

Abstract
Thymic-derived CD25+ CD4+ T regulatory cells (Tregs) suppress immune responses, including transplantation. Here we evaluated the ability of dendritic cells (DCs) to expand alloantigen-specific Tregs in the mixed leukocyte reaction (MLR) that develops from polyclonal populations of T cells. The allogeneic DCs, when supplemented with IL-2 in the cultures, were much more effective than bulk spleen cells in expanding the numbers of Tregs. Likewise, DCs and not spleen cells were effective in sustaining expression of the transcription factor Foxp3 in Tregs, but neither IL-2 nor CD80/86 was required for this effect in the cultures. On a per-cell basis, the DC-expanded, but not unexpanded, Tregs were more potent suppressors of a fresh MLR by CD25− CD4+ T cells. Suppression was 3- to 10-fold more active for MLRs induced by the original alloantigens than for third-party stimulators. When DC-expanded Tregs were introduced into sublethally irradiated hosts, the T cells suppressed graft-versus-host-disease induced by CD25− CD4+ T cells. Again, suppression was more active against the same mouse strain that provided the DCs to expand the Tregs. Therefore, alloantigen-selected Tregs are more effective suppressors of responses to major transplantation antigens, and these Tregs can be expanded from a polyclonal repertoire by DCs.
Keywords: transplantation, graft-versus-host disease, tolerance, immune regulation
Naturally occurring CD25+ CD4+ regulatory or suppressor T cells (Tregs) represent 5–10% of CD4+ T cells in mice and are also found in human blood (1–4). Tregs maintain immunological self tolerance, preventing autoimmunity (1, 2). Tregs also control immune responses to tumors, infections, allergens, and transplants (5–10). The development of Tregs requires the transcription factor Foxp3, which currently is their most selective marker (11–15). It is important to understand how antigen-presenting cells (APCs) control the function of Tregs and to establish whether antigen-specific Tregs are the major mediators of tolerance in vivo.
Recent reports have shown that Tregs proliferate and retain their antigen-dependent suppressive functions when the APCs are antigen-loaded mature dendritic cells (DCs) (16–18). When Tregs specific for a pancreatic islet β cell antigen are stimulated by DCs together with IL-2, the expanded antigen-specific T cells regulate the development of autoimmune diabetes in nonobese diabetic mice and do so much more effectively than polyclonal populations (18). From the perspective of suppressing unwanted immune reactions, preferential expansion of antigen-specific Tregs will also avoid complications likely to be incurred if therapeutic T cells were contaminated with Tregs that suppress resistance to infections and tumors.
We will show here that DCs expand alloantigen-specific Tregs from polyclonal starting populations, which initially have little specific suppressive activity. The DCs prove to be much more effective than a standard source of spleen APCs in expanding Tregs and maintaining high Foxp3 expression. When function is tested, DC-expanded Tregs exert more potent and antigen-specific suppression of transplantation immunity.
Results
Allogeneic DCs Are More Effective than Spleen Cells in Expanding Tregs.
Previously, we found that antigen-bearing DCs could expand Tregs from TCR transgenic mice (16), and that the specific Tregs were more potent suppressors of autoimmunity than polyclonal populations (18). To determine whether DCs could preferentially expand antigen-specific Tregs from polyclonal populations, we turned to the MLR and cultured CD25+ CD4+ T cells from BALB/c H-2d mice with allogeneic B6 H-2b DCs. Others had found that Tregs could expand with spleen cells as APCs, and that these suppressed graft-versus-host disease (GVHD) (19–21), skin graft rejection in nude mice (22), and allogeneic bone marrow transplants (23). However, we noted that the expansion of CD25+ CD4+ T cells in the presence of IL-2 was much weaker with splenic APCs than DCs, both spleen CD11c+ DCs and bone marrow-derived DCs (BM-DCs) that had been matured with or without LPS stimulation. This was the case whether T cell responses were monitored by 3H-thymidine uptake (Fig. 1A) or expansion of cell numbers (typically 2- to 5-fold; data not shown). The expansion of Tregs increased progressively as the dose of IL-2 was increased from 20 to 500 units/ml (data not shown). We also purified CD62L+ or CD62L− fractions of Tregs. The allogeneic DCs induced strong proliferation of both CD62L+ and CD62L− Treg fractions from naïve mice, and proliferation again depended upon the dose of IL-2 (Fig. 1B). The T cells also responded to syngeneic BM-DCs (Fig. 1B), which is consistent with prior data that antigen-specific TCR transgenic Tregs respond to mature DCs without added antigen (16, 18). Therefore, mature DCs and IL-2 effectively expand Tregs from naïve mice in the MLR.
