressed Sequence Tags) of rumen Ciliates were sequenced from 10 species of protozoa. Vectors and linkers were automatically filtered out from the data. First the vector pieces were detected using a Blastn search against the pBKCMV vector sequence on a Paracel genematcher machine using the same parameters as VecScreen (-q -5 -G 3 -E 3 -e 700 -Y 1.75e12 -F mD) [46].
Regions showing similarity to the pBKCMV vector were trimmed from the ends of the sequences. If there was remaining vector in the middle, it was removed and the longer of the two remaining fragments was kept. Subsequently, sequences that had less than 100 bases after trimming were discarded. After this filtering procedure, the dataset contained 4335 sequences.
Furthermore, the sequences were analyzed for contamination from other organisms by aligning against the EMBL database using Megablast [47] (version 2.2.4 [Aug-26-2002]) (identity ≥94% and a match length ≥40% of the sequence). This procedure removed 11 contaminated sequences (mainly by Escherichia coli). The remaining 4324 sequences varied in size between 100 and 896 nucleotides (nt), with an average length of 527 nt. By homology with known proteins we could get an estimate of the percentage of the genes covered by the Ciliate ESTs. The homology covered from 3% to 100% of the length of a known gene in another species, with a mean value of 57%. The 4324 sequences are distributed as follows: 715 sequences from P. multivesiculatum, 595 from Epi. ecaudatum, 543 from Eud. maggii, 1062 from Ent. caudatum, 27 from Ent. simplex, 10 from Dip. affine,151 from M. medium, 546 from I. prostoma, 84 from I. intestinalis and 591 from Das. ruminantium.
Data storage and clustering
The sequences and the results of the analyses performed were stored in a relational MySQL database. In order to minimize the redundancy of the data, we aligned the dataset to itself using SWN (Smith-Waterman (SW) comparison at the nucleotide level). Sequences pairs having an identity ≥97% over at least 100 bp were considered to be from the same gene and were put in the same cluster. Sequences sharing less or no sequence similarity can be in the same cluster as long as they are linked via other sequences.
Function assignment
Function assignment of the sequences was performed on three levels:
1) The protein domain structure was predicted by comparing the Hidden Markov Models (HMM) from PFAM (Protein Families Database) [48] with our dataset, using an e-value threshold of 0.01. Analysis of the O-Glycosides hydrolases was further performed with the CDD (Conserved Domain Database)[49] and SMART (Simple Modular Architecture Research Tool) [50].
2) The cDNAs were assigned to COGs (Clusters of Orthologous Groups of genes, [51]) based on sequence similarity to existing COGs in a Cognitor-like procedure [52]. The COG assignment yields a higher resolution prediction of the molecular function than a gene family assignment derived from PFAM. In addition, the COGs provide a classification of cellular roles for all the molecular protein functions that we used to classify the EST data. The COG database contains a set of all proteins and protein fragments (for fusion proteins) from a set of 66 complete unicellular genomes (COG) and a set of 7 complete eukaryotic genomes (KOG). All proteins are compared to the COG and KOG sets using SWX (SW comparison of DNA translated in the six reading frames against a protein database). We use a conservative variant of the Cognitor to match cDNAs to a COG: when the three first significant (e < 0.01) hits of the query sequence match the same COG, then this COG is assigned to that region of the query sequence. In the results we used the notation "KOG/COG" because we used the information from both sets. We first examine the KOG (eukaryotic set); in cases where no KOG was attributed to the EST sequence, the COG information was used.
3) Sequences were compared to version 42.8 of the Swissprot database (January 2004) to annotate each sequence according to a restricted vocabulary. This comparison was performed with SWX. Sequences with significant matches (e-value < 1.10–5) were annotated according to their most significant hit.
In addition, the CAZy database which contains the description of enzymes degrading, modifying, or creating glycosidic bonds[53], and the KEGG database which contains a description of enzymes and their pathway [54] were used in the analysis.
All sequence comparisons were run on a Paracel Genematcher using the Smith-Waterman [55] algorithm for pairwise sequence comparisons and Hidden Markov Models for sequence-to-profile comparisons. The output was subsequently analyzed in a Linux environment using Perl scripts.
