

Abstract
Tissue factor (TF) plays an important role in hemostasis, inflammation, angiogenesis, and the pathophysiology of atherosclerosis and cancer. In this article we uncover a mechanism in which protein S, which is well known as the cofactor of activated protein C, specifically inhibits TF activity by promoting the interaction between full-length TF pathway inhibitor (TFPI) and factor Xa (FXa). The stimulatory effect of protein S on FXa inhibition by TFPI is caused by a 10-fold reduction of the Ki of the FXa/TFPI complex, which decreased from 4.4 nM in the absence of protein S to 0.5 nM in the presence of protein S. This decrease in Ki not only results in an acceleration of the feedback inhibition of the TF-mediated coagulation pathway, but it also brings the TFPI concentration necessary for effective FXa inhibition well within range of the concentration of TFPI in plasma. This mechanism changes the concept of regulation of TF-induced thrombin formation in plasma and demonstrates that protein S and TFPI act in concert in the inhibition of TF activity. Our data suggest that protein S deficiency not only increases the risk of thrombosis by impairing the protein C system but also by reducing the ability of TFPI to down-regulate the extrinsic coagulation pathway.
Keywords: anticoagulant, venous thrombosis, extrinsic coagulation
Tissue factor (TF) is a multifunctional protein that is not only involved in hemostasis and thrombosis (1) and atherosclerosis (2), but also participates in cell signaling activities (3, 4) that play an important role in inflammation (5) and angiogenesis (6, 7). Historically, TF was identified as the protein component from tissue extracts responsible for the initiation of blood coagulation. Upon exposure to blood, TF binds the circulating coagulation factor VIIa (FVIIa). The resulting phospholipid-bound TF/FVIIa complex converts the zymogen factor X (FX) into the active serine protease, factor Xa (FXa). Together with its cofactor factor Va, FXa subsequently incorporates into the prothrombinase complex and activates prothrombin to thrombin.
Coagulation is finely tuned, and during thrombin formation several anticoagulant reactions are initiated to prevent systemic activation of coagulation. Impaired activity of the anticoagulant systems results in a hypercoagulable state and increases the risk of venous thrombosis (8). This article deals with two natural anticoagulant proteins, TF pathway inhibitor (TFPI) and protein S, deficiencies of which are associated with venous thrombosis (9, 10).
TFPI is a Kunitz-type inhibitor that inhibits TF/FVIIa-initiated coagulation (11) via a two-step feedback mechanism through formation of a bimolecular FXa/TFPI complex that subsequently interacts with TF/FVIIa, yielding an inactive quaternary complex and resulting in termination of TF/FVIIa-catalyzed FX activation (12).
Protein S is an essential component of the protein C pathway that down-regulates thrombin formation (13). Activated protein C (APC) is a serine protease that inhibits thrombin generation via inactivation of the coagulation factors Va and VIIIa. Protein S is a cofactor in these reactions that enhances the anticoagulant activity of APC up to 20-fold (14, 15).
Protein S can also down-regulate thrombin generation in the absence of APC via a mechanism that is as yet not fully understood. Because protein S directly inhibits prothrombin activation in model systems, it is generally thought that protein S exerts its anticoagulant activity in the absence of APC via direct interactions with FXa, factor Va, and phospholipids (16–18). However, we have recently shown that the APC-independent anticoagulant effect of protein S in plasma is particularly observed at low TF concentrations (19), which suggests that protein S may have a direct effect on TF-mediated FX activation. This article describes an interplay between TFPI and protein S in the inhibition of TF activity.
Results
Effect of Protein S and TFPI on Thrombin Formation.
In plasma, in which coagulation was initiated with TF, inhibition of protein S with polyclonal antibodies considerably increased thrombin generation (Fig. 1A). This effect of protein S was independent of APC because all experiments in plasma were performed in the presence of inhibiting antibodies against APC. Calculation of the area under the thrombin-generation curves, which yields the so-called endogenous thrombin potential (ETP), indicated that protein S inhibited thrombin (IIa) generation 2.5-fold from 735 nM IIa·min in the absence of protein S to 285 nM IIa·min in the presence of protein S. To explore whether the effect of protein S is limited to its free form, plasma was saturated with C4b-binding protein (C4BP). Addition of a molar excess of purified C4BP to free protein S in plasma resulted in a 40% decrease of the anticoagulant activity of protein S, independent of the concentration of TF used for initiation of coagulation (Table 1).
