

Abstract
Although the wild-type prion protein (PrP) is abundant and widely expressed in various types of tissues and cells, its physiological function(s) remain unknown, and PrP knockout mice do not exhibit overt and undisputed phenotypes. Here we showed that PrP is expressed on the surface of several bone marrow cell populations successively enriched in long-term (LT) hematopoietic stem cells (HSCs) using flow cytometry analysis. Affinity purification of the PrP-positive and -negative fractions from these populations, followed by competitive bone marrow reconstitution assays, shows that all LT HSCs express PrP. HSCs from PrP-null bone marrow exhibited impaired self-renewal in serial transplantation of lethally irradiated mouse recipients both in the presence and absence of competitors. When treated with a cell cycle-specific myelotoxic agent, the animals reconstituted with PrP-null HSCs exhibit increased sensitivity to hematopoietic cell depletion. Ectopic expression of PrP in PrP-null bone marrow cells by retroviral infection rescued the defective hematopoietic engraftment during serial transplantation. Therefore, PrP is a marker for HSCs and supports their self-renewal.
Keywords: bone marrow, serial transplantation, hematopoiesis, reconstitution
The difficulty in purification and study of hematopoietic stem cells (HSCs) is hampered by our limited understanding of the proteins that are specifically expressed on their surface (1–3). Furthermore, few HSC markers are of functional significance for HSC activity (4). We characterized a population of mouse fetal liver cells that express the CD3 antigen (CD3+) and support the ex vivo expansion of HSCs, and showed that they express abundant amounts of prion protein (PrP) (5). PrP is a highly conserved glycoprotein tethered to cell membranes by a glycosylphosphatidylinositol (GPI) anchor that is expressed on hematopoietic cells as well as in many tissues including brain, heart, and muscle (6–8). Although it is well established that PrP is the primary component of infectivity in prion diseases (9), its normal function(s) remains obscure. Several roles for PrP have been suggested, including copper uptake, cell signaling, cell survival, protection against oxidative stress, cell adhesion, and differentiation (10, 11). However, PrP-null mice exhibit no consistent, overt, phenotype other than resistance to infection with prions (12, 13). Here we demonstrate that PrP is a surface marker for HSCs and is required for their self-renewal, as judged by successive bone marrow transplantations.
Results
PrP Is a Marker for Long-Term (LT) HSCs.
Preliminary studies showed that 40% of adult mouse bone marrow (BM) cells express PrP on their surface. More than 80% of these PrP+ cells were erythroid cells as they expressed the glycophorin-related surface protein Ter119 (Ter119+) (data not shown). In Fig. 1 the expression of the PrP protein was monitored on the surface of WT mouse BM cell populations that were progressively enriched for HSCs. One way to enrich HSCs is isolation of the side population (SP) fraction of adult BM cells, which is identified by low accumulation of Hoechst dye 33342 (14). Fig. 1A, plot 4 shows that PrP was expressed on 47.1 ± 5.4% of SP cells (n = 6).
Fig. 1.

PrP is expressed on bone marrow populations enriched in HSC activity. (A) Freshly isolated BM cells were stained with Hoechst dye 33342, and the SP fraction was gated (gates 1 and 2) to analyze the expression of PrP. In plot 1, forward scatter (FSC) and side scatter (SSC) is used to gate on hematopoietic cells. Hoechst Red and Hoechst Blue (plots 2 and 2a, gate 2 was set as 0.02% of total cells) represent two fluorescence emission wavelengths used to detect the SP cells. Plot 2a is an expansion of the gate 2 region of plot 2. PrP-null BM SP cells served as a negative control for PrP antibody staining (plot 3). WT BM SP cells were stained for PrP (plots 4–12) together with isotype control (plot 4), Endoglin (plot 5), Sca-1 (plot 6), CD43 (plot 7), CD44 (plot 8), CD49D (plot 9), CD49E (plot 10), CD11A (plot 11), or CD62L (plot 12). (B) Total BM cells were stained with anti-Endoglin followed sequentially by anti-rat-PE/CY5.5, a mixture of biotinylated lineage-specific antibodies, and streptavidin–APC, anti-PrP-FITC, and anti-Sca-1-PE. Plot 1 shows the gate of FSC and SSC channels. The lowest 5% of APC-stained cells (i.e., Lin−) were gated (plot 2). Plot 3 shows the staining of the gated Lin− cells with Sca-1 and Endoglin, and plot 4 shows the staining of the gated Lin− Sca-1+ Endoglin+ cells with PrP. Plot 5 shows the staining of the gated Lin− Sca-1+ Endoglin− cells with PrP.
All HSCs in the SP population express Endoglin (4), an ancillary TGF-β receptor. Of the Endoglin+ SP cells, 56.4 ± 12.0% also expressed PrP (Table 1, which is published as supporting information on the PNAS web site, and Fig. 1A, plot 5). Sca-1 is another well established surface marker for HSCs (15). Of the Sca-1+ SP cells, 50.9 ± 12.4% also expressed PrP (Table 1 and Fig. 1A, plot 6). Furthermore, virtually all of the PrP+ SP cells also expressed other cell adhesion molecules that are expressed on HSCs or early hematopoietic progenitors, including CD43, CD44, CD49D, and CD49E (16, 17) (Table 1 and Fig. 1A, plots 7–10, respectively). In contrast, CD11A and CD62L, antigens not expressed on HSCs (16), were expressed on only 27% [=13/(13 + 34)] and 9% [=4/(4 + 41)], respectively, of the PrP+ SP cells (Fig. 1A, plots 11 and 12).