Fig. 1.

Allogeneic DCs together with IL-2 expand Tregs. (A) BALB/c CD25+CD4+ T cells (104) were cultured with 104 B6 BM-DCs, selected as CD86 high mature DCs with or without LPS stimulation. Spleen CD11c+ DCs or 105 splenic APCs (104) were also tested. The DCs were added to T cells without (open) or with (closed) 100 units/ml rhIL-2. (B) As in A, 104 BALB/c CD62L+ or CD62L− CD25+ CD4+ T cells were cultured with 104 B6 or BALB/c BM-DCs and IL-2 for 7 d. A and B are one of three similar results.
Phenotype of Tregs, Including Foxp3 Expression, After Expansion with Allogeneic DCs.
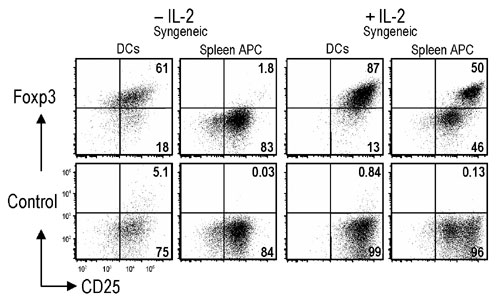
We then verified that the typical markers of Tregs were maintained when DCs were used to expand CD62L+ or CD62L− CD25+ CD4+ T cells for 1 or 2 wk in the MLR. The T cells expressed high levels of CD25, GITR, and CD62L (in the case of CD62L+CD25+ fraction), but low CD103 (Fig. 6, which is published as supporting information on the PNAS web site). We then tested Foxp3 expression. Staining with anti-Foxp3 was observed primarily in CD25+ CD4+ T cells in fresh spleen, as expected (Fig. 2A). When we assessed Foxp3 expression in Thy1.1+ CD25+ CD4+ T cells from B6 mice that had been cultured 7 days with allogeneic APCs from Thy 1.2+ BALB/c mice, we noted that DCs sustained Foxp3 expression, which was increased in the presence of IL-2 (Fig. 2B). Spleen APCs in contrast poorly sustained Foxp3 (Fig. 2B). Similar results were seen when DCs and spleen APCs were used to expand syngeneic Tregs (Fig. 7, which is published as supporting information on the PNAS web site). To test the need for CD28 costimulation in Foxp3 expression, we showed that CD80/86−/− allogeneic DCs expanded CD25+ Tregs, although not as much as wild-type DCs (Fig. 2C). Nonetheless, the T cells maintained Foxp3 expression when the DCs were CD80/86-deficient (Fig. 2D). Thus DC-expanded Tregs maintain their phenotype, including high Foxp3.
Fig. 2.

Foxp3 expression on expanded Tregs. (A) Expression of Foxp3 relative to isotype control antibody on CD4+ spleen cells labeled for CD25. (B) B6 Thy1.1+ CD25+ CD4+ T cells (104) were cultured with 104 BM-DCs or 105 spleen cells from BALB/c mice with or without 500 units/ml IL-2 for 7 d. The cultures were gated on CD4+ and Thy1.1+ cells. (C) Viable cell numbers at 7 d and fold increases compared with cell numbers at day 0. (D) As in B, but Thy1.1+ BALB/c CD25+ CD4+ T cells were cultured with DCs from B6 control or CD80/CD86−/− mice with or without 500 units/ml IL-2 for 7 d. One of three similar results.
Antigen-Specific MLR Suppression by Tregs Expanded by Allogeneic DCs.