Fig. 1.

Thrombin generation in plasma. (A) Thrombin generation was initiated in plasma (in the presence of APC-inhibiting antibodies) with 1.4 pM TF, 10 μM phospholipid vesicles, and 16 mM CaCl2 (final concentrations) and followed continuously with the fluorogenic substrate I-1140 (Z-Gly-Gly-Arg-7-amino-4-methylcoumarin·HCl). •, normal plasma with protein S; ○, normal plasma without protein S; ▴, TFPI-depleted plasma with protein S; ▵, TFPI-depleted plasma without protein S. A typical experiment is shown. (B) ETP values of TFPI-depleted plasma reconstituted with varying amounts of full-length TFPI (circles) or TFPI1–161 (triangles) in the presence of protein S (closed symbols) or absence of protein S (open symbols). The averages of two independent experiments are shown.
Table 1.
Effect of C4BP on the inhibition of thrombin generation by TFPI and protein S
| TF, pM | Inhibition of ETP by protein S, % | Inhibition of ETP by protein S–C4BP, % | Activity of protein S–C4BP complex, % |
|---|---|---|---|
| 3.5 | 12.6 | 7.4 | 59 |
| 1.4 | 41.1 | 23.0 | 56 |
| 0.7 | 57.1 | 33.4 | 58 |
ETPs were determined at varying concentrations of TF. The inhibitory effect of protein S–C4BP complex on the ETP was determined by preincubating normal pooled plasma with saturating amounts of purified C4BP (see Materials and Methods).
Next, the APC-independent effect of protein S was determined in TFPI-depleted plasma. Thrombin generation in TFPI-depleted plasma was increased compared with normal pooled plasma, which likely reflects increased FXa generation because of the lack of inhibition of the TF/FVIIa complex. In contrast to normal plasma, antibodies against protein S had no effect on thrombin generation in TFPI-depleted plasma. This finding indicates that protein S does not express APC-independent anticoagulant activity in the absence of TFPI (Fig. 1A), which led to the hypothesis that protein S enhances the ability of TFPI to down-regulate FXa and thrombin formation during TF-initiated coagulation.
To gain more insight into the interaction between TFPI and protein S, TFPI-depleted plasma was reconstituted with varying amounts of recombinant full-length TFPI or a truncated form of TFPI (TFPI1–161) that lacks the Kunitz-3 domain and the C terminus (Fig. 1B). In plasma that contained protein S, thrombin generation decreased with increasing concentrations of full-length TFPI (IC50 of ≈1.7 nM) and was fully inhibited at 3.1 nM TFPI, whereas TFPI1–161 did not show an inhibitory effect. In the absence of protein S, neither full-length TFPI nor TFPI1–161 affected the ETP.
Inhibition of TF/FVIIa by TFPI and Protein S.
TFPI inhibits extrinsic coagulation via a feedback mechanism that requires the presence of FXa, the product of extrinsic FX activation (12). The first step, in which TFPI binds to and inhibits FXa, is rate-limiting (20). The second step, in which FXa/TFPI reacts with TF/FVIIa and forms an inactive quaternary complex, has been reported to proceed at near diffusion-limited rate (20). Because protein S did not inhibit thrombin generation in TFPI-depleted plasma and TFPI lost its anticoagulant activity in the absence of protein S, we hypothesized that protein S stimulates the inhibition of TF/FVIIa-catalyzed FX activation by TFPI.