To further test PrP expression on the HSC enriched population, we took advantage of two other features of HSCs. First, HSCs are lineage negative (Lin−), that is, they do not express surface markers of differentiated erythroid, myeloid, or lymphoid cell lineages. Second, all HSCs do express the surface markers Sca-1 and Endoglin (4) and thus are Lin−Sca-1+Endoglin+. Plot 3 in Fig. 1B shows that Lin−Sca-1+Endoglin+ cells comprised 0.03% (0.05 × 0.006) of total BM cells; of these cells, 85.7% expressed surface PrP (plot 4). In contrast, of the non-HSC Lin−Sca-1+Endoglin− cell population (4), only 22.8% expressed surface PrP (plot 5). Because 1 in 18 Lin−Sca-1+Endoglin+ cells is a LT HSC (4), the expression of PrP on Lin−Sca-1+ Endoglin+ cells, together with PrP coexpression on SP stained Sca-1+ and Endoglin+ cells, suggested that PrP might be a surface marker for LT HSCs.
The ability to reconstitute the hematopoietic system of lethally irradiated mice is the hallmark of LT HSCs. To determine whether LT HSCs indeed express surface PrP, we affinity-purified fractions from enriched BM HSC populations that do or do not express PrP on their surface, followed by reconstitution assays (Fig. 2). To examine the HSC activity of the donor cells in reconstitution analyses, lethally irradiated recipient mice were cotransplanted with both the donor cells to be tested and WT BM competitors. The competitor cells serve as an internal control and as a supply of hematopoietic cells until the transplanted stem cells can generate sufficient mature lymphoid and myeloid cells for survival. Donor and recipient mice are genetically identical except for the CD45 surface protein that is found on nucleated peripheral blood cells and is not involved in hematopoiesis or stem cell activity; donor cells carried the marker CD45.2, whereas recipient mice and supportive cells expressed CD45.1.
Fig. 2.

All LT repopulating bone marrow HSC cells express surface PrP. A total of 1 × 105 PrP− or 2 × 104 PrP+ CD45.2 donor BM cells (A), 2 × 104 PrP+ CD45.2 donor BM cells (B), 500 sorted Lin−Sca-1+PrP− or 100 Lin−Sca-1+PrP+ CD45.2 donor BM cells (C), or 500 isolated SP PrP− or 250 SP PrP+ donor BM cells (D) were mixed with 1 × 105 competitor CD45.1 cells and transplanted into lethally irradiated CD45.1 mice (n = 4–5). (A) Donor CD45.2 contribution at 4 weeks and 6 months after transplant. (B) Multilineage contribution at 6 months after transplant. (C) Donor contribution at 3 weeks and 4 months after transplant. (D) Donor contribution at 4 months after transplant.
In the study in Fig. 2A, total adult CD45.2 BM cells were sorted according to their cell surface expression of PrP. PrP+ and PrP− fractions were mixed separately with 1 × 105 CD45.1 total BM cells and injected into lethally irradiated CD45.1 recipients. A total of 1 × 105 PrP− donor cells exhibited a small measure of reconstitution at 4 weeks after transplantation, which primarily measures short-term (ST) HSC activity (Fig. 2A, bar 1). In contrast, at 6 months after transplant, there were no peripheral blood cells derived from these donor PrP− cells (bar 3), showing that BM cells that do not express the prion protein lack LT HSC activity. In contrast, 2 × 104 PrP+ BM cells supported both ST and LT engraftment, as assayed by reconstitution of peripheral blood cells (Fig. 2A, bars 2 and 4). PrP+ donor cells repopulated both lymphoid and myeloid compartments (Fig. 2B).
Next, HSC enriched Lin−Sca-1+ BM cells were sorted according to their expression of PrP. Of Lin− cells, 0.8% and 5.2% are Sca-1+PrP+ and Sca-1+PrP−, respectively (data not shown). Competitive transplantation of 500 isolated Lin−Sca-1+PrP− cells resulted in significant repopulation after 3 weeks (Fig. 2C, bar 1) but none after 4 months (bar 3). In contrast, injection of only 100 Lin−Sca-1+PrP+ CD45.2 donor BM cells resulted in significant reconstitution after both 3 weeks and 4 months (bars 2 and 4, respectively). Each of the five recipient mice were engrafted with frequencies of 5.1%, 1.4%, 27.6%, 2.5%, and 1.4% of peripheral blood cells. Thus, starting with a purified Lin−Sca-1+ HSC population, the subfraction of cells that express PrP on their surface contain all LT HSCs.
Fig. 2D further shows that all LT HSCs express PrP on their surface. SP cells were sorted based on PrP expression. SP cells that did not express PrP contained no LT HSC activity, whereas those that expressed PrP had significant activity (Fig. 2D). Thus, adult BM LT HSC activity resides in the PrP+ but not in the PrP− fractions of both Lin−Sca1+ and SP populations. In the transplantation experiments in Fig. 2 A and C, we used 5-fold more PrP− cells than PrP+ cells. From the data in Fig. 2A, we conclude that the presence of surface PrP protein distinguishes those Lin− Sca-1+ cells that contain the LT HSCs from those that do not. The ability of PrP+ SP cells to repopulate all recipients coupled with no detectable LT HSC activity in the PrP−SP population (Fig. 2D) similarly suggests that all LT HSCs express PrP. We conclude that PrP is a marker for LT HSCs. Like other LT HSC markers, PrP expression is not exclusive to that cell type.