Best Hit
We performed a SWX search against all complete proteomes (see below; e-value < 0.01) to find in which species we encountered the most similar sequence (Best Hit).
Including other ciliate genomes
Within the set of 148 predicted proteomes, Plasmodium falciparum (Apicomplexa), is evolutionarily the closest organism to the Ciliates. In order to distinguish events that could have occurred in the Ciliates before their colonization of the rumen environment from more recent events in the rumen we added the nucleotide data from the non-rumen ciliate Tetrahymena thermophila (Tetrahymena Genome Database) to our analyses.
First a tSWX (a SW search with translated nucleotides against a translated nucleotides database) comparison against the T. thermophila genome was performed. (nov. 2003, TIGR; E-value threshold 1.10–5, match length > 30 amino acids). The T. thermophila matching sequences were retrieved and translated in the 6 frames. The frame that matched the rumen ciliate was included in the tree (see below).
Phylogenetic analyses
To derive a phylogenetic tree for each cluster of ESTs we used the following procedure: First the longest sequence of the cluster was translated into six reading frames and used as a query to search the published genomes. Homologous sequences were retrieved using SWP (SW at the protein level) with an e-value threshold of 1.10–5, T. thermophila homologs were added when available, and the EST was aligned with its homologs using ClustalW [56]. Subsequently phylogenetic trees were constructed using Neighbor-Joining as implemented in ClustalW (excluding positions with gaps and using 1000 bootstraps). The multiple alignments are based on the Ciliate EST sequences compared with the full length sequences of the other organisms. Trees are constructed solely based on the homologous positions after selecting the conserved positions and removing the gaps from the alignment The minimal number of conserved positions used to construct the tree is 58 amino acids (except for one xylanase that was much shorter but was based a on manual check of the alignment and of the sequences present). The maximal length was 369 amino acids, the mean length was 172.5 amino acids.
Those ciliate sequences that clustered within a bacterial partition of the tree were considered HGT candidates. To automatically detect such cases, an algorithm examined the species composition of the "tree neighbourhood" of the ciliate sequence. By requiring that the second-smallest partition containing the ciliate sequence (Figure 3) is fully composed of bacterial sequences (plus our ciliate sequence) we assured that the ciliate sequence clustered "within" the bacterial sequences. In principle we do not know the root of the tree, and it is therefore hard to argue that when a query sequence clusters within a set of Bacterial sequences it is actually derived from a Bacterial sequence. The principle of selecting the second-smallest partition is that we assume that the root of the tree lies outside that (small) partition.
Figure 3.
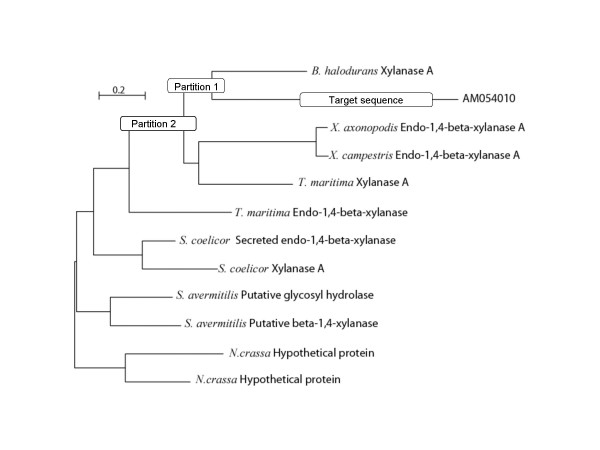
A likely horizontally transferred gene in Entodinium. The tree contains the target sequence (AM054010) and its homologuous proteins. To determine whether the target sequence is HGT candidate, an algorithm examines the presence or absence of Bacteria, Archaea and Eukaryotes in the second smaller partition containing the target sequence. In the example, the smallest second partition containing the target sequence contains also the sequences from Bacillus halodurans, Xanthomonas axonopodis, Xanthomonas campestris and Thermotoga maritima, which are all Bacteria. AM054010 was therefore retained as a HGT candidate.