This hypothesis was tested in a model system containing purified proteins. FX activation by TF/FVIIa was followed in time in the absence and presence of TFPI and/or protein S (Fig. 2). In the absence of TFPI, TF/FVIIa-catalyzed FX activation was linear in time and was not affected by protein S. In the presence of TFPI, the generation of FXa progressively decreased and was fully inhibited after 2 min. When both TFPI and protein S were present, virtually no FXa was generated, which indicates that protein S indeed accelerates the inhibition of TF/FVIIa-catalyzed FX activation by TFPI.
Fig. 2.

Inhibition of TF/FVIIa-catalyzed FX activation by full-length TFPI and protein S. Activation of 160 nM FX by 1 pM TF/FVIIa was followed in reaction mixtures that contained 15 μM phospholipids, 3 mM Ca2+, no TFPI and no protein S (♦), 100 nM protein S (■), 1 nM TFPI (•), and 1 nM TFPI and 100 nM protein S (▴). Averages of three independent measurements ± SD are shown.
Inhibition of FXa by TFPI and Protein S.
Theoretically, protein S can accelerate the inhibition of TF/FVIIa-catalyzed FX activation by TFPI by stimulating the formation of the FXa/TFPI and/or the FXa/TFPI/TF/FVIIa (quaternary) complex. Because the formation of the quaternary complex is very fast and diffusion-limited (20) it is unlikely that this step is affected by protein S. Hence, we quantified the effect of protein S on FXa/TFPI complex formation by measuring progress curves of FXa inhibition by TFPI. These progress curves were analyzed according to a slow tight-binding mechanism that describes the inhibition of FXa by TFPI (21) (Eq. 1).
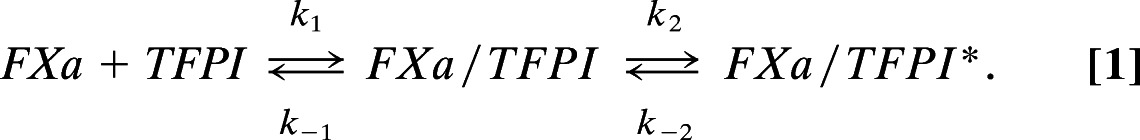 |
In this mechanism enzyme (FXa) and inhibitor (TFPI) are in rapid equilibrium and form a complex (FXa/TFPI) with a dissociation constant Ki (Ki = k−1/k1 = [FXa]·[TFPI]/[FXa/TFPI]). The FXa/TFPI complex subsequently slowly isomerizes into a tight complex (FXa/TFPI*), which at final equilibrium results in an overall dissociation constant Ki* that is much lower than Ki (Ki* = [FXa]·[TFPI]/[FXa/TFPI+FXa/TFPI*]).
Rate constants and dissociation constants for the interaction between TFPI and FXa were determined in reaction mixtures that contained FXa, TFPI, and the FXa-specific chromogenic substrate S2222 for monitoring the loss of FXa activity in time. For the mechanism presented in Eq. 1, the progress curves of S2222 conversion by FXa are described by the integrated rate equation (2) (22):
in which At and A0 are the absorbance values at time t and time 0; vo and vs are the initial velocity and final steady-state velocities of S2222 conversion, respectively, and kobs is the apparent rate constant for the transition from vo to vs.
S2222 conversion by FXa was inhibited upon addition of TFPI (1.5 nM), and this inhibition was strongly potentiated by protein S (Fig. 3A). In the absence of TFPI, protein S had no effect on S2222 conversion by FXa, indicating that protein S enhances the inhibition of FXa by TFPI. Estimates of free FXa concentrations, obtained from the first derivative of the curves (Fig. 3A), showed that the addition of TFPI resulted in an immediate decrease of FXa activity by ≈10% (Fig. 3B). This finding indicates that a rapid binding equilibrium was attained in which ≈10% of the FXa present was incorporated in the FXa/TFPI complex. The further decrease of free FXa with time reflects the slow isomerization of FXa/TFPI into the tight FXa/TFPI* complex, which causes a continuous re-establishment of equilibrium until finally >95% of FXa ended up in a complex with TFPI. In the presence of protein S, the fraction of FXa that was rapidly inhibited by TFPI increased to ≈60% with a similar final equilibrium. This result demonstrates that protein S primarily stimulates the formation of the FXa/TFPI complex and has less effect on the isomerization of FXa/TFPI into FXa/TFPI*.