PrP Is Important for Renewal of HSCs Under Stress.
We asked whether PrP expression had a functional role in HSCs by using mice ablated for PrP. The PrP-null and WT control mice used in the first studies (Figs. 3 and 4) were backcrossed onto the C57BL/6J background four times (termed N4). First, we analyzed the nature and number of hematopoietic cells from WT and PrP-null littermates (18). The hematocrits, hemoglobin levels, and total red and white blood cell levels in peripheral blood were similar in the PrP-null and control mice (Table 2, which is published as supporting information on the PNAS web site). Furthermore, BM cells were reacted with various lineage-specific antibodies, and the cells were analyzed by flow cytometry; we found no differences in staining profiles between the two types of animals (data not shown), consistent with prior observations (19). We assayed PrP-null and WT BM cells for colony-forming units (CFUs) to look for defects in progenitor numbers or activities. PrP-null and control BM had similar numbers of multipotential granulocyte/erythroid/monocyte/megakaryocyte progenitors (CFU-GEMM), granulocyte/monocyte progenitors (CFU-GM), erythroid progenitors (BFU-E, burst-forming unit), and B lymphoid (CFU-Pre-B) progenitors (Fig. 3A). In addition, the surface expression of seven important hematopoietic surface antigens on BM SP cells isolated from PrP-null and WT mice was similar, as detected by flow cytometry analysis (Table 3, which is published as supporting information on the PNAS web site). Thus, PrP-deficient BM has normal levels of progenitors and terminally differentiated hematopoietic cells.
Fig. 3.

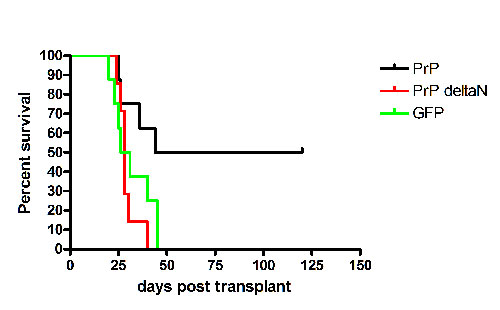
Ectopic expression of PrP rescues the impaired self-renewal of PrP-null stem cells during serial transplantation. (A) Colony assays for erythroid, lymphoid, and myeloid progenitors in PrP-null and WT BM. Total BM cell populations were plated in methylcellulose medium M3434 (StemCell Technologies) for quantifying CFU-GEMM, CFU-GM, and BFU-E colonies, and in M3630 (StemCell Technologies) for quantifying CFU-Pre-B colonies. (B) Serial transplantation of PrP-null and WT BM with competitor bone marrow cells. A total of 2 × 106 donor CD45.2 PrP-null or littermate control BM cells were mixed with 2 × 106 CD45.1 WT BM cells and transplanted into lethally irradiated CD45.1 recipients (n = 6). The extent of chimerism in peripheral blood (bars 1 and 2) was analyzed 4 months after transplant. In the experiment in bars 3 and 4, BM cells from the primary transplanted mice were pooled, and 2 × 106 cells were injected directly into each of five lethally irradiated CD45.1 recipients. The fraction of donor CD45.2 cells in the peripheral blood of these transplanted mice was analyzed 4 months later. The process was repeated for the tertiary transplants (bars 5 and 6). This is a combined result of three independent experiments from a total of initial six null or wild-type control mice. ∗, Significantly different from bar 3 value, P < 0.005; ∗∗, significantly different from bar 5 value, P < 0.005. (C) Serial transplantation of PrP-null and WT BM cells without competitors; rescue of HSC activity in PrP-null cells by PrP expression. A total of 1 × 106 PrP-null or WT BM CD45.2 cells, pooled from three donors, were transplanted into lethally irradiated CD45.1 recipients without competitors. Recipients were monitored daily for survival for >30 days (bars 1 and 2, n = 6). These mice were killed after 4 months. From them, 5 × 105 BM cells were collected and transplanted into new irradiated recipients (bars 3 and 4, n = 7). The process was repeated an additional time for tertiary transplants (bars 5 and 6, n = 12). In parallel, 1 × 106 PrP-null BM cells isolated from the surviving secondary transplant recipients, as shown in bar 3, were infected by retroviruses encoding GFP, PrP, or PrP Δ23-72, and injected into irradiated recipients (bars 7–9, n = 7–8). Plotted is the fraction of surviving mice 50 days after each bone marrow transplant. See Fig. 6 for details of animal survival. (D) A total of 1 × 106 BM cells from the secondary transplanted mice shown in bars 3 and 4 of C were transplanted into the lethally irradiated recipients. Survival data were plotted as Kaplan–Meier curves (n = 11 for each group, P < 0.0001, log-rank test). (E) Competitive transplantation demonstrates impaired renewal of PrP-null HSC activity during successive bone marrow transplants. Here, 5 × 105 PrP-null or WT BM collected from primary transplanted mice 4 months after transplant (without competitors, as in C, bars 1 and 2) were mixed with 5 × 105 CD45.1 freshly isolated BM cells and transplanted into lethally irradiated recipients. Peripheral blood engraftment at 6 weeks and 5 months after transplant is shown (n = 4). ∗, Significantly different from bar 1 value, P < 0.005; ∗∗, significantly different from bar 3 value, P < 0.05.