Subsequently for the selected cases we obtained high quality trees with a different methodology. We used Muscle [57] to obtain the sequence alignments, and for the positions without gaps we obtained phylogenies using Maximum Likelihood as calculated by MrBayes [58,59], using four gamma-distributed rate categories plus invariant positions and the Poisson amino acid similarity matrix. Again the "second-smallest partition" algorithm was used to obtain HGT candidates.
For the HGT candidates identified here, the annotations (See previous paragraph) were manually examined based on their phylogenetic tree, to assure that sequences were orthologous to annotated proteins, and their domain compositions.
Proteomes
A set of 148 complete proteomes was used, which included 16 archaeal, 116 bacterial and 16 eukaryotic proteomes. (A complete list of the proteomes included can be found in Additional file 1.)
List of abbreviations
HGT: Horizontal Gene Transfer; BH: Best Hit; FCA: SSP: Second Smallest Partition; GH: Glycosyl Hydrolase; COG: Cluster of Orthologous Group; SW: Smith Waterman; ESTs: Expressed Sequence Tags.
Authors' contributions
GR and MAH designed the bioinformatic experiment.
GR performed computational sequence analysis and wrote the manuscript.
JHPH and MAH initiated and coordinated the study.
TM, JPJ, DM and EN established ciliate cell lines for cDNA librairies construction.
CJN and NRMcE coordinated and participated in the generation of the cDNA libraries and the production of the EST sequences.
NN and NAT participated in the growth of clones for sequencing.
FMMcI created cDNA libraries.
AT and MM generated financial support for the sequencing of clones. KU and TN sequenced the clones.
GR, BED, NRMcE, TM, MAH, JHPH, JPJ participated in drafting the manuscript.
All authors read and approved the final manuscript.
Supplementary Material
Distribution of the complete proteomes over the various taxa. Eukaryotic proteomes are: Plasmodium falciparum, Guillardia theta, Candida albicans, Encephalitozoon cuniculi, Neurospora crassa, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Anopheles gambiae, Caenorhabditis elegans, Drosophila melanogaster, Danio rerio (zebrafish), Fugu rubripes, Homo sapiens, Mus musculus, Rattus norvegicus, and Arabidopsis thaliana.
Classification of the 148 HGT candidates. Annotation was given on the basis of the complete analysis (homology with known proteins, homology to proteins of a KOG/COG, PFAM domains and confirmed from the tree). CCD: Complex carbohydrates degradation, F: Fermentation, G: Glycolysis, PD: protein degradation, NM: Nitrogen metabolism. Composition of the Smith Waterman comparison results (Hit; E-value < 1·10–5) or second-smallest partition (SSP): B: only Bacteria; AB: Bacteria and Archaea; A: only Archaea.
Acknowledgments
Acknowledgements
This work was supported by the European projects CIMES (CIliates as Monitors for Environmental Safety) and ERCULE (European Rumen ciliates CULturE collection).
Contributor Information
Guénola Ricard, Email: g.ricard@cmbi.ru.nl.
Neil R McEwan, Email: nrm@aber.ac.uk.
Bas E Dutilh, Email: dutilh@cmbi.ru.nl.
Jean-Pierre Jouany, Email: jouany@clermont.inra.fr.
Didier Macheboeuf, Email: dmache@clermont.inra.fr.
Makoto Mitsumori, Email: mitsumori@affrc.go.jp.
Freda M McIntosh, Email: fmm@rri.sari.ac.uk.
Tadeusz Michalowski, Email: t.michalowski@ifzz.pan.pl.
Takafumi Nagamine, Email: nagt@shirakawa.ne.jp.
Nancy Nelson, Email: n.nelson@hrsu.mrc.ac.uk.
Charles J Newbold, Email: cjn@aber.ac.uk.
Eli Nsabimana, Email: nsabima@hotmail.com.
Akio Takenaka, Email: akio@affrc.go.jp.
Nadine A Thomas, Email: N.Thomas@macaulay.ac.uk.
Kazunari Ushida, Email: k_ushida@kpu.ac.jp.