Fig. 3.

Influence of protein S on FXa inhibition by full-length TFPI. (A) Conversion of 0.5 mM S2222 by 0.2 nM FXa was monitored in reaction mixtures containing 10 μM phospholipids, 3 mM CaCl2, and either no protein S (dashed line) or 160 nM protein S (dotted line). Without TFPI, S2222 conversion by FXa was linear in time (with or without protein S present, solid line). At the time indicated 1.54 nM TFPI was added. The absorbance data were fitted to Eq. 2. (B) First derivatives of the fitted curves representing the change in free FXa with time. A typical experiment is shown.
The effect of protein S on FXa inhibition by TFPI was further explored by measuring progress curves of FXa inhibition at varying TFPI concentrations (0–3.9 nM) both in the absence and presence of 100 nM protein S. Fitting the experimental data to Eq. 2 yielded values for vo, vs, and kobs at each TFPI concentration from which the rate constants k+2 and k−2 and the dissociation constants Ki and Ki* were calculated (see Materials and Methods).
Both in the presence and absence of protein S, the initial velocity vo decreased with increasing concentrations of TFPI (Fig. 4A). However, the decrease of vo was more pronounced in the presence of protein S, supporting the concept that protein S promotes the formation of the FXa/TFPI complex. The dissociation constant Ki of this complex decreased from 4.4 nM in the absence of protein S to 0.5 nM in the presence of protein S (Table 2). The final equilibrium rate (vs) also decreased with increasing concentrations of TFPI, but protein S had much less effect on vs than on vo (Fig. 4B). From the variation of vs as a function of the TFPI concentration, Ki* values were calculated that were 0.05 nM in the absence of protein S and 0.02 nM in the presence of protein S, respectively (Table 2). Comparison of k+2 determined in the absence of protein S (k+2 = 2.5 min−1) and presence of protein S (k+2 = 0.72 min−1) indicated that protein S actually slowed down the transition of FXa/TFPI into FXa/TFPI*. The reverse reaction described by k−2 was not influenced by protein S (Table 2).
Fig. 4.

Effect of protein S on vo and vs calculated from time courses of FXa inhibition by TFPI. Progress curves of S2222 conversion by FXa were measured at varying concentrations of TFPI in the absence and presence of protein S. Fitting of the progress curves to Eq. 2 yielded values for vo (A) and vs (B) as function of the TFPI concentration. Final concentrations were 0.2 nM FXa, 10 μM phospholipid vesicles (20:60:20 DOPS/DOPC/DOPE), 3 mM CaCl2, 0.5 mM S2222, 0–3.9 nM TFPI, and no protein S (○) or 100 nM protein S (•). The average of two independent experiments is shown.
Table 2.
Kinetic constants for the inhibition of FXa by TFPI with or without protein S
| Addition | TFPI type | Ki, nM | Ki*, nM | k−2, min−1 | k+2, min−1 |
|---|---|---|---|---|---|
| None | TFPIfl | 8.3 | 0.08 | 0.018 | 1.84 |
| Protein S | TFPIfl | 10.9 | 0.12 | 0.009 | 0.75 |
| PL | TFPIfl | 4.4 | 0.05 | 0.030 | 2.49 |
| PL + protein S | TFPIfl | 0.5 | 0.02 | 0.028 | 0.72 |
| PL | TFPI1–161 | 39.0 | 0.21 | 0.010 | 1.88 |
| PL + protein S | TFPI1–161 | 42.3 | 0.10 | 0.013 | 5.52 |
PL, phospholipid vesicles 20:60:20 DOPS/DOPC/DOPE; TFPIfl, full–length TFPI; TFPI1–161, TFPI that lacks the third Kunitz domain and the C terminus.