Fig. 4.

PrP-null HSCs are more sensitive than normal to myelotoxic injury. (A) A total of 5 × 105 CD45.2 PrP-null or wild-type BM cells, pooled from three littermates, were transplanted into lethally irradiated CD45.1 recipients. One month after transplant, 11 recipient mice from each group were treated with 150 mg/kg 5-FU i.p. weekly for 2 weeks. The survival of these two groups was analyzed by using a log-rank nonparametric test (P = 0.0307, n = 11 in each group) and expressed as Kaplan–Meier survival plot. (B) 5-FU (150 mg/kg) was administered i.p. to three wild-type CD45.2 mice. After 3 days, the BM of these treated mice was pooled and fractionated according to PrP expression. A total of 1 × 105 PrP+ or 1 × 106 PrP× BM cells were transplanted, together with 2 × 105 competitor CD45.1 wild-type BM cells, into lethally irradiated CD45.1 recipients. The level of chimerism in the peripheral blood of the recipients was analyzed 4 months later (n = 5).
To determine whether PrP has a role in activity of HSCs, we used competitive reconstitution assays (20). Fig. 3B shows the competitive repopulation results pooled from three independent experiments. A total of 2 × 106 PrP-null or WT CD45.2 BM cells were mixed with an equal number of competitor CD45.1 BM cells and transplanted into lethally irradiated CD45.1 recipients. If the PrP-null or control WT donor CD45.2 BM cells functioned equivalently to the CD45.1 competitor, one would expect 50% of the peripheral blood cells in the recipient to express the CD45.2 protein. Indeed, both WT and PrP-null donor cells exhibited ≈50% reconstitution of peripheral blood 4 months after transplant (Fig. 3B, bars 1 and 2). This finding suggests that freshly isolated BM from PrP-null and WT mice have similar HSC numbers and activities. We tested the serial engraftment capacity of PrP-null HSCs by pooling BM cells from primary transplanted recipients and transplanting them into lethally irradiated secondary CD45.1 recipients. After 4 months, peripheral blood of the secondary recipients was analyzed for expression of the donor CD45.2 marker (Fig. 3B, bars 3 and 4). Cells derived from original WT CD45.2 BM comprised 60 ± 4% of the nucleated peripheral blood cells. In contrast, cells derived from the original PrP-null CD45.2 BM comprised only 29 ± 7% of the peripheral blood, a significantly lower number (P < 0.005, t test) than observed with WT donors. The lineage profiles of the primary and secondary transplanted PrP-null BM were normal (data not shown). Tertiary transplantations were carried out in the same manner. Again cells derived from the original PrP-null BM showed significantly less engraftment than their WT counterparts (Fig. 3B, bars 5–6, P < 0.05, t test). Because the contribution of PrP-null HSCs to the reconstitution of lethally irradiated recipients steadily decreased with each transplantation, relative to WT HSCs, we infer that PrP-null HSCs have impaired self-renewal capabilities as analyzed by LT serial transplantation.
Because the competitor CD45.1 cells used in the transplants in Fig. 3B may have had untoward effects on the CD45.2 WT or PrP-null cells, we carried out serial transplantation without competitors. To this end 1 × 106 BM cells isolated from three PrP-null or WT littermates were transplanted into lethally irradiated CD45.1 recipients. As expected all of the recipients survived (Fig. 3C, bars 1 and 2). Four months later, 5 × 105 BM cells from these primary recipients were pooled and transplanted into secondary recipients, and the same procedure was repeated in tertiary transplantations. After the secondary transplant, 86% of the mice receiving WT BM survived, whereas only 57% of those receiving PrP-null BM did (Fig. 3C, bars 3 and 4). A more striking difference in survival between PrP-null and WT repopulated mice was noted after tertiary transplantation: 67% of transplanted mice receiving WT BM survived, whereas none of the mice transplanted with null BM did (Fig. 3C, bars 5 and 6). In all cases, flow cytometry analysis of the transplanted mice confirmed that the recipient BM derived exclusively from the donor cells (data not shown). The defect in the ability of PrP-null BM to repopulate during serial transplantation was confirmed in a separate experiment in which a higher number of cells (1 × 106 cells) from the secondary transplant were used to transplant lethally irradiated tertiary recipients. The survival curve confirmed the dramatically lower repopulation potential of PrP-null BM after the tertiary transplantation (Fig. 3D, P < 0.0001, log-rank test).
