

Abstract
Nex1/MATH-2 is a neurogenic basic Helix-Loop-Helix (bHLH) transcription factor that belongs to the NeuroD subfamily. Its expression parallels that of the GAP-43 gene and peaks during brain development, when neurite outgrowth and synaptogenesis are highly active. We previously observed a direct correlation between the levels of expression of Nex1 and GAP-43 proteins, which resulted in extensive neurite outgrowth and neuronal differentiation of PC12 cells in the absence of nerve growth factor. Since the GAP-43 gene is a target for bHLH regulation, we investigated whether Nex1 could regulate the activity of the GAP-43 promoter. We found that among the members of the NeuroD subfamily, Nex1 promoted maximal activity of the GAP-43 promoter. The Nex1-mediated activity is restricted to the conserved E1–E2 cluster located near the major transcription start sites. By electrophoretic mobility shift assay and site-directed mutagenesis, we showed that Nex1 binds as homodimers and that the E1 E-box is a high affinity binding site. We further found that Nex1 released the ME1 E-protein-mediated repression in a concentration dependent manner. Thus, the E1–E2 cluster has a dual function: it can mediate activation or repression depending on the interacting bHLH proteins. Finally, a series of N-terminal and C-terminal deletions revealed that Nex1 transcriptional activity is linked to two distinct transactivation domains, TAD1 and TAD2, with TAD1 being unique to Nex1. Together, our results suggest that Nex1 may engage in selective interactions with components of the core transcriptional machinery whose assembly is dictated by the architecture of the GAP-43 promoter and cellular environment.
Keywords: basic helix-loop-helix transcription factor, GAP-43 promoter, neuritogenesis, NeuroD subfamily, neuronal differentiation, TBP-associated factors
In the age of cell replacement therapy for CNS disorders and injury, the emerging concept indicates that a specific combination of extrinsic signaling and intrinsic factors is critical to the programming of neural stem cells toward specific lineages. Thus, many studies have focused on the elucidation of the molecular steps leading to the establishment of the neuronal fate and the diverse genetic programs controlling the final neurochemical phenotype of mature neurons.
A useful approach in elucidating the distinct differentiation pathways is to unravel the network of transcription factors. A wealth of evidence indicates that the basic helix-loop-helix (bHLH) transcription factors are critical intrinsic determinants of cell fate, differentiation, and maintenance during neurogenesis (reviewed by Bertrand et al. 2002). These transcription factors bind DNA as dimers and recognize the consensus sequence, CANNTG,called an E-box (reviewed by Massari and Murre, 2000). The neurogenic bHLH factors identified in the mammalian nervous system have been classified into two groups based on sequence homology and temporal expression pattern (reviewed by Lee 1997; Guillemot 1999). The determination factors, such as Mash-1, MATH-1, Neurogenin-1, and Neurogenin-2, are transiently expressed in proliferative neural multipotent progenitor cells of the developing brain and are believed to be critical factors in determining various cell lineages (reviewed in Brunet and Ghysen 1991). In contrast, the differentiation factors are expressed later in committed immature neuronal progenitor cells and post-mitotic neurons and regulate the sequential steps leading to terminal differentiation by activating specific genes that contribute to a mature neuronal phenotype (reviewed in Lee 1997; Goridis and Brunet 1999). Members of the NeuroD subfamily, such as NeuroD, NeuroD2 (NDRF), and Nex-1 (MATH-2) display characteristics of differentiation factors as they exhibit a restricted embryonic expression pattern that parallels overt neuronal differentiation and synaptogenesis and are expressed in post-mitotic neurons throughout adulthood (Lee et al. 1995; Shimizu et al. 1995; Schwab et al. 1998).
Unraveling the dynamic interplay between transcription factors and their target genes is the key to understanding their role in mediating the progressive steps leading to neuronal fate and differentiation. In our quest to dissect the bHLH transcriptional network, we recently observed a direct correlation between the level of expression of the Nex1 and GAP-43 proteins, which may be in part responsible for the extensive neurite outgrowth of PC12–Nex1 cells in the absence of nerve growth factor (NGF) (Uittenbogaard and Chiaramello 2002a). A wealth of evidence indicates that GAP-43 expression is critical to the establishment of axonal outgrowth during the initiation and remodeling of neural connections (Zuber et al. 1989; Benowitz and Perrone-Bizzozero 1991; Aigner et al. 1995; Strittmatter et al. 1995; Maier et al. 1999). Recent studies have shown that the GAP-43 gene plays a broader role during neuronal differentiation by controlling the transition from proliferating neuroblasts to fully differentiated neurons (Mani et al. 2000,2001).
Given the pivotal roles of the GAP-43 gene,elucidating its regulation is critical to the understanding of the molecular mechanisms controlling neuronal differentiation, which is the main objective of this study. The regulation of the GAP-43 gene is complex, as it involves transcriptional and post-transcriptional regulatory mechanisms (Basi et al. 1987; Federoff et al. 1988; Perrone-Bizzozero et al. 1991, 1993). At the post-transcriptional level, the increased expression of the GAP-43 protein is the result of an increased stability of the GAP-43 mRNA through the action of neuronal-specific RNA-binding proteins (Kohn et al. 1996; Irwin et al. 1997; Anderson et al. 2000; Mobarak et al. 2000). At the transcriptional level, we and others have shown that the expression of the GAP-43 gene is under the control of bHLH transcription factors (Chiaramello et al. 1996; Kinney et al. 1996; McCormick et al. 1996). The GAP-43 promoter region contains seven E-boxes (E1 to E7) that are organized in two clusters, a distal cluster (E3 to E7) and a proximal cluster (E1 and E2), the latter being critical to the modulation of the GAP-43 promoter activity (Chiaramello et al. 1996). Our initial transcriptional studies have identified the ME1 E-protein as a down-regulator of the GAP-43 gene. This negative regulation was further substantiated with ME1-overexpressing PC12 cells displaying an inverse correlation between the levels of ME1 and GAP-43 expression, even in the presence of NGF (Chiaramello et al. 1996).
To further decipher the molecular mechanisms underlying the transcriptional regulation of GAP-43 expression, we sought to determine whether Nex1 directly modulates the activity of the GAP-43 promoter and has the potential to suppress the ME1-mediated repression. In this study, we show that the Nex-1-mediated activation of the GAP-43 promoter solely occurs through the proximal E1–E2 cluster. Furthermore, among the members of the NeuroD subfamily, Nex1 is the most potent regulator of the GAP-43 gene. By electrophoretic mobility shift assay (EMSA) and site-directed mutagenesis, we demonstrate that Nex1 binds to both E-boxes as homodimers and that only the E1 E-box is a high-affinity Nex1-binding site. In addition, we show that Nex1 releases the ME1-mediated repression in a concentration-dependent manner, indicating that the transcriptional regulation of the GAP-43 gene is the result of a competition between distinct bHLH proteins occurring at the conserved proximal E-box cluster. Finally, a series of N- and C-terminal deletions of the Nex1 protein reveal that the main determinants for the Nex1 transactivation functions lie within two structurally distinct transactivation domains, TAD1 and TAD2, with TAD1 being unique to Nex1.