To look for antigen-specific suppression, Tregs from H-2b Thy 1.1+ B6 mice were cultured with H-2d BALB/c or H-2s SJL allogeneic DCs. Both DC-expanded and freshly isolated Tregs were then added to carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled, Thy-1.2+, and CD25− CD4+ T cells from B6 mice. We tested whether the DC expanded Tregs would suppress a primary MLR, monitored by CFSE dilution and by numbers of Thy-1.2+ T cells. If the Tregs had been expanded with BALB DCs, they suppressed the MLR to BALB spleen APCs even at a 1:27 ratio, whereas SJL expanded Tregs (which are called “third party”) required a 1:3 ratio to show some suppression, (Fig. 3A and C). In “criss-cross” experiments, we found that Tregs expanded with SJL DCs suppressed the MLR to SJL spleen APCs, again in small numbers, 1:27 (Fig. 3 B and C). When freshly isolated Tregs were tested, some suppression of recovered T cell numbers was seen but only at a 1:3 suppressor-to-responder ratio (Fig. 3 A–C). Therefore, not only do DCs expand the numbers of CD25+ Tregs in the MLR, but also the expanded cells are far more active in suppressing a fresh MLR and in an antigen-specific manner.
Fig. 3.

Allogeneic-DC expanded Tregs suppress the MLR in an antigen-specific manner. CD62L+ CD25+ CD4+ T cells from Thy1.1+ B6 mice were freshly isolated or expanded with BALB/c or SJL-DCs. These were mixed with 105 CFSE-labeled, Thy 1.2+, B6, and CD25− CD4+ T cells in various ratios and stimulated with 105 irradiated spleen cells from BALB/c (A) or SJL mice (B). Five days later, CFSE dilution was analyzed by FACS. The displayed cells were gated on live Thy1.2+ cells, and the percent of divided CFSE-low live Thy1.2+ cells is shown inside the plot. (C) As in B, but numbers of live Thy1.2+ responder cells per culture. One of two similar experiments.
Tregs Expanded with Syngeneic DCs and IL-2 Weakly Suppress the alloMLR.
As mentioned above (Figs. 2 and 7), Tregs expanded by syngeneic DCs and IL-2 express high levels of Foxp3, so we tested the suppressive function of Tregs from B6 mice that had been cultured with allogeneic BALB or syngeneic B6 DCs plus IL-2 (Fig. 4). Allogeneic DC-expanded Tregs suppressed the MLR to BALB/c spleen APCs strongly, even at suppressor-to-responder ratios of 1:27 or 1:81 (Fig. 4 A and B), whereas syngeneic DC-expanded Tregs showed strong suppression only at 1:3 (Fig. 4 A and B). When we examined the MLR to third-party spleen APCs, both third-party and syngeneic DC-expanded Tregs showed weak suppression at higher 1:3 suppressor-to-responder ratios (Fig. 4 A and C). These results extend those in Fig. 3 that DC-expanded Tregs are enriched for antigen-specific suppression of the MLR.
Fig. 4.

Syngeneic DC-expanded Tregs are weak and antigen-nonspecific suppressors. (A) As in Fig. 3, CD25+ CD4+ T cells from CD45.2 B6 mice were expanded with allogeneic (BALB) or syngeneic (B6) DCs and added to 105 CFSE-labeled, CD45.1+ CD25− CD4+ B6 responder T cells in various ratios. These were stimulated with 3 × 105 irradiated BALB/c (Left) or CBA (Right) spleen cells. The displayed cells were gated on live responder CD45.1+ cells, and the percent live CD45.1+ divided cells is shown inside the plot. The numbers of live CD45.1+ cells per culture are also shown for BALB/c (B) and CBA (C) MLR data. One of five similar experiments.
Antigen-Specific Suppression of GVHD by DC-Expanded Tregs.