Because the PrP-null mice used in these studies were only backcrossed four times into the C57BL/6J background, it remained possible that some genetic difference linked to the PrP locus was responsible for the observed differences in serial transplantation. Thus, in the experiment depicted in bars 7–9 of Fig. 3C, we used a retroviral vector to introduce PrP into the PrP-null BM cells isolated from the secondary transplanted recipients. The efficiency of infection was 30–50% (Fig. 5, which is published as supporting information on the PNAS web site); specifically, 1 × 106 cells were infected and transplanted into lethally irradiated recipients. Consistent with the data in bar 5, none of the mice transplanted with BM infected by the control GFP vector survived (Fig. 3C, bar 7). In contrast, half of the mice transplanted with the same BM but expressing exogenous PrP survived (Fig. 3C, bar 8 and Fig. 6, which is published as supporting information on the PNAS web site, P < 0.05, log-rank test). In another control experiment, the same BM cells were infected with a retrovirus encoding a mutant PrP with deletion of amino acids 23–72, the segment that contains the N-terminal octapeptide repeats (11). None of the mice transplanted with BM expressing this mutant PrP protein survived (Fig. 3C, bar 9, and Fig. 6). This deletion might also remove amino acids that may be essential for the internalization or the proper signal peptide processing (21, 22), and further study is needed to characterize the nature of PrP sequence critical for supporting PrP’s HSC engraftment. However, this complementation experiment demonstrates that full-length PrP supports hematopoietic engraftment during long-term transplantation. It will be interesting to further study whether the hematopoietic rescue is PrP dose dependent, and whether overexpression of PrP in WT bone marrow gains better serial engraftment capacity.
The competitive reconstitution experiment in Fig. 3E confirms our conclusion that the defects observed in the PrP-null BM during serial transplantations indeed are due to defects in HSC renewal. BM from the initially transplanted mice used in bars 1 and 2 of Fig. 3C was mixed with 5 × 105 freshly isolated BM cells from WT CD45.1 mice and transplanted into irradiated WT CD45.1 recipients. Consistent with impaired survival of HSCs, the PrP-null cells showed 4.4 ± 0.8% repopulation as short-term HSC activity (6 weeks) and 2.2 ± 0.5% repopulation as LT HSC activity (5 months). Both figures were significant lower than that obtained from their WT counterparts (23.8 ± 4.0% and 6.1 ± 1.0% respectively, Fig. 3E, P < 0.05, t test).
As noted, the experiments depicted in Fig. 3 were performed by using PrP-null and WT mice that were backcrossed to C57 BL/6J background four times. To confirm the notion that PrP is important for hematopoietic engraftment under stress, we also used mice backcrossed six times (termed N6). Similar HSC frequencies (Fig. 7 A and B, which is published as supporting information on the PNAS web site) and activities (Fig. 7C, bars 1 and 2) were seen in freshly isolated N6 PrP-null and WT BM cells as well as in purified BM SP cells. In parallel, we infected freshly isolated PrP-null BM cells with PrP/GFP or control GFP retrovirus and collected the GFP-positive cells by flow cytometric sorting. Again, there was no significant difference in HSC activities between the PrP- and GFP-infected BM measured by competitive reconstitution (Fig. 7C, bars 3 and 4). These results confirm that PrP is not required for the HSC activity in normal unstressed mice.
We further compared the HSC activities of N6 PrP-null and WT BM after the stress of serial transplantation. In Fig. 3E, we transplanted N6 PrP-null or WT BM cells without competitors into irradiated recipients. Three months later, the transplanted BM cells were collected and their HSC activities were measured by competitive reconstitution. PrP-null BM cells showed 2.2 ± 0.9% engraftment, significantly lower than that from WT cells, which had 5.9 ± 1.4% engraftment (Fig. 7D, bars 1 and 2, P < 0.05, t test). In parallel, we infected N6 PrP-null BM cells with either a PrP/GFP or control GFP retrovirus population, isolated the GFP-positive population, and then transplanted the cells without competitors into irradiated recipients. Three months later, the transplanted BM cells were collected and their HSC activities were measured in secondary transplants by competitive reconstitution (Fig. 7D, bars 3 and 4). Consistent with the results in bars 1 and 2, the expression of PrP increased the HSC activity of prion null BM (bar 3, 1.8 ± 0.4% engraftment by control GFP infected cells; bar 4, 4.8 ± 1.9% engraftment by PrP infected cells, P < 0.05, t test). We conclude that although the absence of PrP does not affect HSC activity in normal unstressed mice, it is important for renewal of HSC activity that occurs under the stress of serial bone marrow transplantation.
To further test the notion that PrP deficiency leads to a defect in the stress response of hematopoietic cells, we treated mice with 5-fluorouracil (5-FU), which is toxic to cycling cells and accelerates the entry of HSCs into the cell cycle (23). Isogenic WT mice reconstituted with either PrP-null or control BM were treated with 5-FU at 1 month after transplantation. The survival of mice repopulated with PrP-null BM was significantly lower than those reconstituted with wild-type cells (Fig. 4A, P < 0.05, log-rank test). Repeated experiments showed similar results (data not shown).
We verified that PrP was still expressed on the surface of wild-type HSCs after 5-FU stress; in this experiment, BM cells from 5-FU-treated wild-type mice were sorted into PrP+ and PrP− fractions. Competitive repopulation assays showed that, as in normal BM, PrP+ cells contain all of the HSC activity (Fig. 4B). The 5-FU-treated mice used in the study in Fig. 4A were WT in all tissues except for their hematopoietic system, which was of either wild-type or PrP-null origin. Thus, the difference in survival after 5-FU treatment is due only to the presence or absence of the PrP on hematopoietic stem or early progenitor cells. Therefore, consistent with its role of supporting sustained LT HSC self-renewal in serial transplantation, PrP protects hematopoietic cells from exhaustion by toxic agents such as 5-FU.