Materials and methods
Expression vector constructs
The eukaryotic expression plasmid pCDNA3/His/Nex1 was as described in Uittenbogaard and Chiaramello (2002a) and the eukaryotic expression plasmids pRcCMV/ME1 and pRcCMV/Id2 were described in Chiaramello et al. (1995b). The NeuroD cDNA was subcloned into the BamHI and XhoI restriction sites of pcDNA3 (InVitrogen, Carlsbad, CA, USA) to generate the eukaryotic expression plasmid pcDNA3/NeuroD. The NeuroD2 cDNA was subcloned into the ApaI restriction site of pcDNA3 to generate the eukaryotic expression plasmid pcDNA3/NeuroD2. The G2minCAT and the G4minCAT constructs as well as all GAP-43-CAT constructs (GHC, GXC, GHC/E1m, GHC/E2m) were described in Chiaramello et al. (1996). All the N-terminal and C-terminal deletions of Nex1 were generated by PCR using 5′ primers carrying the BamHI restriction site and 3′ primers carrying the XbaI restriction site and cloned into the pcDNA3.1-His vector series to produce polyhistidine-carrying recombinant proteins upon transfection. The Nex1-Mut#1 was obtained by deleting the first 41 amino acids and was cloned in frame into the eukaryotic expression vector pcDNA3.1/HisA (InVitrogen) to generate pcDNA3.1/HisA-Nex1Mut#1. The Nex1-Mut#2 was obtained by deleting the first 72 amino acids and was cloned in frame into the eukaryotic expression vector pcDNA3.1/HisC (InVitrogen) to generate pcDNA3.1/HisC-Nex1-Mut#2. The Nex1-Mut#3 was obtained by deleting the last 98 amino acids and was cloned in frame into the eukaryotic expression vector pcDNA3.1/HisA to generate pcDNA3.1/HisA-Nex1-Mut#3. Finally, the double N-terminal and C-terminal Nex1-Mut#4 mutant was created by deleting the first 41 amino acids and the last 174 amino acids and was cloned in frame into the eukaryotic expression vector pcDNA3.1/HisA to generate pcDNA3.1/HisA-Nex1-Mut#4.
For bacterial expression of the truncated recombinant Nex1, the BamH–EcoRI fragment containing the bHLH motif flanked by 18 amino acids upstream and 90 amino acids downstream was subcloned in frame into the pLEX bacterial expression vector (InVitrogen) to generate pLEX/Nex1-mini plasmid.
Cell culture and chloramphenicol acetyl transferase (CAT) assays
The mouse N18 neuroblastoma cells were described in Amano et al. (1972) and were grown in Dulbecco's modified Eagle's medium as described in Chiaramello et al. (1996). N18 cells were transfected using the Fugene™6 reagent (Roche Applied Sciences, Indianapolis, IN, USA) as recommended by the manufacturer's specifications, and a 60–70% transfection efficiency was constantly achieved with 12 μg of total DNA. Reporter CAT-vectors were transfected in the presence of various expression vectors, when necessary. Amounts of each DNA construct are indicated in the respective graphs. The total amount of transfected DNA was kept constant by the addition of carrier DNA. In addition, to normalize transfection efficiencies, cells were cotransfected with plasmid pRcRSVlacZ (0.5 μg). To measure the transcriptional activity of the GAP-43 promoter, CAT assay was performed following the harvesting and lysing of N18 cells as described in Chiaramello et al. (1996). Relative CAT activity was determined by at least four independent experiments, which were quantified by PhosphorImager analysis (Amersham Biosciences, Piscataway, NJ, USA).
Protein purification
The bacterial expression vector pLEX/Nex1-mini was transformed into the E. coli strain GI724 (InVitrogen) and cells were grown according to the manufacturer's recommendations. The GI724 cells were grown to mid-log phase at 30 °C in induction medium and induced by adding tryptophan to a final concentration of 100 μg/mL. The cells were then grown at 37 °C for 2 h. The cells were centrifuged and the polyhistidine-containing recombinant Nex1 protein was purified by Ni2+-NTA chromatography (BD Biosciences, Clontech, Palo Alto, CA,USA) as described in Chiaramello et al. (1995b). The Nex1 recombinant protein was suspended in a buffer containing 10% glycerol, 20 mm HEPES pH 7.2, 1 mm EDTA, 0.1 m NaCl, 1 mm dithiothreitol, and 1 mm phenylmethanesulfonyl fluoride.
Electrophoretic mobility shift assay (EMSA)
The sequences of the top strands of oligonucleotides (Sigma Genosys, The Woodlands, TX, USA) used as probes or competitors in EMSA analysis were as follows: wild-type E1 E-box, 5′-TAGACCTTACAGTTGCTGCTAACTGCCCTG-3′; mutated E1 E-box, 5′-TAGACCTTATAGTTCCTGCTAACTGCCCTG-3′, wild-type E2 E-box, 5′-GAGAAATGCATATGCGGTGAGCAATAG-3′; and mutated E2 E-box, 5′-GAGAAATGTATATCCGGTGAGCAATAG-3′. The wild-type E-box sequence is underlined and the mutated E-box sequence is in boldface. The complementary strand of each oligonucleotide was annealed, and the double-stranded probe (wild-type E1 or E2) was labeled by kinase treatment using [γ32P]ATP (Amersham Biosciences) and T4 kinase (Roche Applied Sciences). Unlabeled competitor DNA was prepared by annealing complementary oligonucleotides. DNA-binding reactions consisted of 60 ng of purified recombinant Nex1-mini protein incubated with 40 fmol of labeled oligonucleotide E1 or E2 and 100 ng of poly(deoxyinosinic-deoxycytidylic) (Roche Molecular Biochemicals) for 20 min at room temperature in the presence of binding buffer (10 mm Tris-HCl pH 7.5, 50 mm KCl, 0.1 mm EDTA, 1 mm dithiothreitol, 1 mm MgCl2, and 5% glycerol). For competition experiments, the purified recombinant protein Nex1-mini was incubated with either the E1 or E2 probe in the presence of various excess of either unlabeled specific (wild-type E1 or E2) or unlabeled non-specific (mutated E1 or E2) competitors. The products of the DNA-binding reactions were separated on a 5% native gel containing 0.5 × Tris/boric acid/EDTA (45 mm Tris, 45 mm boric acid, and 1 mm EDTA). Gels were dried and visualized by PhosphorImager analysis (Amersham Biosciences).
Results
Nex1 specifically activates the GAP-43 promoter
Following our previous observations that overexpression of Nex1 induced neurite outgrowth accompanied by an increased expression of the GAP-43 protein in untreated PC12 cells, we investigated whether Nex1 could up-regulate the GAP-43 promoter activity through the two clusters of E-boxes. N18 neuroblastoma cells were chosen to perform transient CAT assays, based on the fact that they express high levels of endogenous GAP-43 RNA and that they allow transcriptional modulation of the GAP-43 promoter in a similar manner to PC12 cells (Chiaramello et al. 1996). N18 cells were transfected with the full length GAP-43 GHC carrying the seven E-boxes alone or in combination with the eukaryotic expression plasmid pcDNA3/Nex1. Figure 1 clearly indicates that Nex1 up-regulates the GAP-43 promoter activity by a 12-fold factor. Since NeuroD2 was shown to activate the GAP-43 promoter in P19 cells (McCormick et al. 1996), we compared the levels of Nex1-mediated activation of the GAP-43 promoter to those obtained with the other two members of the NeuroD subfamily, NeuroD and NeuroD2 in N18 cells. We found that NeuroD2 modestly stimulated the GAP-43 promoter activity by a two-fold factor, whereas NeuroD did not have any effect on the GAP-43 promoter activity (Fig. 1). Thus, under this cellular context, Nex1 is the only member of the NeuroD subfamily to induce maximal activation of the GAP-43 promoter. It is noteworthy to mention that the Nex1-mediated activation of GAP-43 promoter appears to be neuronal-specific, as we did not observed a similar activation in non-neuronal cells, such as the African green monkey kidney CV-1 cells (data not shown).
Fig. 1.