Johannes HP Hackstein, Email: j.hackstein@science.ru.nl.
Martijn A Huynen, Email: huynen@cmbi.ru.nl.
References
- Doolittle WF. You are what you eat: a gene transfer ratchet could account for bacterial genes in eukaryotic nuclear genomes. Trends Genet. 1998;14:307–311. doi: 10.1016/S0168-9525(98)01494-2. [DOI] [PubMed] [Google Scholar]
- Martin W, Herrmann RG. Gene transfer from organelles to the nucleus: how much, what happens, and Why? Plant Physiol. 1998;118:9–17. doi: 10.1104/pp.118.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Nelson WC, Ketchum KA, McDonald L, Utterback TR, Malek JA, Linher KD, Garrett MM, Stewart AM, Cotton MD, Pratt MS, Phillips CA, Richardson D, Heidelberg J, Sutton GG, Fleischmann RD, Eisen JA, White O, Salzberg SL, Smith HO, Venter JC, Fraser CM. Evidence for lateral gene transfer between Archaea and Bacteria from genome sequence of Thermotoga maritima. Nature. 1999;399:323. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- Worning P, Jensen LJ, Nelson KE, Brunak S, Ussery DW. Structural analysis of DNA sequence: evidence for lateral gene transfer in Thermotoga maritima. Nucleic Acids Res. 2000;28:706–709. doi: 10.1093/nar/28.3.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Lage JL, Feller G, Janecek S. Horizontal gene transfer from Eukarya to bacteria and domain shuffling: the alpha-amylase model. Cell Mol Life Sci. 2004;61:97–109. doi: 10.1007/s00018-003-3334-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Vallve S, Romeu A, Palau J. Horizontal gene transfer of glycosyl hydrolases of the rumen fungi. Mol Biol Evol. 2000;17:352–361. doi: 10.1093/oxfordjournals.molbev.a026315. [DOI] [PubMed] [Google Scholar]
- Devillard E, Newbold CJ, Scott KP, Forano E, Wallace RJ, Jouany JP, Flint HJ. A xylanase produced by the rumen anaerobic protozoan Polyplastron multivesiculatum shows close sequence similarity to family 11 xylanases from gram-positive bacteria. FEMS Microbiol Lett. 1999;181:145–152. doi: 10.1111/j.1574-6968.1999.tb08837.x. [DOI] [PubMed] [Google Scholar]
- Scholl EH, Thorne JL, McCarter JP, Bird DM. Horizontally transferred genes in plant-parasitic nematodes: a high-throughput genomic approach. Genome Biol. 2003;4:R39. doi: 10.1186/gb-2003-4-6-r39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftus B, Anderson I, Davies R, Alsmark UC, Samuelson J, Amedeo P, Roncaglia P, Berriman M, Hirt RP, Mann BJ, Nozaki T, Suh B, Pop M, Duchene M, Ackers J, Tannich E, Leippe M, Hofer M, Bruchhaus I, Willhoeft U, Bhattacharya A, Chillingworth T, Churcher C, Hance Z, Harris B, Harris D, Jagels K, Moule S, Mungall K, Ormond D, Squares R, Whitehead S, Quail MA, Rabbinowitsch E, Norbertczak H, Price C, Wang Z, Guillen N, Gilchrist C, Stroup SE, Bhattacharya S, Lohia A, Foster PG, Sicheritz-Ponten T, Weber C, Singh U, Mukherjee C, El-Sayed NM, Petri WAJ, Clark CG, Embley TM, Barrell B, Fraser CM, Hall N. The genome of the protist parasite Entamoeba histolytica. Nature. 2005;433:865–868. doi: 10.1038/nature03291. [DOI] [PubMed] [Google Scholar]
- Morrison M. Do ruminal bacteria exchange genetic material? J Dairy Sci. 1996;79:1476–1486. doi: 10.3168/jds.S0022-0302(96)76507-4. [DOI] [PubMed] [Google Scholar]
- Mercer DK, Melville CM, Scott KP, Flint HJ. Natural genetic transformation in the rumen bacterium Streptococcus bovis JB1. FEMS Microbiol Lett. 1999;179:485–490. doi: 10.1111/j.1574-6968.1999.tb08767.x. [DOI] [PubMed] [Google Scholar]