Discussion
Previous work showed that several types of blood cells express PrP, albeit at vastly different levels: lymphocytes, dendritic cells, monocytes, granulocytes, erythrocytes, platelets, certain lymphoid precursors, and CD34+ cells, which in humans are an enriched stem/progenitor cell population (7, 24–27). However, it was unknown whether PrP is expressed on HSCs with repopulating activity, and there was no indication of a possible function of PrP in any of these cells. We demonstrated that PrP is located on the surface of HSCs and supports their engraftment during serial transplantation. Thus, using antibodies to PrP, together with other HSC markers, it may be possible to devise novel protocols for purifying human HSCs.
Other GPI-anchored proteins, including Sca-1 (20) and possibly CD59 (28) are also expressed on HSCs. Similar to PrP, Sca-1 is also required for HSC self-renewal (20). Like other GPI-anchored proteins (29), PrP has been reported to localize to lipid rafts in the plasma membrane (30), and it might regulate certain signaling proteins that are also concentrated in these domains. Indeed, PrP has been reported to be involved in activation of Fyn tyrosine kinase (31) and to interact with laminin, the laminin receptor, and stress-inducible protein 1, as well as other proteins (10). Like the GPI-anchored α-subunit of the ciliary neurotrophic factor receptor (32), PrP might be the coreceptor for a hormone affecting HSC activity, possibly concentrating this as yet unidentified molecule on the cell surface and/or presenting it to the signaling receptor(s). In this function, PrP might protect HSCs from apoptosis or sustain their long-term self-renewal. Alternatively, PrP might interact with proteins in the BM extracellular matrix or on the surface of stromal cells, and possibly support retention of transplanted HSCs within the BM microenvironment. Our work opens an avenue of investigation that may illuminate the details of the normal function of PrP in cell biology.
Materials and Methods
Mouse Strains and Genotyping.
CD45.1 and CD45.2 C57BL/6 mice were purchased from The Jackson Laboratory or the National Cancer Institute. The PrP knockout mice (18) were provided by R. Race and B. Chesebro (Rocky Mountain Laboratories, National Institute of Allergy and Infectious Diseases, Hamilton, MT) and backcrossed to C57 BL/6J CD45.2 mice at least four or six times (indicated in text and figure legends) to obtain PrP-null and WT control littermates. To genotype mice, DNA was extracted from proteinase K-digested tail tips by using a DNeasy kit according to the manufacturer’s instructions (Qiagen, Valencia, CA). The PrP and/or neomycin (neo) insert was amplified in four-primer PCR using primers 5′-TCATCCCACGATCAGGAAGATGAG-3′ and 5′-ATGGCGAACCTTGGCTACTGGCTG-3′ for PrP and 5′-TTGAGCCTGGCGAACAGTTC-3′ and 5′-GATGGATTGCACGCAGGTTC-3′ for the neomycin insert. The cycling conditions were 94°C for 3 min, followed by 30 cycles of 94°C for 30 s, 62°C for 30 s, and 72°C for 1 min, followed by a final extension of 72°C for 10 min.
Flow Cytometry.
Donor BM cells were isolated from 7- to 10-week-old mice. Anti-PrP mAb (SAF-83; Cayman Chemical, Ann Arbor, MI) was FITC-conjugated by using the Quick-Tag FITC conjugation kit (Roche Diagnostics). Its specificity was verified by its inability to bind to PrP-null cells and its specific binding to cell lines expressing transfected prion protein (data not shown).
SP cells were stained with Hoechst dye 33342 as described (14). In Fig. 1A, Hoechst-stained cells were then costained with anti-PrP-FITC and phycoerythrin (PE)-conjugated anti-Sca-1, CD43, CD44, CD49D, CD49E, CD11A, or CD62L antibodies (BD Pharmingen). When Endoglin was detected, Hoechst 33342 stained cells were subsequently stained with anti-Endoglin mAb (BD Pharmingen), anti-rat-PE, and anti-PrP-FITC. In Fig. 1B, the cells were stained first with anti-Endoglin mAb (BD Pharmingen) followed by anti-rat-PE/CY5.5 (eBioscience, San Diego, CA), a biotinylated antibody mixture recognizing multiple hematopoietic lineage markers (StemCell Technologies, Vancouver), as well as streptavidin-APC, anti-PrP-FITC, and anti-Sca-1-PE (BD Pharmingen). In Fig. 2, cells were stained with anti-PrP-FITC (Fig. 2 A and B), or biotinylated multilineage antibody mixture followed by streptavidin-APC, anti-PrP-FITC, and anti-Sca-1-PE (Fig. 2C). In Fig. 2D, the staining was the same as in Fig. 1A, plot 4.
The peripheral blood analysis after reconstitution was performed as described (5). Peripheral blood cells were stained with anti-CD45.2-FITC and anti-CD45.1-PE. Anti-Thy1.2-PE, anti-B220-PE, anti-Mac-1-PE, anti-Gr-1-PE, and anti-Ter119-PE monoclonal antibodies (BD Pharmingen) were used for detecting specific hematopoietic lineages. FACS analyses were performed on a FACSCalibur instrument. Cells were sorted in a MoFlo cell sorter. The purity of sorted cells was typically >90%.
Reconstitution Analysis.
The reconstitution protocol was essentially as described (5). Briefly, the indicated numbers of CD45.2 donor cells were injected directly, or after mixing with 1 × 105, 2 × 105, or 5 × 105 (as indicated) freshly isolated CD45.1 competitor BM cells, intravenously into a group of 6- to 9-week-old CD45.1 mice that had been irradiated with a total dose of 10 Gy. To measure reconstitution of transplanted mice, peripheral blood or BM was collected by at the indicated times after transplant and the presence of CD45.1+ and CD45.2+ cells in lymphoid and myeloid compartments were measured. The calculation of competitive repopulating units in limiting dilution experiments was conducted as described (5, 33), using l-calc software (StemCell Technologies).