We then assessed suppressive function in vivo in a GVHD model. We first used bm12 recipients and B6 donors, which differ at three amino acids in the MHC II peptide binding regions (8, 20). Freshly isolated CD25− CD4+ B6 T cells (105) were injected into sublethally irradiated bm12 hosts to initiate GVHD. We coadministered 105 B6 Tregs that had been expanded with DCs from either antigen-specific bm12 or third-party CBA mice (Fig. 5A, open triangles and squares, respectively). All recipients of CD25− CD4+ T cells alone lost weight quickly and died by day 20. In contrast, mice receiving antigen-specific Tregs (expanded with bm12 DCs) survived 2 wk longer, whereas recipients of third-party CBA DC-expanded Tregs survived slightly longer (Fig. 5A). Next, we induced GVHD across a fully MHC-mismatched BALB and B6 combination. Again, we observed a 2-wk retardation of GVHD with DC-expanded, alloreactive Tregs (Fig. 5B, open triangles). Although syngeneic DC-expanded Tregs expressed Foxp3 at high levels (see Fig. 7), all recipients of syngeneic DC-expanded Tregs lost weight and died by day 25 (Fig. 5B, open diamonds). We further compared B6 Tregs that had been expanded in vitro with either BALB/c H-2d DCs or CBA H-2k DCs (third party). Tregs expanded with BALB DCs were more effective than Tregs expanded with third-party DCs (Fig. 5C). Therefore, when Tregs are expanded from polyclonal populations with allogeneic DCs, the T cells specifically suppress transplant reactions across MHC barriers in vivo.
Fig. 5.

Allogeneic DC-expanded Tregs suppress GVHD in an antigen-specific manner. (A) Naïve B6 CD25− CD4+ T cells (105) (effectors alone, closed circles) were adoptively transferred into irradiated bm12 recipients with or without 105 CD62L+ CD25+ CD4+ T cells that had been expanded with bm12-DC (alloantigen-specific, open triangles) or CBA-DC (third-party, open squares). Percent of surviving mice is shown (n = 5). (B) Naïve B6 CD25− CD4+ T cells (2 × 105) (effector-alone, closed circles) were adoptively transferred into irradiated BALB/c recipients with or without 2 × 105 CD62L+ CD25+ CD4+ T cells that had been expanded with BALB DC (alloantigen-specific, open triangles) or B6-DC (syngeneic, antigen-nonspecific, open diamonds) (n = 5). (C) As in B, but CD62L+ CD25+ CD4+ T cells that had been expanded with BALB DC-expanded (alloantigen-specific, open triangles), or CBA DC-expanded (third party, open squares) were cotransferred (n = 6). A and B are each representative of two experiments, whereas C is a single experiment.
Discussion
To understand the control of Tregs, it is important to address APC requirements. In the thymus, mature DCs can expand and differentiate Tregs (24). In the periphery, mature DCs also expand functional Tregs (16–18). The latter may be important during infection, when DCs are presenting microbial antigens, because there is likely to be concomitant presentation of self and environmental antigens. For example, when DCs present microbial antigen at a mucosal surface during a cytopathic infection, the DCs are capturing dying infected self tissues and environmental proteins. Because Tregs suppress autoimmunity and chronic inflammatory disease, the repertoire of Tregs likely contains cells reactive with harmless self and environmental antigens. The capacity of maturing DCs to expand Tregs should then reduce the risk of auto- and environmental immunity. In tumors, DCs produce TGF-β to expand Tregs (25). DCs also influence the formation IL-10-producing Tr1-type Tregs (26, 27) and natural killer T cells (28), including Tr1 cells that suppress allergy (29). DCs therefore control several suppressive forms of immune silencing or tolerance.
We have now addressed the capacity of DCs to expand antigen-specific Tregs from a polyclonal repertoire. Since the pioneering research of Dye and North (30) on the capacity of suppressor T cells to regulate antitumor immunity, it has been known that antigen-specific Tregs are pivotal in suppressing responses by other effector T cells (30). We have assessed alloreactive Tregs, where the existence of alloantigen-specific suppressors has been controversial. Some studies find that these cells do not suppress transplantation immunity in an antigen-specific way, whereas others report contrary evidence (reviewed in refs. 9 and 10).