- Zagulski M, Nowak JK, Le Mouel A, Nowacki M, Migdalski A, Gromadka R, Noel B, Blanc I, Dessen P, Wincker P, Keller AM, Cohen J, Meyer E, Sperling L. High coding density on the largest Paramecium tetraurelia somatic chromosome. Curr Biol. 2004;14:1397–1404. doi: 10.1016/j.cub.2004.07.029. [DOI] [PubMed] [Google Scholar]
- Williams AG, Coleman GS. The rumen Protozoa. In: Brock TD, Wisconsin-Madison U, editor. Brock/Springer Series in Contemporary Bioscience. Springer-Verlag New York Inc; 1992. pp. 192–210. [Google Scholar]
- Wereszka K, McIntosh FM, Michalowski T, Jouany JP, Nsabimana E, Macheboeuf D, McEwan NR, Newbold CJ. A cellulase produced by the rumen anaerobic protozoan epidinium ecaudatum has an unusual pH optimum. Endocytobiosis and Cell Research. 2004;15:561–569. [Google Scholar]
- Newbold CJ, McEwan NR, Calza RE, Chareyron EN, Duval SM, Eschenlauer SC, McIntosh FM, Nelson N, Travis AJ, Wallace RJ. An NAD(+)-dependent glutamate dehydrogenase cloned from the ruminal ciliate protozoan, Entodinium caudatum. FEMS Microbiol Lett. 2005;247:113–121. doi: 10.1016/j.femsle.2005.04.034. [DOI] [PubMed] [Google Scholar]
- Baldauf SL, Roger AJ, Wenk-Siefert I, Doolittle WF. A kingdom-level phylogeny of eukaryotes based on combined protein data. Science. 2000;290:972–977. doi: 10.1126/science.290.5493.972. [DOI] [PubMed] [Google Scholar]
- Sankar M, Delgado O, Mattiasson B. Isolation and characterization of solventogenic, cellulase-free xylanolytic Clostridia from cow rumen. Water Sci Technol. 2003;48:185–188. [PubMed] [Google Scholar]
- Russell JB, Rychlik JL. Factors that alter rumen microbial ecology. Science. 2001;292:1119–1122. doi: 10.1126/science.1058830. [DOI] [PubMed] [Google Scholar]
- Edwards JE, McEwan NR, Travis AJ, Wallace JR. 16S rDNA library-based analysis of ruminal bacterial diversity. Antonie van Leeuwenhoek. 2004;86:263. doi: 10.1023/B:ANTO.0000047942.69033.24. [DOI] [PubMed] [Google Scholar]
- Nelson KE, Zinder SH, Hance I, Burr P, Odongo D, Wasawo D, Odenyo A, Bishop R. Phylogenetic analysis of the microbial populations in the wild herbivore gastrointestinal tract: insights into an unexplored niche. Environ Microbiol. 2003;5:1212–1220. doi: 10.1046/j.1462-2920.2003.00526.x. [DOI] [PubMed] [Google Scholar]
- Koski LB, Golding GB. The closest BLAST hit is often not the nearest neighbor. J Mol Evol. 2001;52:540–542. doi: 10.1007/s002390010184. [DOI] [PubMed] [Google Scholar]
- Doi RH, Kosugi A. Cellulosomes: plant-cell-wall-degrading enzyme complexes. Nat Rev Microbiol. 2004;2:541–551. doi: 10.1038/nrmicro925. [DOI] [PubMed] [Google Scholar]
- Jouany JP, Senaud J. [Effect of rumen ciliates on the digestion of different carbohydrates in sheep. I.--Utilization of cell wall carbohydrates (cellulose and hemicellulose) and of starch] Reprod Nutr Dev. 1982;22:735–752. [PubMed] [Google Scholar]
- Abou Akkada AR, Howard BH. The biochemistry of rumen protozoa: 3 - The carbohydrate metabolism of Entodinium. Biochem J. 1960;76:445–451. doi: 10.1042/bj0760445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushida K, Jouany JP. Fiber digesting capacities of five genera of rumen ciliates. Proc Soc Nutr Physiol. 1994;3:168. [Google Scholar]