Colony Assays.
PrP-null or WT BM cells were diluted to 2 × 105 per ml in Iscove’s modified Dulbecco’s medium (IMDM) with 2% FBS, and then were seeded into methylcellulose medium M3434 (StemCell Technologies) for CFU-GEMM, CFU-GM, and BFU-E colony formation, or into M3630 (StemCell Technologies) for CFU-pre-B colonies, according to the manufacturer’s protocols.
Retrovirus Infection.
Mouse PrP (Δ23–72) was constructed by ligating the PCR products of PrP1–23 and 72–254 and inserting into XZ201 (a MSCV-IRES-GFP vector, gift from Xiaowu Zhang, Whitehead Institute, Cambridge, MA). Mouse PrP or PrP (Δ23–72) cDNA was cloned upstream of the internal ribosomal entry site (IRES) in XZ201. The retroviral plasmids were transfected by using lipofectamine 2000 (Invitrogen) into BOSC packaging cells. The resulting retroviral supernatant was collected 48 h later and was used for infection. To this end, BM cells were resuspended in viral supernatants (2 × 105 cells per ml) with 6 μg/ml polybrene and centrifuged at 720 × g for 90 min before culturing for 24 h in StemSpan (StemCell Technologies) in the presence of 10 μg/ml heparin, 10 ng/ml SCF, 20 ng/ml TPO, 20 ng/ml IGF-2, and 10 ng/ml FGF-1 (33). Cells were then resuspended in viral supernatant for another round of infection. Cells were then used directly for transplantation or cultured for another 4 days before sorting.
5-FU Challenge.
In Fig. 4A, 5 × 105 PrP-null or wild-type BM cells were used to reconstitute 10 Gy-irradiated CD45.1 C57BL/6 mice; there were 11 mice in each group. At 1 month after transplant, 5-FU was administered i.p. at a dose of 150 mg/kg weekly for 2 weeks. The survival rates of the two groups were analyzed by using a log-rank test (prism, GraphPad, San Diego). In Fig. 4B, a single dose of 150 mg per kg of body weight of 5-FU was injected i.p. into CD45.2 C57BL/6 mice (n = 3). After 3 days, the BM was pooled and subjected to FACS staining and sorting before competitive repopulation (n = 5).
Supplementary Material
Acknowledgments
We thank Drs. R. Race and B. Chesebro for providing the PrP knockout mice and Dr. R. Barron (J. Manson laboratory, Institute for Animal Health, Edinburgh, U.K.) for the genotyping protocol. We are grateful to M. Kaba for the excellent technical assistance, A. Topolszki for help with our mouse colony, and G. Paradis and M. Jennings in the MIT flow cytometry core facility for cell sorting. We thank Drs. X. Zhang, M. J. Liao, W. Tong, and J. Ma as well as members of the Lindquist laboratory for reagents and helpful discussion. C.C.Z. is a Leukemia and Lymphoma Society Fellow. H.F.L. was supported by the Cambridge/MIT Institute and the Engineering Research Centers Program of the National Science Foundation under NSF Award EEC 9843342 through the Biotechnology Process Engineering Center at the Massachusetts Institute of Technology.
Glossary
Abbreviations:
- HSC
hematopoietic stem cell
- PrP
prion protein
- GPI
glycosylphosphatidylinositol
- BM
bone marrow
- SP
side population
- LT
long term
- 5-FU
5-fluoracil.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Jordan C. T., McKearn J. P., Lemischka I. R. Cell. 1990;61:953–963. doi: 10.1016/0092-8674(90)90061-i. [DOI] [PubMed] [Google Scholar]
- 2.Rebel V. I., Miller C. L., Thornbury G. R., Dragowska W. H., Eaves C. J., Lansdorp P. M. Exp. Hematol. 1996;24:638–648. [PubMed] [Google Scholar]
- 3.Spangrude G. J., Heimfeld S., Weissman I. L. Science. 1988;241:58–62. doi: 10.1126/science.2898810. [DOI] [PubMed] [Google Scholar]
- 4.Chen C. Z., Li L., Li M., Lodish H. F. Immunity. 2003;19:525–533. doi: 10.1016/s1074-7613(03)00265-6. [DOI] [PubMed] [Google Scholar]
- 5.Zhang C. C., Lodish H. F. Blood. 2004;103:2513–2521. doi: 10.1182/blood-2003-08-2955. [DOI] [PubMed] [Google Scholar]
- 6.Aguzzi A., Klein M. A., Montrasio F., Pekarik V., Brandner S., Furukawa H., Kaser P., Rockl C., Glatzel M. Ann. N. Y. Acad. Sci. 2000;920:140–157. doi: 10.1111/j.1749-6632.2000.tb06916.x. [DOI] [PubMed] [Google Scholar]
- 7.Vostal J. G., Holada K., Simak J. Transfus. Med. Rev. 2001;15:268–281. doi: 10.1053/tmrv.2001.26957. [DOI] [PubMed] [Google Scholar]