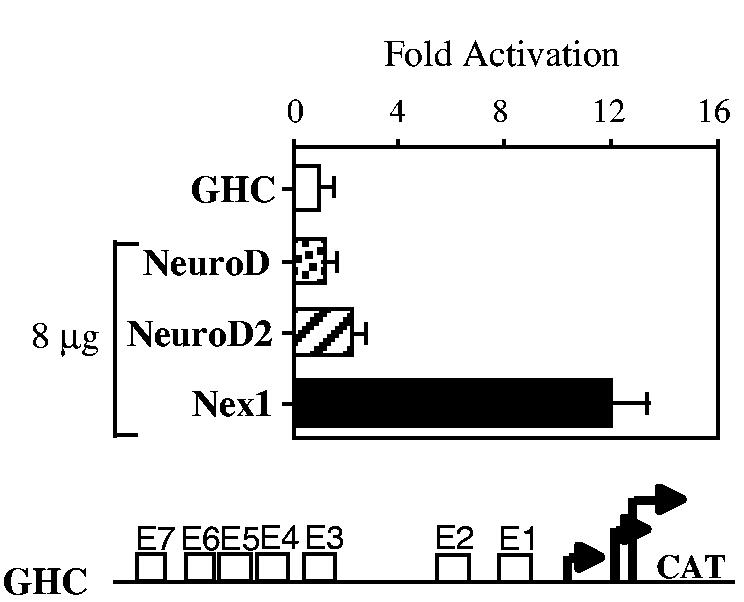
Transcriptional properties of the members of the NeuroD subfamily on the GAP-43 promoter. N18 cells were cotransfected with the reporter-promoter plasmid GHC (1 μg) and eukaryotic expression plasmid containing either NeuroD, NeuroD2, or Nex1 (8 μg). CAT activities are corrected for transfection efficiency and expressed as fold activation relative to the value obtained by transfection of the GHC plasmid alone. Relative CAT activities were presented as means ± SD and represented results of at least four independent experiments performed in duplicate. The reporter-promoter GAP-43/CAT construct (GHC) is illustrated below the graph.
Nex1 activates the GAP-43 promoter through the conserved proximal E-box cluster
In order to determine whether Nex1 regulation of the GAP-43 promoter activity requires both the distal and proximal clusters or only one of them, we used two distinct GAP-43 reporter-promoter constructs, the GAP-43 full length construct GHC and the GAP-43 5′-deletion construct GXC carrying only the proximal E1–E2 cluster. We found that the E1–E2 proximal cluster was sufficient to induce maximal activation of the GAP-43 promoter upon Nex1 expression and that the presence of the distal cluster did not contribute to additional transcriptional activity (Fig. 2a). To confirm the distinct regulatory roles of these two clusters, we used the G2minCAT and G4minCAT reporter-promoter plasmids, which contains either the proximal E1–E2 cluster or the distal cluster E4–E7 cluster, upstream of the minimal herpes simplex virus thymidine kinase (tk) promoter, respectively (Chiaramello et al. 1996). Nex1 only stimulated the promoter activity of the G2minCAT construct (Fig. 2b). In contrast, the G4minCAT promoter activity was not modulated by Nex1 (Fig. 2b). Therefore, these results offer additional support to the conclusion that Nex1 modulates the GAP-43 promoter activity through the conserved E1–E2 proximal cluster.
Fig. 2.

Nex1 activates the GAP-43 promoter through the E1–E2 cluster. (a) The proximal E1–E2 cluster of the GAP-43 promoter is sufficient for maximal Nex1-mediated regulation. N18 cells were cotransfected with the reporter-promoter plasmids (1 μg) GXC or GHC alone (open bars) or with the Nex1 expression plasmid (4 μg) (filled bars). Relative CAT activities were presented as means ± SD and represented results of at least four independent experiments performed in duplicate. A map of the GXC and GHC plasmids is shown on the left of the graph. (b) Nex1 also regulates the activity of the E1–E2 cluster in the context of the chimeric thymidine kinase promoter. N18 cells were cotransfected with the reporter-promoter plasmids (1 μg) G2minCAT or G4minCAT alone (open bars) or with the Nex1 expression plasmid (4 μg) (filled bars). Relative CAT activities were presented as means ± SD and represented results of at least four independent experiments performed in duplicate. A map of the G2minCAT and G4minCAT plasmids is shown on the left of the graph. (c) Mutation of the E1 E box alters the Nex1-mediated regulation of the GAP-43 promoter. N18 cells were cotransfected with the reporter-promoter plasmids (1 μg) GHC, GHC/E1m, or GHC/E2m alone (open bars) or with the Nex1 expression plasmid (4 μg) (filled bars). Relative CAT activities were presented as means ± SD and represented results of at least four independent experiments performed in duplicate. A map of the GHC/E1m and GHC/E2m plasmids is shown on the left of the graph.
To identify which E-box of the proximal cluster is critical to the modulation of the GAP-43 promoter activity by Nex1, we used the full length GHC/E1m and GHC/E2m constructs that carry mutations in either E1 or E2 E-box, respectively, as described in Chiaramello et al. (1996). We found that mutation of the E1 E-box sequence resulted in a 50% decrease of the Nex1-mediated transactivation of GAP-43 promoter, whereas mutation of the E2-box sequence did not affect the regulation of the GAP-43 promoter activity by Nex1 (Fig. 2c). These results indicate that Nex1 regulates the GAP-43 promoter activity predominantly through the E1 E-box. However, the E2 E-box by itself permits partial Nex1-mediated activation of GAP-43 promoter.
Nex1 preferentially binds to the E1 E-box of the GAP-43 promoter
To investigate the mechanism underlying the transcriptional regulation of the GAP-43 promoter activity by Nex1, we determined the Nex1 DNA-binding characteristics by EMSA, using oligonucleotides carrying the E1 or E2 E-box sequence, CAGTTG and CATATG, respectively. Recombinant Nex1 protein was expressed in bacteria and purified by nickel affinity chromatography. As depicted in Fig. 3, Nex1 produced a sequence-specific retarded complex with both E1 and E2 sequences (Figs 3a and b, lane 2). When an excess of unlabeled competitor carrying a mutated E1 or E2 E-box sequence, TAGTTC, was added to the binding reaction, no competition was observed (Fig. 3a, lanes 7–10, and Fig. 3b, lanes 6–8). Thus,Nex1 specifically binds to the E-boxes of the proximal cluster of the GAP-43 promoter as homodimers. To evaluate the strength of Nex1 DNA-binding activity toward the E1 and E2 E-boxes, we performed a competition analysis using different amounts of unlabeled specific oligonucleotides. We found that the Nex1-E1 complex could be successfully competed only in the presence of 200-fold excess unlabeled E1 oligonucleotide (Fig. 3a, lanes 2–6). In contrast, a 25-fold excess of unlabeled E2 oligonucleotide was enough to significantly disrupt the Nex1-E2 complex (Fig. 3b, lanes 2–5). Thus, Nex1 preferentially recognizes the E1 E-box sequence, which is in agreement with the transcriptional data obtained from the transient CAT assays using the GHC/E1m and GHC/E2m reporter-promoter plasmids.
Fig. 3.