Our results suggest that a critical variable is the degree to which Tregs have responded to mature DCs that bear specific transplantation antigens. Naïve Tregs from lymphoid tissues suppress the MLR weakly. However, mature DCs, together with IL-2, stimulate a 2- to 5-fold expansion of CD25+ CD4+ T cells in the MLR. At the same time, there is a 3- to 10-fold expansion in the capacity of the expanded Tregs (on a per-cell basis) to suppress transplant immunity in vitro and in vivo in an antigen-specific manner. In other words, if H-2d DC from BALB/c mice are used to expand Tregs from H-2b B6 mice, the DC-expanded Tregs suppress the response of B6 CD25− CD4+ T cells ≈10 times better if the stimulators are BALB/c rather than third-party H-2s SJL mice (Fig. 3). Reciprocally, if the Tregs are expanded with SJL DCs, they suppress the MLR to SJL much better than to BALB.
We also find that mature DCs, but not spleen cells, are effective APCs to expand Tregs and increase their specific suppressive activity. In prior studies, Tregs expanded with splenic APCs were unable to exert antigen-specific suppression in vitro (19, 21), except for one recent report (22). Presumably, bulk populations of splenic leukocytes lack sufficient mature DCs to clonally select and expand antigen-specific Tregs.
The recent availability of effective antibodies to Foxp3, a critical transcription factor for the development of Treg (11–15), has allowed us to study the function of APCs in maintaining Foxp3 protein at the single-cell level. Mature DCs sustain Foxp3 during 7-d coculture with syngeneic or allogeneic CD25+ CD4+ T cells, whereas Foxp3 is not sustained when spleen cells are used as APCs. Foxp3 expression in the DC-Treg cocultures does not require the addition of IL-2 or expression of CD80/86 costimulatory molecules by DCs.
To assess Treg function in vivo, we have used an allogeneic GVHD model. Lethal GVHD is induced in irradiated recipients with CD25− CD4+ T cells reactive exclusively to allogeneic MHC class II products or to fully allogeneic strain differences. Freshly isolated Treg are inactive in suppressing GVHD, but DC-expanded alloreactive Tregs prolong survival at least 2 wk. Again, allospecificity is evident because Tregs expanded with syngeneic or third-party DCs are less effective in suppressing GVHD. However, alloreactive Tregs do not fully block GVHD, possibly because we have tested only a single limiting cell dose of Tregs given once at the initiation of GVHD. In sum, we find that freshly isolated naturally occurring Tregs suppress alloreactive T cell responses weakly, but DCs can expand antigen-specific Treg numbers and function from polyclonal populations, suggesting a path to specific immune-silencing therapies.
Materials and Methods
Mice.
Six- to 8-wk specific pathogen-free female C57BL/6 (“B6”) H-2b, BALB/c H-2d, CBA H-2k, B6 CD45.1 H-2b, B6.PL-Thy1.1/Cy H-2b, SJL H-2s, bm12 H-2b, and CD80/CD86−/− mice were purchased from Taconic Farms (Germantown, NY) or The Jackson Laboratory. BALB/c Thy1.1+ mice were kindly provided by Maria and Juan Lafaille (New York University, New York). All mice were used according to the guidelines of our institutional animal care and use committee.
Antibodies.
Biotin, phycoerythrin (PE), FITC, PerCP, or APC-anti-CD25 (7D4,PC61), CD4 (H129.19), CD62L (MEL-14), CD86 (GL1),CD45.1, Thy1.1, Thy1.2, isotype control mAbs, streptavidin PE, and purified anti-CD16/CD32 (2.4G2) were from BD PharMingen. rhIL-2 was from Chiron, CFSE from Molecular Probes, and anti-mouse PE-Foxp3 (FJK-16s) staining kit from eBioscience (San Diego, CA).
T Cells.
Spleen and lymph node cells were stained with anti-CD25, CD4, and CD62L mAbs. CD25+ cells were positively selected with MACS beads (Miltenyi Biotech, Gladbach, Germany). CD62L+ or CD62L−, CD25+ CD4+ T cells were then purified to >98% on a FACS Vantage (BD Biosciences Clontech). CD25− CD4+ T cells were negatively separated by MACS (>90%).
DCs.
BM-DCs were derived with granulocyte/macrophage (or granulocyte–macrophage) colony-stimulating factor (16). On day 5, LPS (Sigma-Aldrich) was added at 50 ng/ml for 16 h to increase the yield of CD86-high mature DCs, but functionally similar DCs, for expanding CD25+CD4+ T cells, could be obtained without LPS by disrupting the cultures at day 5 and transferring to new wells (16). Spleen CD11c+ DCs were selected with anti-CD11c beads (Miltenyi Biotech) (16).