- 8.Aguzzi A. Haematologica. 2000;85:3–10. [PubMed] [Google Scholar]
- 9.Prusiner S. B. Proc. Natl. Acad. Sci. USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lasmezas C. I. Br. Med. Bull. 2003;66:61–70. doi: 10.1093/bmb/66.1.61. [DOI] [PubMed] [Google Scholar]
- 11.Roucou X., Gains M., LeBlanc A. C. J. Neurosci. Res. 2004;75:153–161. doi: 10.1002/jnr.10864. [DOI] [PubMed] [Google Scholar]
- 12.Weissmann C., Flechsig E. Br. Med. Bull. 2003;66:43–60. doi: 10.1093/bmb/66.1.43. [DOI] [PubMed] [Google Scholar]
- 13.Kaeser P. S., Klein M. A., Schwarz P., Aguzzi A. J. Virol. 2001;75:7097–7106. doi: 10.1128/JVI.75.15.7097-7106.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodell M. A., Brose K., Paradis G., Conner A. S., Mulligan R. C. J. Exp. Med. 1996;183:1797–1806. doi: 10.1084/jem.183.4.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okada S., Nakauchi H., Nagayoshi K., Nishikawa S., Miura Y., Suda T. Blood. 1992;80:3044–3050. [PubMed] [Google Scholar]
- 16.Orschell-Traycoff C. M., Hiatt K., Dagher R. N., Rice S., Yoder M. C., Srour E. F. Blood. 2000;96:1380–1387. [PubMed] [Google Scholar]
- 17.Scott L. M., Priestley G. V., Papayannopoulou T. Mol. Cell. Biol. 2003;23:9349–9360. doi: 10.1128/MCB.23.24.9349-9360.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manson J. C., Clarke A. R., Hooper M. L., Aitchison L., McConnell I., Hope J. Mol. Neurobiol. 1994;8:121–127. doi: 10.1007/BF02780662. [DOI] [PubMed] [Google Scholar]
- 19.Genoud N., Behrens A., Miele G., Robay D., Heppner F. L., Freigang S., Aguzzi A. Proc. Natl. Acad. Sci. USA. 2004;101:4198–4203. doi: 10.1073/pnas.0400131101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito C. Y., Li C. Y., Bernstein A., Dick J. E., Stanford W. L. Blood. 2003;101:517–523. doi: 10.1182/blood-2002-06-1918. [DOI] [PubMed] [Google Scholar]
- 21.Sunyach C., Jen A., Deng J., Fitzgerald K. T., Frobert Y., Grassi J., McCaffrey M. W., Morris R. EMBO J. 2003;22:3591–3601. doi: 10.1093/emboj/cdg344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor D. R., Watt N. T., Perera W. S., Hooper N. M. J. Cell Sci. 2005;118:5141–5153. doi: 10.1242/jcs.02627. [DOI] [PubMed] [Google Scholar]
- 23.Cheng T., Rodrigues N., Shen H., Yang Y., Dombkowski D., Sykes M., Scadden D. T. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 24.Liu T., Li R., Wong B. S., Liu D., Pan T., Petersen R. B., Gambetti P., Sy M. S. J. Immunol. 2001;166:3733–3742. doi: 10.4049/jimmunol.166.6.3733. [DOI] [PubMed] [Google Scholar]
- 25.Dodelet V. C., Cashman N. R. Blood. 1998;91:1556–1561. [PubMed] [Google Scholar]
- 26.Risitano A. M., Holada K., Chen G., Simak J., Vostal J. G., Young N. S., Maciejewski J. P. Exp. Hematol. 2003;31:65–72. doi: 10.1016/s0301-472x(02)01011-1. [DOI] [PubMed] [Google Scholar]
- 27.Starke R., Harrison P., Mackie I., Wang G., Erusalimsky J. D., Gale R., Masse J. M., Cramer E., Pizzey A., Biggerstaff J., Machin S. J. Thromb. Haemost. 2005;3:1266–1273. doi: 10.1111/j.1538-7836.2005.01343.x. [DOI] [PubMed] [Google Scholar]
- 28.Hill B., Rozler E., Travis M., Chen S., Zannetino A., Simmons P., Galy A., Chen B., Hoffman R. Exp. Hematol. 1996;24:936–943. [PubMed] [Google Scholar]
- 29.Horejsi V., Drbal K., Cebecauer M., Cerny J., Brdicka T., Angelisova P., Stockinger H. Immunol. Today. 1999;20:356–361. doi: 10.1016/s0167-5699(99)01489-9. [DOI] [PubMed] [Google Scholar]
- 30.Naslavsky N., Stein R., Yanai A., Friedlander G., Taraboulos A. J. Biol. Chem. 1997;272:6324–6331. doi: 10.1074/jbc.272.10.6324. [DOI] [PubMed] [Google Scholar]
- 31.Mouillet-Richard S., Ermonval M., Chebassier C., Laplanche J. L., Lehmann S., Launay J. M., Kellermann O. Science. 2000;289:1925–1928. doi: 10.1126/science.289.5486.1925. [DOI] [PubMed] [Google Scholar]
- 32.Stahl N., Boulton T. G., Ip N., Davis S., Yancopoulos G. D. Braz. J. Med. Biol. Res. 1994;27:297–301. [PubMed] [Google Scholar]
- 33.Zhang C. C., Lodish H. F. Blood. 2005;105:4314–4320. doi: 10.1182/blood-2004-11-4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}