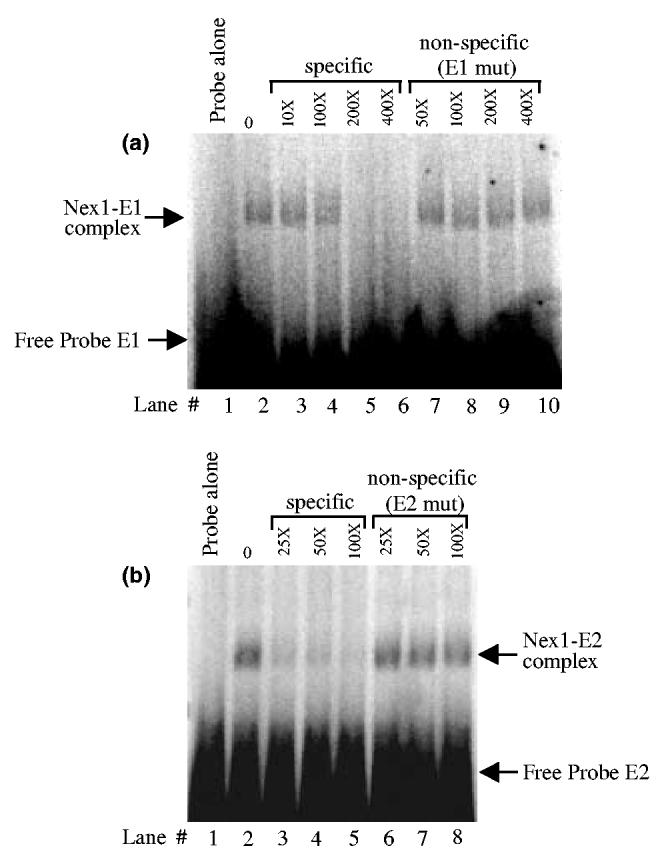
Nex1 binds to the E1 and E2 E-boxes as homodimers. (a) Nex1 binds to the E1 E-box with high affinity. EMSA was performed with 60 ng of purified recombinant Nex1 protein in the absence (lane 2) or presence of increasing amounts of unlabeled specific competitor (lanes 3–6) or unlabeled non-specific competitor in which the E1 E-box was mutated (lanes 7–10). Lane 1 shows the probe E1 alone. The DNA-protein complexes were resolved by native polyacrylamide gel electrophoresis. The free probe E1 is indicated by an arrow. (b) Nex1 binds poorly to the E2 E-box. EMSA was performed as described in (a) in the absence (lane 2) or presence of increasing amounts of unlabeled specific competitor (lanes 3–5) or unlabeled non-specific competitor in which the E1 E-box was mutated (lanes 6–8). Lane 1 shows the probe E2 alone. The free probe E2 is indicated by an arrow.
To investigate further the notion that Nex1 regulates the GAP-43 promoter activity as homodimers, we challenged Nex1 transcriptional activities in the presence of the dominant-negative HLH regulator Id2. The Id proteins lack the DNA-binding motif and thus form non-functional heterodimeric complexes through their helix-Loop-Helix motif (Murre et al. 1989). It is known that the Id proteins essentially sequester the ubiquitous bHLH E-proteins (ME1 and E12), rather than the tissue-specific bHLH proteins, such as Mash-1 and NeuroD, leaving their homodimeric DNA-binding activities unaffected (Benezra et al. 1990; Sun et al. 1991; Jen et al. 1992). We transfected N18 cells with either Nex1 or Id2 alone or with both Nex1 and Id2. We observed that Nex1-mediated transactivation was not sensitive to Id2, implying that Nex1 does not appear to heterodimerize with an endogenous E-protein to activate the GAP-43 promoter (Fig. 4).
Fig. 4.
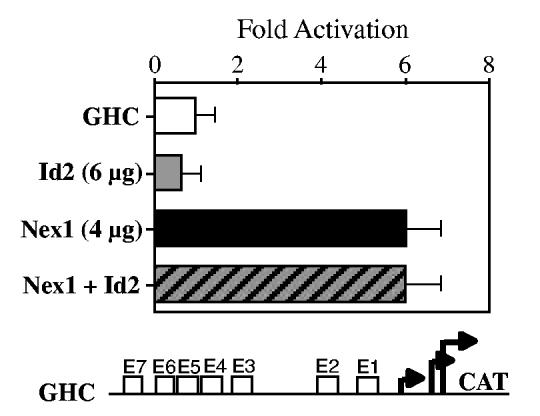
Nex1-mediated regulation of the GAP-43 promoter is insensitive to the dominant negative regulator Id2. N18 cells were transfected with the reporter-promoter plasmid GHC alone (1 μg) or in the presence of eukaryotic expression plasmid containing either Id2 (6 μg) (gray bar) or Nex1 (4 μg) (filled bar) or a combination of both plasmids (hatched bar). CAT activities are corrected for transfection efficiency and expressed as fold activation relative to the value obtained by transfection of the GHC plasmid alone. Relative CAT activities were presented as means ± SD and represented results of at least four independent experiments performed in duplicate. The reporter-promoter GAP-43/CAT construct (GHC) is illustrated below the graph.
Nex1 releases the ME1-mediated repression of the GAP-43 promoter
In view of our previous observations that the activity of the GAP-43 promoter is influenced by the outcome of a protein competition ocurring at the E1–E2 cluster (Chiaramello et al. 1996), we explored the possibility that Nex1 might release the ME1-mediated repression of the GAP-43 promoter. Increasing amounts (0–12 μg) of the Nex1 expression plasmid were cotransfected with a constant amount (8 μg) of ME1 expression plasmid into N18 cells. As shown in Fig. 5, in the presence of 8 μg of ME1 expression plasmid, 4 μg of the Nex1 expression plasmid restored full GAP-43 promoter activity. Furthermore, we found that Nex1 released the ME1-mediated repression of the GAP-43 promoter activity in a concentration-dependent manner. Thus, these transcriptional results clearly demonstrate that Nex1 does not require dimerization with the ME1 E-protein to activate the GAP-43 promoter. These results are in accordance with our in vitro DNA-binding data combined with our results showing a lack of Id2 effect on the Nex1/E1–E2 cluster complex.
Fig. 5.
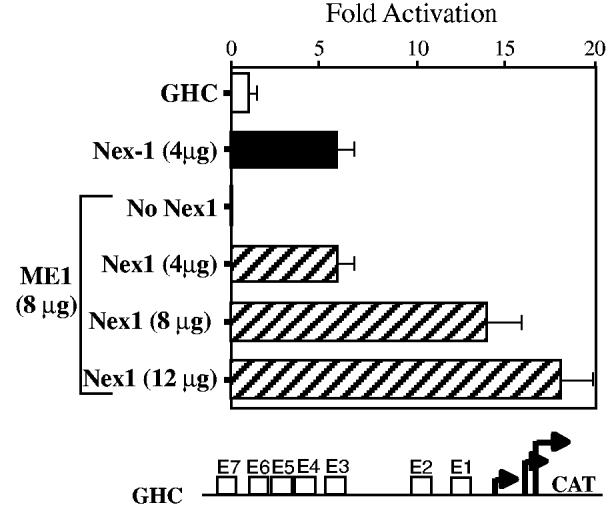
Nex1 releases the ME1-mediated repression of the GAP-43 promoter in a concentration-dependent manner. N18 cells were transfected with the reporter-promoter plasmid GHC alone (open bar) or in the presence of Nex1 expression plasmid (4 μg) (filled bar) or in the presence of a constant amount of ME1 expression plasmid (8 μg) and various amounts of Nex1 expression plasmid (0–12 μg) (hatched bars). CAT activities are corrected for transfection efficiency and expressed as fold activation relative to the value obtained by transfection of the GHC plasmid alone. Relative CAT activities were presented as means ± SD and represented results of at least four independent experiments performed in duplicate. The reporter-promoter GAP-43/CAT construct (GHC) is illustrated below the graph.
Nex1 activates the GAP-43 promoter through two distinct transactivation domains
To explore Nex1 transcriptional functions further, we initially subjected the full-length amino acid sequence of Nex1 to prediction of secondary structures associated with transcriptional activities using both the Chou–Fasman and Robson–Garnier algorithms (Chou and Fasman 1978; Garnier et al. 1978). Both the Chou–Fasman and Robson–Garnier analyses predict an α-helical structure rich in acidic amino acids (25%), called TAD1 that maps in the first 40 amino acids, and a series of beta turns rich in proline and serine residues, called TAD2 that maps between amino acids 232 and 270. Both secondary structures have been found associated with transcriptional activities (Ptashne 1988). Using the BLAST sequence alignment algorithm, we found that only the TAD1 domain is specific to the NeuroD subfamily member Nex1. In contrast, the TAD2 is common to the three members, NeuroD, NeuroD2 and Nex1 (McCormick et al. 1996). In addition, the three members share a glutamic acid rich domain located near the bHLH motif.