- Boxma B, de Graaf RM, van der Staay GW, van Alen TA, Ricard G, Gabaldon T, van Hoek AH, Moon-van der Staay SY, Koopman WJ, van Hellemond JJ, Tielens AG, Friedrich T, Veenhuis M, Huynen MA, Hackstein JH. An anaerobic mitochondrion that produces hydrogen. Nature. 2005;434:74–79. doi: 10.1038/nature03343. [DOI] [PubMed] [Google Scholar]
- Lloyd D, Ralphs JR, Harris JC. Giardia intestinalis, a eukaryote without hydrogenosomes, produces hydrogen. Microbiology. 2002;148:727–733. doi: 10.1099/00221287-148-3-727. [DOI] [PubMed] [Google Scholar]
- Nixon JE, Field J, McArthur AG, Sogin ML, Yarlett N, Loftus BJ, Samuelson J. Iron-dependent hydrogenases of Entamoeba histolytica and Giardia lamblia: activity of the recombinant entamoebic enzyme and evidence for lateral gene transfer. Biol Bull. 2003;204:1–9. doi: 10.2307/1543490. [DOI] [PubMed] [Google Scholar]
- Dolezal P, Vanacova S, Tachezy J, Hrdy I. Malic enzymes of Trichomonas vaginalis: two enzyme families, two distinct origins. Gene. 2004;329:81–92. doi: 10.1016/j.gene.2003.12.022. [DOI] [PubMed] [Google Scholar]
- Yarlett N, Coleman GS, Williams AG, Lloyd D. Hydrogenosomes in known species of rumen entodiniomorphid protozoa. FEMS Microbiol Lett. 1984;21:15–19. doi: 10.1016/0378-1097(84)90171-X. [DOI] [Google Scholar]
- Bapteste E, Moreira D, Philippe H. Rampant horizontal gene transfer and phospho-donor change in the evolution of the phosphofructokinase. Gene. 2003;318:185–191. doi: 10.1016/S0378-1119(03)00797-2. [DOI] [PubMed] [Google Scholar]
- Mertens E. Pyrophosphate-dependent phosphofructokinase, an anaerobic glycolytic enzyme? FEBS Lett. 1991;285:1–5. doi: 10.1016/0014-5793(91)80711-B. [DOI] [PubMed] [Google Scholar]
- Siebers B, Klenk HP, Hensel R. PPi-dependent phosphofructokinase from Thermoproteus tenax, an archaeal descendant of an ancient line in phosphofructokinase evolution. J Bacteriol. 1998;180:2137–2143. doi: 10.1128/jb.180.8.2137-2143.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace RJ, Onodera R, Cotta MA. Metabolism of nitrogen-containing compounds. In: Hobson PNSCS, editor. The Rumen Microbial Ecosystem. London , Chapman and Hall; 1997. pp. 283–328. [Google Scholar]
- Koonin EV, Makarova KS, Aravind L. Horizontal gene transfer in prokaryotes: quantification and classification. Annu Rev Microbiol. 2001;55:709–742. doi: 10.1146/annurev.micro.55.1.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syvanen M. Horizontal gene transfer: evidence and possible consequences. Annu Rev Genet. 1994;28:237–261. doi: 10.1146/annurev.ge.28.120194.001321. [DOI] [PubMed] [Google Scholar]
- Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- Skovorodkin IN, Zassoukhina IB, Hojak S, Ammermann D, Gunzl A. Minichromosomal DNA replication in the macronucleus of the hypotrichous ciliate Stylonychia lemnae is independent of chromosome-internal sequences. Chromosoma. 2001;110:352–359. doi: 10.1007/s004120100154. [DOI] [PubMed] [Google Scholar]
- Prescott DM. Genome gymnastics: unique modes of DNA evolution and processing in ciliates. Nat Rev Genet. 2000;1:191–198. doi: 10.1038/35042057. [DOI] [PubMed] [Google Scholar]
- Lawrence JG, Ochman H. Amelioration of bacterial genomes: rates of change and exchange. J Mol Evol. 1997;44:383–397. doi: 10.1007/PL00006158. [DOI] [PubMed] [Google Scholar]
- Koski LB, Morton RA, Golding GB. Codon bias and base composition are poor indicators of horizontally transferred genes. Mol Biol Evol. 2001;18:404–412. doi: 10.1093/oxfordjournals.molbev.a003816. [DOI] [PubMed] [Google Scholar]