Treg Expansion.
Sorted Tregs (104) were cultured with 104 allogeneic or syngeneic DCs (irradiated with 15 Gy) and 20–500 units/ml rhIL-2 in 96-well round bottomed plates. For 3H-TdR assays, 1 μCi/well (1 Ci = 37 GBq) NEN Life Science) was added for the last 12 h in triplicate. A DC-to-T cell ratio of 1:1 was superior to 1:3 and 1:10 for expanding Tregs. After 1- to 2-wk expansion, DCs were depleted with anti-CD11c MACS microbeads to provide >98% Tregs. We did not try to scale up Treg production with larger culture vessels or repeated stimulations.
MLR.
Fresh CD25− CD4+ T cells (105) were responders to one to three × 105 whole spleen cell stimulators (15 Gy). Responders from CD45.1+ or Thy1.2+ B6 mice were first labeled with 1 μM CFSE. To these, we added graded numbers of suppressors, either fresh or DC-expanded Tregs. After 5 d of culture in 96-well round-bottom plates (Corning Costar), CFSE dilution was assessed on live CD45.1+ or Thy1.2+ cells (dead cells gated out with TOPRO-3 iodide, Molecular Probes) and analyzed with flow jo software (Tree Star, Ashland, OR).
GVHD.
BALB/c or bm12 mice were irradiated sublethally and 6–8 h later, transferred with alloreactive B6 CD25− CD4+ T cells (8, 20) with or without Tregs (expanded by syngeneic, allogeneic, or third-party DCs). The drinking water had 1.1 mg/ml neomycin sulfate and 1,000 units/ml polymyxin B sulfate (Sigma).
Supplementary Material
Acknowledgments
We thank Maria and Juan Lafaille for Thy1.1+ BALB/c mice, Klara Velinzon for expert sorting, Judy Adams for help with graphics, and Priscilla Toy for technical help. S.Y. was the recipient of a Uehara Memorial Foundation fellowship. R.M.S. and J.V.R. are supported by Grants AI-051573 from the National Institutes of Health and 7-2005-1160 from the Juvenile Diabetes Research Foundation.
Abbreviations
- Tregs
CD25+ CD4+ regulatory T cells
- MLR
mixed leukocyte reaction
- DCs
dendritic cells
- BM-DCs
bone marrow-derived DCs
- APCs
antigen-presenting cells
- CFSE
carboxyfluorescein diacetate succinimidyl ester
- GVHD
graft-versus-host disease.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Sakaguchi S. Annu. Rev. Immunol. 2004;22:531–562. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 2.Shevach E. M. Nat. Rev. Immunol. 2002;2:389–400. doi: 10.1038/nri821. [DOI] [PubMed] [Google Scholar]
- 3.Dieckmann D., Plottner H., Berchtold S., Berger T., Schuler G. J. Exp. Med. 2001;193:1303–1310. doi: 10.1084/jem.193.11.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levings M. K., Sangregorio R., Sartirana C., Moschin A. L., Battaglia M., Orban P. C., Roncarolo M.-G. J. Exp. Med. 2002;196:1335–1346. doi: 10.1084/jem.20021139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimizu J., Yamazaki S., Sakaguchi S. J. Immunol. 1999;163:5211–5218. [PubMed] [Google Scholar]
- 6.Belkaid Y., Rouse B. T. Nat. Immunol. 2005;6:353–360. doi: 10.1038/ni1181. [DOI] [PubMed] [Google Scholar]
- 7.Hoffmann P., Ermann J., Edinger M., Fathman C. G., Strober S. J. Exp. Med. 2002;196:389–399. doi: 10.1084/jem.20020399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor P. A., Noelle R. J., Blazar B. R. J. Exp. Med. 2001;193:1311–1318. doi: 10.1084/jem.193.11.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wood K. J., Sakaguchi S. Nat. Rev. Immunol. 2003;3:199–210. doi: 10.1038/nri1027. [DOI] [PubMed] [Google Scholar]