As a first assessment, we focused on both the TAD1 and TAD2 domains by generating several N-terminal and C-terminal deletions by PCR without disturbing the nuclear localization domain and the bHLH motif (Fig. 6). Mutant #1 did not include the TAD1 domain, whereas mutant #2 carried a more extensive deletion removing both the TAD1 and the glutamate-rich domain. In mutant #3, the proline-rich domain TAD2 was deleted, whereas in mutant #4,both the TAD1 and TAD2 domains were deleted. These mutants were cloned in frame in the eukaryotic expression vector pcDNA3.1/His and their transcriptional activities were tested on two reporter-promoter constructs, the GAP-43 reporter-promoter plasmid (GHC) and the chimeric reporter-promoter plasmid G2minCAT, carrying the critical E1–E2 cluster upstream of the TK promoter. We compared the transcriptional activity of the wild-type Nex1 protein with various Nex1 truncated proteins in transfected N18 cells. By Western blot, we confirmed that the wild-type and truncated proteins were expressed at comparable levels using a monoclonal antibody against the his-tag epitope of the pcDND3/His vector (data not shown). Deletion of the TAD1 domain abolished Nex1 transcriptional activity on the GAP-43 promoter (Fig. 6, left panel). Further N-terminal deletions (Δ72) that included the stretch of 12 glutamic acid residues did not result in more severe loss of Nex1 transcriptional activity. Removal of the TAD2 domain also resulted in loss of Nex1 transcriptional activity on the GAP-43 promoter (Fig. 6, left panel). As expected, deletion of both the TAD1 and TAD2 domains eliminated most of the Nex1 transcriptional activity. Similar results were obtained on the chimeric reporter-promoter G2minCAT using the same series of truncated mutants (Fig. 6, right panel). These results suggest that retention of both the TAD1 domain and the TAD2 domain is required for full transcriptional activities of Nex1. Thus, these transactivation domains are distinct and both appear to be required for the regulation of the GAP-43 gene.
Fig. 6.

Nex1 activates the GAP-43 promoter through two distinct transactivation domains. The Nex1 mutants used for this analysis are schematically represented at the top of the figure. Both TAD1 and TAD2 domains are indicated by a shaded box, the nuclear localization domain (NL) is indicated by a hatched box and the bHLH motif as well as the stretch of 12 glutamic acid residues are indicated by open boxes. The activity of the wild-type Nex1 protein and each Nex1 mutant was challenged on the two reporter-promoter plasmids, GHC on the left and G2minCAT on the right. Each plasmid is illustrated at the bottom of the figure. N18 cells were transfected with either the reporter-promoter plasmid alone (1 μg) (open bars) or in the presence of wild-type or mutant Nex1 (4 μg) (filled bars). CAT activities are corrected for transfection efficiency and expressed as fold activation relative to the value obtained by transfection of the GHC plasmid alone. Relative CAT activities were presented as means ± SD and represented results of at least four independent experiments performed in duplicate.
Discussion
The present work clearly shows that Nex1 is a key regulator of the GAP-43 gene. We demonstrate that Nex1-mediated activation depends on the presence of the conserved E1–E2 cluster proximal to the multiple transcriptional start sites of the GAP-43 promoter. Nex1 transcriptional activity is dependent on two distinct transactivation domains, TAD1 and TAD2, with TAD1 being unique to Nex1. Furthermore, we observe that the ability of Nex1 to modulate the activity of the GAP-43 promoter appears to require a neural-specific environment suggesting a restrictive selection of protein–protein interactions involving the basal transcription machinery and other enhancer specific transcription factors. Finally, the experiments described in this study indicate that the E1–E2 cluster plays a dual regulatory role in the native context of the GAP-43 promoter: it can function as an activator or repressor element, depending on the interacting bHLH proteins. Thus, this conserved cluster most likely plays a critical role in regulating the expression of the GAP-43 gene during neural development.
Nex1 activates the GAP-43 promoter directly via the conserved E1–E2 cluster
In this study, we examined the transcriptional regulation of the GAP-43 gene by the differentiation factor Nex1, as the expression of both genes peaks during brain development, when neurite outgrowth and synaptogenesis are highly active (Schwab et al. 1998). The GAP-43 protein was shown to modulate key events of neuronal differentiation, such as the transition from proliferative neuroblasts to differentiated neurons and axonal outgrowth (Benowitz and Routtenberg 1997; Mani et al. 2000, 2001). In a PC12 cell system, we showed that overexpression of Nex1 led to increased expression of the GAP-43 protein and extensive neurite outgrowth in the absence of NGF (Uittenbogaard and Chiaramello 2002a). A similar correlation between GAP-43 expression and neurite outgrowth was also observed in PC12 cells stably transfected with the GAP-43 gene (Yankner et al. 1990). Our transcriptional analysis shows that the Nex1-mediated activation mainly occurs through the conserved E1–E2 cluster, which is located in the P2 promoter region of the GAP-43 promoter. Both the rat and human GAP-43 promoters contain two distinct promoter regions, P1 (mapping between −750 and −407) and P2 located within the first −233 bp from the translational start codon (Grabczyk et al. 1990; Örtoft et al. 1993). Only the P2 promoter region is activated during neuronal differentiation of P19-EC cells and in 8-day-old rat brain (Eggen et al. 1994). Interestingly, Nex1 does not activate the distal cluster (E3–E4), which is located in the P1 promoter region. The fact that the activity of this cluster is not regulated by Nex1 when cloned upstream of the chimeric thymidine kinase (tk) promoter suggests that Nex1 might not target these E-boxes. In contrast, Nex1 up-regulates the activity of the proximal cluster E1–E2, when cloned upstream of the tk promoter, in a manner similar to the homologous GAP-43 promoter. From our site-directed mutagenesis and DNA-binding studies on the E1–E2 cluster, it is clear that Nex1 predominantly regulates the GAP-43 promoter activity through the E1 E-box. However, the E2 E-box by itself permits a partial Nex1-mediated activation of the GAP-43 promoter. These two E-boxes differ in sequence and position; the E1 E box (CAGTTG) is located proximal to the major transcription start sites, whereas the E2 E-box (CATATG) is located distal to the E1 E-box.
Thus, the transcriptional analysis described in this study combined with the morphological and genetic characteristics of PC12-Nex1 cells imply that the increased expression of the GAP-43 protein is most likely the result of a direct transcriptional regulation of the GAP-43 gene under the control of Nex1. However, it is known that the regulation of the GAP-43 gene also occurs at the post-transcriptional level through the modulation of the GAP-43 mRNA stability involving several RNA-binding proteins induced upon NGF exposure (Kohn et al. 1996; Irwin et al. 1997). Overexpression of the RNA-binding protein HuD in untreated PC12 cells results in increased stability of GAP-43 mRNA and therefore increased expression of the GAP-43 protein, accompanied with spontaneous neurite outgrowth (Tsai et al. 1997; Anderson et al. 2000; Mobarak et al. 2000). Thus, the fact that Nex1 is an important effector of the NGF pathway (Uittenbogaard and Chiaramello 2002a) raises the possibility that Nex1 may also indirectly control the stability of the GAP-43 mRNA by up-regulating the expression of RNA-binding protein encoding genes.
The transactivation domain TAD1 unique to Nex1 may be responsible for maximal activation of the GAP-43 promoter
Our deletion analysis shows that Nex1 harbors two transactivation domains, TAD1 and TAD2, which are both required for GAP-43 promoter activation. These two transactivation domains differ in their location, amino acid preponderance, and secondary structure. TAD1 resides in the first 40 amino acids, whereas TAD2 lies in the last 98 amino acids. TAD1 is rich in acidic amino acids and is characterized by an α-helical segment on its C-terminal side, whereas TAD2 is rich in proline and serine residues. Interestingly, amino-acid sequence alignment analysis reveals that TAD1 is specific to Nex1, whereas TAD2 is found in the C-terminal portion of all three members of the NeuroD subfamily. Several recent studies have demonstrated that the TAD2 motif is essential to NeuroD and NeuroD2-mediated transactivation (Sharma et al. 1999; Konishi et al. 2000). Thus, the differential transcriptional modulation of the GAP-43 promoter activity may stem from the presence of distinct transactivation domains in Nex1 and NeuroD2 sequences, which may result in differential interactions with neighboring transcription factors and cofactors of the general transcriptional machinery.