The stimulatory effect of protein S on FXa inhibition by TFPI required the presence of anionic phospholipids (Table 2). In the absence of phospholipid, protein S hardly influenced the Ki (8.3 nM without protein S and 10.9 nM with protein S, Table 2). The finding that TFPI was a relatively poor inhibitor of FXa in reaction mixtures containing calcium ions but no phospholipids is in agreement with literature (21). The Ki values determined for FXa inhibition by TFPI1–161 explain the observed lack of inhibitory activity of TFPI1–161 on thrombin formation in plasma (Fig. 1). The fact that protein S had no effect on complex formation between FXa and TFPI1–161 (Table 2) indicates that the Kunitz 3 and/or the C-terminal domain of TFPI are involved in protein S-dependent stimulation of TFPI activity. The effect of protein S on initial FXa/TFPI complex formation in a model system using fixed amounts of purified FXa was half-maximal at 45 nM protein S (data not shown) and reached optimal levels around the free protein S concentration present in plasma (150 nM). However, in a more physiologic plasma model system in which FXa was generated by TF/FVIIa (19), a dose-dependent decrease of the ETP was observed with increasing concentrations of protein S over the whole possible range (0–100%) of protein S concentrations in plasma. In this respect, any change of protein S concentration in plasma will be able to affect the regulation of thrombin generation.
Discussion
The results presented in this study provide insight in the mechanism through which TF activity is regulated in plasma. Protein S inhibits TF activity by enhancing the interaction between TFPI and FXa, thereby accelerating the feedback inhibition of the extrinsic TF/FVIIa pathway by TFPI. This observation not only underscores the important role of protein S in the down-regulation of coagulation, but also provides a mechanistic basis of the APC-independent anticoagulant activity of protein S in plasma (19).
Since the first report on the inhibition of prothrombin activation by protein S in the absence of APC (23), this inhibition was explained by direct interactions of protein S with the components of the prothrombinase complex factor Va, FXa, and phospholipids (16–18). Since then, in purified protein S preparations in vitro-generated protein S multimers have been identified that bind with a high affinity to phospholipids (Kd < 1 nM) and account for the effective inhibition of prothrombin activation by protein S in model systems (24). However, protein S multimers are absent in plasma (24), and it was proposed that the APC-independent inhibition of thrombin generation in plasma by protein S is not caused by competition between protein S and other coagulation factors for binding to procoagulant membrane surfaces (19).
We demonstrate that the APC-independent inhibition of thrombin generation by protein S in plasma is also not explained by inhibition of prothrombin activation through direct interactions of protein S with FXa and factor Va. The observations that protein S does not inhibit thrombin generation in TFPI-deficient plasma (Fig. 1A) and that TFPI is a very poor inhibitor of thrombin generation in the absence of protein S (Fig. 1B) led to the hypothesis that protein S acts as a cofactor of TFPI in the inhibition of TF/FVIIa-catalyzed FX activation. The partial activity (60%) of the protein S–C4BP complex is not yet understood and can originate from a change in phospholipid-binding affinity of the complex or sterical hindrance by C4BP when protein S is in complex with C4BP.
Experiments in a model system confirmed that protein S enhances the inhibition of TF/FVIIa-catalyzed FX activation by TFPI (Fig. 2). The inhibition of FX activation by TFPI involves the formation of a FXa/TFPI complex that slowly isomerizes into a tight FXa/TFPI* complex (21) that subsequently forms an inactive quaternary complex with TF/FVIIa (20). Detailed kinetic analysis showed that protein S enhances the formation of the FXa/TFPI complex and has a minor effect on the subsequent isomerization step.
The stimulatory effect of protein S on FXa inhibition by TFPI is caused by a 10-fold reduction of the Ki of the FXa/TFPI complex, which decreased from 4.4 nM in the absence of protein S to 0.5 nM in the presence of protein S. We were not able to test whether protein S may also have an effect on the formation of the quaternary complex. However, because the formation of this complex is very fast and diffusion-limited (20), it is unlikely that this step is affected by protein S.