- 10.Graca L., Chen T. C., Moine A. L., Cobbold S. P., Howie D., Waldmann H. Trends Immunol. 2005;26:130–135. doi: 10.1016/j.it.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 11.Hori S., Nomura T., Sakaguchi S. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- 12.Fontenot J. D., Gavin M. A., Rudensky A. Y. Nat. Immunol. 2003;4:330–336. [PubMed] [Google Scholar]
- 13.Khattri R., Cox T., Yasayko S. A., Ramsdell F. Nat. Immunol. 2003;4:337–342. [PubMed] [Google Scholar]
- 14.Wan Y. Y., Flavell R. A. Proc. Natl. Acad. Sci. USA. 2005;102:5126–5131. doi: 10.1073/pnas.0501701102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fontenot J. D., Rasmussen J. P., Williams L. M., Dooley J. L., Farr A. G., Rudensky A. Y. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 16.Yamazaki S., Iyoda T., Tarbell K., Olson K., Velinzon K., Inaba K., Steinman R. M. J. Exp. Med. 2003;198:235–247. doi: 10.1084/jem.20030422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fehervari Z., Sakaguchi S. Int. Immunol. 2004;16:1769–1780. doi: 10.1093/intimm/dxh178. [DOI] [PubMed] [Google Scholar]
- 18.Tarbell K. V., Yamazaki S., Olson K., Toy P., Steinman R. M. J. Exp. Med. 2004;199:1467–1477. doi: 10.1084/jem.20040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen J. L., Trenado A., Vasey D., Klatzmann D., Salomon B. L. J. Exp. Med. 2002;196:401–406. doi: 10.1084/jem.20020090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor P. A., Lees C. J., Blazar B. R. Blood. 2002;99:3493–3499. doi: 10.1182/blood.v99.10.3493. [DOI] [PubMed] [Google Scholar]
- 21.Trenado A., Charlotte F., Fisson S., Yagello M., Klatzmann D., Salomon B. L., Cohen J. L. J. Clin. Invest. 2003;112:1688–1696. doi: 10.1172/JCI17702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishimura E., Sakihama T., Setoguchi R., Tanaka K., Sakaguchi S. Int. Immunol. 2004;16:1189–1201. doi: 10.1093/intimm/dxh122. [DOI] [PubMed] [Google Scholar]
- 23.Joffre O., Gorsse N., Romagnoli P., Hudrisier D., Van Meerwijk J. P. Blood. 2004;103:4216–4221. doi: 10.1182/blood-2004-01-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watanabe N., Wang Y. H., Lee H. K., Ito T., Cao W., Liu Y. J. Nature. 2005;436:1181–1185. doi: 10.1038/nature03886. [DOI] [PubMed] [Google Scholar]
- 25.Ghiringhelli F., Puig P. E., Roux S., Parcellier A., Schmitt E., Solary E., Kroemer G., Martin F., Chauffert B., Zitvogel L. J. Exp. Med. 2005;202:919–929. doi: 10.1084/jem.20050463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dieckmann D., Bruett C. H., Ploettner H., Lutz M. B., Schuler G. J. Exp. Med. 2002;196:247–253. doi: 10.1084/jem.20020642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levings M. K., Gregori S., Tresoldi E., Cazzaniga S., Bonini C., Roncarolo M. G. Blood. 2005;105:1162–1169. doi: 10.1182/blood-2004-03-1211. [DOI] [PubMed] [Google Scholar]
- 28.Kojo S., Seino K., Harada M., Watarai H., Wakao H., Uchida T., Nakayama T., Taniguchi M. J. Immunol. 2005;175:3648–3655. doi: 10.4049/jimmunol.175.6.3648. [DOI] [PubMed] [Google Scholar]
- 29.Akbari O., Freeman G. J., Meyer E. H., Greenfield E. A., Chang T. T., Sharpe A. H., Berry G., DeKruyff R. H., Umetsu D. T. Nat. Med. 2002;8:1024–1032. doi: 10.1038/nm745. [DOI] [PubMed] [Google Scholar]
- 30.Dye E. S., North R. J. J. Leuk. Biol. 1984;36:27–37. doi: 10.1002/jlb.36.1.27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}