It has been shown that the GA region located upstream of the E1–E2 cluster (−168 to −118) contributes to the regulation of the GAP-43 promoter activity through the Ets-binding sites (Starr et al. 1994). Moreover, the interactions between the bHLH transcription factors and the Ets-binding proteins have been shown to be critical in regulating the expression of the Ig heavy chain gene (Rivera et al. 1993). Thus, it is plausible that the presence of these Ets-binding sites may be critical to the Nex1-mediated activation of the GAP-43 promoter. In recent years, a number of interactions have been described between transcription factors and the TBP-associated factors (TAFs) of the basal transcription machinery. Furthermore, it is well documented that transactivation domains of sequence-specific activators directly bind to TAFs in order to interface with the basal transcription machinery and mediate specific activation of transcription (reviewed in Lemon and Tjian, 2000). The specificity of these protein–protein interactions is dictated by the motif of transactivation domains, such that different classes of transactivation domains target different TAFs. Acidic activators have been shown to preferentially interact with human TAFII31, whereas proline-rich transcription factors are engaged in specific interactions with hTAFII50 (Thut et al. 1995; Zawel and Reinberg 1995; Uesugi et al. 1997; Uesugi and Verdine 1999). Thus, the requirement of both TAD1 and TAD2 for Nex1 activity supports the notion that both TADs may be selectively engaged in interactions with distinct coactivators of the core transcriptional machinery.
We therefore propose that the strength of the Nex1-mediated activation of the GAP-43 promoter may be attributed to the assembly of different transcription complexes resulting from the adaptation of the GAP-43 promoter architecture to specific cellular environment. Because these promoter studies were conducted in a defined cellular context, such as N18 and P19 cells, we cannot exclude that under different cellular and developmental environment, NeuroD2, NeuroD, or other bHLH factors may efficiently modulate the GAP-43 promoter activity. In this regard, it is noteworthy that Nex1 does not regulate GAP-43 promoter activity in the non-neuronal CV1 cells. Ultimately, the presence of stage-specific or lineage-specific cofactors may determine the outcome of GAP-43 expression.
Implications of the bHLH-mediated regulation of the GAP-43 gene during neural development
The expression of the GAP-43 gene necessitates a complex interplay of activators and repressors in order to achieve a specific spatio-temporal pattern during brain development. The distinct regulatory effects of the bHLH transcription factors Nex1 and ME1 on the GAP-43 promoter activity may be of special relevance. The ME1 and Nex1 genes display both overlapping and non-overlapping patterns of expression during CNS development. Both transcription factors are coexpressed in areas of the brain associated with neuronal plasticity, such as the cerebellum and hippocampus. More specifically, Nex1 and ME1 are found in mature cerebellar granule neurons located in the internal granule layer as well as the pyramidal neurons of the CA1-CA3 layer of the hippocampus (Chiaramello et al. 1995b; Schwab et al. 1998). Our transcriptional analyses reveal that the expression of the GAP-43 gene is the result of a protein competition occurring at the conserved E1–E2 cluster, with ME1 behaving as a repressor and Nex1 as an activator. The fact that Nex1 effectively releases the ME1-mediated repression of the GAP-43 gene at low concentrations in N18 cells suggests that Nex1 may competitively promote in vivo expression of the GAP-43 gene in areas of the brain where Nex1 and ME1 expression overlaps. This notion is further substantiated by our DNA-binding studies revealing that the Nex1–E1 interactions are stronger than ME1–E1 interactions (Chiaramello et al. 1996).
In contrast,during cortex and spinal cord development, the expression of ME1 and Nex1 are mutually exclusive, with ME1 being restricted to the proliferative ventricular zones, and Nex1 limited to the cerebral intermediate zone and cortical plate (Bartholoma and Nave 1994; Shimizu et al. 1995; Schwab et al. 1998; Uittenbogaard and Chiaramello 2002b). Therefore, it has been postulated that ME1 may sustain the undifferentiated state of progenitor cells by repressing the expression of genes promoting terminal differentiation (Uittenbogaard and Chiaramello 2002b). We have shown that ME1 represses the activity of both the GAP-43 p75LNGFR promoters by binding to regulatory E-boxes as homodimers (Chiaramello et al. 1995a, 1996). However, the Nex1 and GAP-43 genes display an overlapping expression profile throughout brain development, with their expression being induced by embryonic day 16 and peaking postnatally around day 5 (Schwab et al. 1998). Our transcriptional analysis combined with the observed coexpression of Nex1 and GAP-43 proteins in Nex1-overexpressing PC12 cells suggests a functional relationship between these two neuronal genes. However, the homozygous Nex1 knockout mice fail to reveal a significant decrease in GAP-43 expression, suggesting that functional redundancy most likely exists among members of the NeuroD subfamily to compensate for the loss of Nex1 activity (Naya et al. 1997; Schwab et al. 1998; Miyata et al. 1999; Schwab et al. 2000; Olson et al. 2001). NeuroD2 (NDRF) may regulate the GAP-43 promoter activity in vivo, as we and others have observed a modest NeuroD2–mediated activation of the GAP-43 gene in PC12 and P19 cells (McCormick et al. 1996). Furthermore, the fact that the NeuroD2 expression pattern overlaps with the GAP-43 gene during brain development lends credence to this hypothesis. In conclusion, the major finding of this study is that the differentiation factor Nex1 regulates the expression of the GAP-43 gene at the transcriptional level, which validates our hypothesis of Nex1 acting as a key regulator of neuritogenesis through transcriptional regulation of specific genes (Uittenbogaard and Chiaramello 2002a). The identification of additional Nex1-regulated target genes will aid in further understanding the molecular mechanisms of action of Nex1 during neurogenesis.
Acknowledgements
We thank Dr Robyn Rufner for her assistance and technical help at the Center for Microscopic Image Analysis. This work was supported in part by NIH grants R01-NS41391 to AC and by P30HD40677.