- Millward-Sadler SJ, Hall J, Black GW, Hazlewood GP, Gilbert HJ. Evidence that the Piromyces gene family encoding endo-1,4-mannanases arose through gene duplication. FEMS Microbiol Lett. 1996;141:183–188. doi: 10.1111/j.1574-6968.1996.tb08382.x. [DOI] [PubMed] [Google Scholar]
- Yan Y, Smant G, Stokkermans J, Qin L, Helder J, Baum T, Schots A, Davis E. Genomic organization of four [beta]-1,4-endoglucanase genes in plant-parasitic cyst nematodes and its evolutionary implications. Gene. 1998;220:61–70. doi: 10.1016/S0378-1119(98)00413-2. [DOI] [PubMed] [Google Scholar]
- Dutilh BE, Huynen MA, Bruno WJ, Snel B. The consistent phylogenetic signal in genome trees revealed by reducing the impact of noise. J Mol Evol. 2004;58:527–539. doi: 10.1007/s00239-003-2575-6. [DOI] [PubMed] [Google Scholar]
- Eschenlauer SCP, McEwan NR, Calza RE, Wallace RJ, Onodera R, Newbold CJ. Phylogenetic position and codon usage of two centrin genes from the rumen ciliate protozoan, Entodinium caudatum. FEMS Microbiology Letters. 1998;166:147. doi: 10.1111/j.1574-6968.1998.tb13196.x. [DOI] [PubMed] [Google Scholar]
- VecScreen http://www.ncbi.nlm.nih.gov/VecScreen/VecScreen.html
- Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7:203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL, Studholme DJ, Yeats C, Eddy SR. The Pfam protein families database. Nucleic Acids Res. 2004;32 Database issue:D138–41. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Marchler GH, Mullokandov M, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Yamashita RA, Yin JJ, Zhang D, Bryant SH. CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res. 2005;33:D192–196. doi: 10.1093/nar/gki069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Copley RR, Schmidt S, Ciccarelli FD, Doerks T, Schultz J, Ponting CP, Bork P. SMART 4.0: towards genomic data integration. Nucl Acids Res. 2004;32:D142–144. doi: 10.1093/nar/gkh088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho PM, Henrissat B. The modular structure of cellulases and other carbohydrate-active enzymes: an integrated database approach. Genetics, Biochemistry and Ecology of Cellulose Degradation. 1999.
- Ogata H, Goto S, Fujibuchi W, Kanehisa M. Computation with the KEGG pathway database. Biosystems. 1998;47:119–128. doi: 10.1016/S0303-2647(98)00017-3. [DOI] [PubMed] [Google Scholar]
- Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Distribution of the complete proteomes over the various taxa. Eukaryotic proteomes are: Plasmodium falciparum, Guillardia theta, Candida albicans, Encephalitozoon cuniculi, Neurospora crassa, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Anopheles gambiae, Caenorhabditis elegans, Drosophila melanogaster, Danio rerio (zebrafish), Fugu rubripes, Homo sapiens, Mus musculus, Rattus norvegicus, and Arabidopsis thaliana.
Classification of the 148 HGT candidates. Annotation was given on the basis of the complete analysis (homology with known proteins, homology to proteins of a KOG/COG, PFAM domains and confirmed from the tree). CCD: Complex carbohydrates degradation, F: Fermentation, G: Glycolysis, PD: protein degradation, NM: Nitrogen metabolism. Composition of the Smith Waterman comparison results (Hit; E-value < 1·10–5) or second-smallest partition (SSP): B: only Bacteria; AB: Bacteria and Archaea; A: only Archaea.