Abbreviations used
- bHLH
basic helix-loop-helix
- CAT
chloramphenicol acetyl transferase
- EMSA
electrophoretic mobility shift assay
- NGF
nerve growth factor
- TAD
transactivation domain
- TAF
TBP-associated factor
- TBP
TATA binding protein
References
- Aigner L, Arber S, Kapfhammer JP, Laux T, Schneider C, Botteri F, Brenner HR, Caroni P. Overexpression of the neural growth-associated protein GAP-43 induces nerve sprouting in the adult nervous system of transgenic mice. Cell. 1995;83:269–278. doi: 10.1016/0092-8674(95)90168-x. [DOI] [PubMed] [Google Scholar]
- Amano T, Richelson E, Nirenberg M. Neurotransmitter synthesis by neuroblastoma clones. Proc. Natl Acad. Sci. USA. 1972;69:258–263. doi: 10.1073/pnas.69.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KD, Morin MA, Beckel-Mitchener A, Mobarak CD, Neve RL, Furneaux HM, Burry R, Perrone-Bizzozero NI. Overexpression of HuD, but not of its truncated form HuD I + II, promotes GAP-43 gene expression and neurite outgrowth in PC12 cells in the absence of nerve growth factor. J. Neurochem. 2000;75:1103–1114. doi: 10.1046/j.1471-4159.2000.0751103.x. [DOI] [PubMed] [Google Scholar]
- Bartholoma A, Nave KA. NEX-1: a novel brain-specific helix-loop-helix protein with autoregulation and sustained expression in mature cortical neurons. Mech. Dev. 1994;48:217–228. doi: 10.1016/0925-4773(94)90061-2. [DOI] [PubMed] [Google Scholar]
- Basi GS, Jacobson RD, Virag I, Schilling J, Skene JH. Primary structure and transcriptional regulation of GAP-43, a protein associated with nerve growth. Cell. 1987;49:785–791. doi: 10.1016/0092-8674(87)90616-7. [DOI] [PubMed] [Google Scholar]
- Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell. 1990;61:49–59. doi: 10.1016/0092-8674(90)90214-y. [DOI] [PubMed] [Google Scholar]
- Benowitz LI, Perrone-Bizzozero NI. The expression of GAP-43 in relation to neuronal growth and plasticity: when, where, how, and why? Prog. Brain Res. 1991;89:69–87. doi: 10.1016/s0079-6123(08)61716-1. [DOI] [PubMed] [Google Scholar]
- Benowitz LI, Routtenberg A. GAP-43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci. 1997;20:84–91. doi: 10.1016/s0166-2236(96)10072-2. [DOI] [PubMed] [Google Scholar]
- Bertrand N, Castro DS, Guillemot F. Proneural genes and the specification of neural cell types. Nat. Rev. Neurosci. 2002;3:517–530. doi: 10.1038/nrn874. [DOI] [PubMed] [Google Scholar]
- Brunet JF, Ghysen A. Deconstructing cell determination: proneural genes and neuronal identity. Bioessays. 1999;21:313–318. doi: 10.1002/(SICI)1521-1878(199904)21:4<313::AID-BIES7>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Chiaramello A, Neuman K, Palm K, Metsis M, Neuman T. Helix-loop-helix transcription factors mediate activation and repression of the p75LNGFR gene. Mol. Cell. Biol. 1995a;15:6036–6044. doi: 10.1128/mcb.15.11.6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaramello A, Soosaar A, Neuman T, Zuber MX. Differential expression and distinct DNA-binding specificity of ME1a and ME2 suggest a unique role during differentiation and neuronal plasticity. Brain Res. Mol. Brain Res. 1995b;29:107–118. doi: 10.1016/0169-328x(94)00236-8. [DOI] [PubMed] [Google Scholar]
- Chiaramello A, Neuman T, Peavy DR, Zuber MX. The GAP-43 gene is a direct downstream target of the basic helix-loop-helix transcription factors. J. Biol. Chem. 1996;271:22035–22043. doi: 10.1074/jbc.271.36.22035. [DOI] [PubMed] [Google Scholar]
- Chou PY, Fasman GD. Empirical predictions of protein conformation. Annu. Rev. Biochem. 1978;47:251–276. doi: 10.1146/annurev.bi.47.070178.001343. [DOI] [PubMed] [Google Scholar]
- Eggen BJ, Nielander HB, de Rensen-Leeuw MG, Schotman P, Gispen WH, Schrama LH. Identification of two promoter regions in the rat B-50/GAP-43 gene. Brain Res. Mol. Brain Res. 1994;23:221–234. doi: 10.1016/0169-328x(94)90229-1. [DOI] [PubMed] [Google Scholar]
- Federoff HJ, Grabczyk E, Fishman MC. Dual regulation of GAP-43 gene expression by nerve growth factor and glucocorticoids. J. Biol. Chem. 1988;263:19290–19295. [PubMed] [Google Scholar]
- Garnier J, Osguthorpe DJ, Robson B. Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J. Mol. Biol. 1978;120:97–120. doi: 10.1016/0022-2836(78)90297-8. [DOI] [PubMed] [Google Scholar]
- Goridis C, Brunet JF. Transcriptional control of neurotransmitter phenotype. Curr. Opin. Neurobiol. 1999;9:47–53. doi: 10.1016/s0959-4388(99)80006-3. [DOI] [PubMed] [Google Scholar]
- Grabczyk E, Zuber MX, Federoff HJ, Ng SC, Pack A, Fishman MC. Cloning and characterization of the rat gene encoding GAP-43. Eur. J. Neurosci. 1990;2:822–827. doi: 10.1111/j.1460-9568.1990.tb00393.x. [DOI] [PubMed] [Google Scholar]
- Guillemot F. Vertebrate bHLH genes and the determination of neuronal fates. Exp. Cell Res. 1999;253:357–364. doi: 10.1006/excr.1999.4717. [DOI] [PubMed] [Google Scholar]
- Irwin N, Baekelandt V, Goritchenko L, Benowitz LI. Identification of two proteins that bind to a pyrimidine-rich sequence in the 3′-untranslated region of GAP-43 mRNA. Nucleic Acids Res. 1997;25:1281–1288. doi: 10.1093/nar/25.6.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jen Y, Weintraub H, Benezra R. Overexpression of Id protein inhibits the muscle differentiation program: in vivo association of Id with E2A proteins. Genes Dev. 1992;6:1466–1479. doi: 10.1101/gad.6.8.1466. [DOI] [PubMed] [Google Scholar]
- Kinney WR, McNamara RK, Valcourt E, Routtenberg A. Prolonged alteration in E-box binding after a single systemic kainate injection: potential relation to F1/GAP-43 gene expression. Brain Res. Mol. Brain Res. 1996;38:25–36. doi: 10.1016/0169-328x(95)00287-3. [DOI] [PubMed] [Google Scholar]
- Kohn DT, Tsai KC, Cansino VV, Neve RL, Perrone-Bizzozero NI. Role of highly conserved pyrimidine-rich sequences in the 3′ untranslated region of the GAP-43 mRNA in mRNA stability and RNA–protein interactions. Brain Res. Mol. Brain Res. 1996;36:240–250. doi: 10.1016/0169-328x(95)00239-o. [DOI] [PubMed] [Google Scholar]
- Konishi Y, Aoki T, Ohkawa N, Matsu-Ura T, Mikoshiba K, Tamura T. Identification of the C-terminal activation domain of the NeuroD-related factor (NDRF) Nucleic Acids Res. 2000;28:2406–2412. doi: 10.1093/nar/28.12.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE. Basic helix-loop-helix genes in neural development. Curr. Opin. Neurobiol. 1997;7:13–20. doi: 10.1016/s0959-4388(97)80115-8. [DOI] [PubMed] [Google Scholar]
- Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science. 1995;268:836–844. doi: 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]
- Lemon B, Tjian R. Orchestrated response: a symphony of transcription factors for gene control. Genes Dev. 2000;14:2551–2569. doi: 10.1101/gad.831000. [DOI] [PubMed] [Google Scholar]
- Maier DL, Mani S, Donovan SL, Soppet D, Tessarollo L, McCasland JS, Meiri KF. Disrupted cortical map and absence of cortical barrels in growth-associated protein (GAP)-43 knockout mice. Proc. Natl Acad. Sci. USA. 1999;96:9397–9402. doi: 10.1073/pnas.96.16.9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani S, Schaefer J, Meiri KF. Targeted disruption of GAP-43 in P19 embryonal carcinoma cells inhibits neuronal differentiation as well as acquisition of the morphological phenotype. Brain Res. 2000;853:384–395. doi: 10.1016/s0006-8993(99)02042-9. [DOI] [PubMed] [Google Scholar]
- Mani S, Shen Y, Schaefer J, Meiri KF. Failure to express GAP-43 during neurogenesis affects cell cycle regulation and differentiation of neural precursors and stimulates apoptosis of neurons. Mol. Cell. Neurosci. 2001;17:54–66. doi: 10.1006/mcne.2000.0931. [DOI] [PubMed] [Google Scholar]
- Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eukaryotic organisms. Mol. Cell. Biol. 2000;20:429–440. doi: 10.1128/mcb.20.2.429-440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick MB, Tamimi RM, Snider L, Asakura A, Bergstrom D, Tapscott SJ. NeuroD2 and neuroD3: distinct expression patterns and transcriptional activation potentials within the NeuroD gene family. Mol. Cell. Biol. 1996;16:5792–5800. doi: 10.1128/mcb.16.10.5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata T, Maeda T, Lee JE. NeuroD is required for differentiation of the granule cells in the cerebellum and hippocampus. Genes Dev. 1999;13:1647–1652. doi: 10.1101/gad.13.13.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobarak CD, Anderson KD, Morin M, Beckel-Mitchener A, Rogers SL, Furneaux H, King P, Perrone-Bizzozero NI. The RNA-binding protein HuD is required for GAP-43 mRNA stability, GAP-43 gene expression, and PKC-dependent neurite outgrowth in PC12 cells. Mol. Biol. Cell. 2000;11:3191–3203. doi: 10.1091/mbc.11.9.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murre C, McCaw PS, Vaessin H, Caudy M, January LY, January YN, Cabrera CV, Buskin JN, Hauschka SD, Lassar AB, et al. Interactions between heterologous helix-loop-helix proteins generate complexes that bind specifically to a common DNA sequence. Cell. 1989;58:537–544. doi: 10.1016/0092-8674(89)90434-0. [DOI] [PubMed] [Google Scholar]
- Naya FJ, Huang HP, Qiu Y, Mutoh H, DeMayo FJ, Leiter AB, Tsai MJ. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997;11:2323–2334. doi: 10.1101/gad.11.18.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JM, Asakura A, Snider L, Hawkes R, Strand A, Stoeck J, Hallahan A, Pritchard J, Tapscott SJ. NeuroD2 is necessary for development and survival of central nervous system neurons. Dev. Biol. 2001;234:174–187. doi: 10.1006/dbio.2001.0245. [DOI] [PubMed] [Google Scholar]
- Örtoft E, Påhlman S, Andersson G, Parrow V, Betsholtz C, Hammerling U. Human GAP-43 gene expression: multiple start sites for initiation of transcription in differentiating human neuroblastoma. Mol. Cell. Neurosci. 1993;4:549–561. doi: 10.1006/mcne.1993.1068. [DOI] [PubMed] [Google Scholar]
- Perrone-Bizzozero NI, Neve RL, Irwin N, Lewis S, Fisher I, Benowitz LI. Post-transcriptional regulation of GAP-43 mRNA levels during neuronal differentiation and nerve regeneration. Mol. Cell. Neurosci. 1991;2:402–409. doi: 10.1016/1044-7431(91)90027-l. [DOI] [PubMed] [Google Scholar]
- Perrone-Bizzozero NI, Cansino VV, Kohn DT. Post-transcriptional regulation of GAP-43 gene expression in PC12 cells through protein kinase C-dependent stabilization of the mRNA. J. Cell Biol. 1993;120:1263–1270. doi: 10.1083/jcb.120.5.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptashne M. How eukaryotic transcriptional activators work. Nature. 1988;335:683–689. doi: 10.1038/335683a0. [DOI] [PubMed] [Google Scholar]
- Rivera RR, Stuiver MH, Steenbergen R, Murre C. Ets proteins: new factors that regulate immunoglobulin heavy-chain gene expression. Mol. Cell. Biol. 1993;13:7163–7169. doi: 10.1128/mcb.13.11.7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab MH, Druffel-Augustin S, Gass P, Jung M, Klugmann M, Bartholomae A, Rossner MJ, Nave KA. Neuronal basic helix-loop-helix proteins (NEX, neuroD, NDRF): spatio-temporal expression and targeted disruption of the NEX gene in transgenic mice. J. Neurosci. 1998;18:408–1418. doi: 10.1523/JNEUROSCI.18-04-01408.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab MH, Bartholomae A, Heimrich B, Feldmeyer D, Druffel-Augustin S, Goebbels S, Naya FJ, Zhao S, Frotscher M, Tsai MJ, Nave KA. Neuronal basic helix-loop-helix proteins (NEX and BETA2/Neuro D) regulate terminal granule cell differentiation in the hippocampus. J. Neurosci. 2000;20:3714–3724. doi: 10.1523/JNEUROSCI.20-10-03714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Moore M, Marcora E, Lee JE, Qiu Y, Samaras S, Stein R. The NeuroD1/BETA2 sequences essential for insulin gene transcription colocalize with those necessary for neurogenesis and p300/CREB binding protein binding. Mol. Cell. Biol. 1999;19:704–713. doi: 10.1128/mcb.19.1.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu C, Akazawa C, Nakanishi S, Kageyama R. MATH-2, a mammalian helix-loop-helix factor structurally related to the product of Drosophila proneural gene atonal, is specifically expressed in the nervous system. Eur. J. Biochem. 1995;229:239–248. doi: 10.1111/j.1432-1033.1995.tb20461.x. [DOI] [PubMed] [Google Scholar]
- Starr RG, Lu B, Federoff HJ. Functional characterization of the rat GAP-43 promoter. Brain Res. 1994;638:211–220. doi: 10.1016/0006-8993(94)90652-1. [DOI] [PubMed] [Google Scholar]
- Strittmatter SM, Fankhauser C, Huang PL, Mashimo H, Fishman MC. Neuronal pathfinding is abnormal in mice lacking the neuronal growth cone protein GAP-43. Cell. 1995;80:445–452. doi: 10.1016/0092-8674(95)90495-6. [DOI] [PubMed] [Google Scholar]
- Sun XH, Copeland NG, Jenkins NA, Baltimore D. Id proteins Id1 and Id2 selectively inhibit DNA binding by one class of helix-loop-helix proteins. Mol. Cell. Biol. 1991;11:5603–5611. doi: 10.1128/mcb.11.11.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thut CJ, Chen JL, Klemm R, Tjian R. p53 transcriptional activation mediated by coactivators TAFII40 and TAFII60. Science. 1995;267:100–104. doi: 10.1126/science.7809597. [DOI] [PubMed] [Google Scholar]
- Tsai KC, Cansino VV, Kohn DT, Neve RL, Perrone-Bizzozero NI. Post-transcriptional regulation of the GAP-43 gene by specific sequences in the 3′ untranslated region of the mRNA. J. Neurosci. 1997;17:1950–1958. doi: 10.1523/JNEUROSCI.17-06-01950.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uesugi M, Verdine GL. The alpha-helical FXXPhiPhi motif in p53: TAF interaction and discrimination by MDM2. Proc. Natl Acad. Sci. USA. 1999;96:14801–14806. doi: 10.1073/pnas.96.26.14801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uesugi M, Nyanguile O, Lu H, Levine AJ, Verdine GL. Induced alpha helix in the VP16 activation domain upon binding to a human TAF. Science. 1997;277:1310–1313. doi: 10.1126/science.277.5330.1310. [DOI] [PubMed] [Google Scholar]
- Uittenbogaard M, Chiaramello A. Constitutive overexpression of the basic helix-loop-helix Nex1/MATH-2 transcription factor promotes neuronal differentiation of PC12 cells and neurite regeneration. J. Neurosci. Res. 2002a;67:235–245. doi: 10.1002/jnr.10119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uittenbogaard M, Chiaramello A. Expression of the bHLH transcription factor Tcf12 (ME1) gene is linked to the expansion of precursor cell populations duirng neurogenesis. Gene Expr. Patterns. 2002b;1:115–121. doi: 10.1016/s1567-133x(01)00022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankner BA, Benowitz LI, Villa-Komaroff L, Neve RL. Transfection of PC12 cells with the human GAP-43 gene: effects on neurite outgrowth and regeneration. Brain Res. Mol. Brain Res. 1990;7:39–44. doi: 10.1016/0169-328x(90)90071-k. [DOI] [PubMed] [Google Scholar]
- Zawel L, Reinberg D. Common themes in assembly and function of eukaryotic transcription complexes. Annu. Rev. Biochem. 1995;64:533–561. doi: 10.1146/annurev.bi.64.070195.002533. [DOI] [PubMed] [Google Scholar]
- Zuber MX, Goodman DW, Karns LR, Fishman MC. The neuronal growth-associated protein GAP-43 induces filopodia in non-neuronal cells. Science. 1989;244:1193–1195. doi: 10.1126/science.2658062. [DOI] [PubMed] [Google Scholar]