We have limited information on the molecular mechanism by which protein S enhances the formation of the FXa/TFPI complex. The fact that protein S only acts as a cofactor of TFPI in the presence of phospholipids suggests that colocalization and/or juxtaposition of protein S, TFPI, and FXa on the phospholipid surface is a prerequisite for the fast protein S-mediated inhibition of FXa by TFPI. Protein S did not stimulate the inhibition of FXa by truncated TFPI (TFPI1–161), a form of TFPI that lacks the Kunitz-3 domain and the C terminus. It was reported that Kunitz-3 and the C terminus of TFPI are involved in the binding of full-length TFPI (25) to cell surfaces and that the C terminus interacts with anionic phospholipids (26) and the Gla domain of FXa (27). Hence, the loss of these interaction sites of TFPI1–161 likely explains why protein S does not stimulate the inhibition of FXa by TFPI1–161 (Table 2) and why truncated TFPI lacks inhibitory activity in plasma (Fig. 1B) (27–29).
The important role of TFPI and protein S in the in vivo regulation of coagulation is illustrated by the observations that mice with a mutant form of TFPI that did not bind FVIIa died intrauterine or during the neonatal period because of consumptive coagulopathy (30) and that homozygous protein S deficiency, which is also lethal if left untreated, presents with a similar phenotype of consumptive coagulopathy (31). Furthermore, population-based studies indicated that low levels of protein S (9) and TFPI (10) are associated with an increased risk of venous thrombosis. In view of the pivotal role of TFPI and protein S in the regulation of coagulation, it is not surprising that our observations have important physiological implications. Considering its effect on the Ki for the inhibition of FXa by TFPI, which decreases from 4.4 nM in the absence of protein S to 0.5 nM in the presence of protein S, protein S brings the TFPI concentration necessary for efficient down-regulation of extrinsic FX activation well within the range of the free TFPI concentration in plasma (0.25–0.5 nM) (32). Using the equations for a simple binding equilibrium, it can be calculated that, during the initiation of coagulation (where [FXa] < [TFPI]), protein S reduces the free FXa concentration from 90% to 45%, and thus increases the concentration of FXa/TFPI complex ≈5-fold. Thus, protein S enhances the down-regulation of thrombin formation by (i) reducing the amount of FXa that can participate in prothrombin and FVII activation and (ii) increasing the amount of FXa/TFPI complex available for inhibition of the TF/FVIIa complex.
The extent of inhibition of the extrinsic coagulation pathway by TFPI depends on the TF concentration, and the amount of FXa that escapes regulation by TFPI linearly increases with the TF concentration (20). This observation indicates that at increasing amounts of TF TFPI will ultimately fail to keep the FXa concentration below the threshold required for thrombin formation (33), which explains why protein S hardly inhibits thrombin generation at high TF concentrations (19). Thus, protein S and TFPI likely play a prominent role in suppressing the procoagulant activities at low TF concentrations, e.g., of the small amounts of TF (≈3 pM) circulating in plasma (34). Our data demonstrate that protein S deficiency affects the two cofactor activities of protein S: the TFPI cofactor activity at low TF concentrations and the APC cofactor activity at high TF concentrations (19). On the basis of our observations we propose that the increased risk of venous thrombosis associated with protein S deficiency may, in part, be explained by an impaired down-regulation of the extrinsic coagulation pathway by TFPI at low protein S concentrations.
In addition to its role in hemostasis, TF is involved in inflammation (5), angiogenesis (6, 7), and tumor metastasis (35), processes that are likely modulated through TF/FVIIa- and TF/FVIIa–FXa-dependent protease-activated receptor (PAR) signaling (3, 4, 36–38). Recently, a selective role for TFPI was proposed in the inhibition of TF signaling through PAR1 and PAR2, in which PAR1 signaling appeared less sensitive to inhibition by TFPI than PAR2 (39). Whether protein S also affects these functions of TF, especially the inhibition of TF-mediated PAR1 signaling by TFPI, remains to be elucidated.
Materials and Methods
Materials.
Hepes buffer was obtained from Sigma, and BSA was from ICN. Fluorogenic substrate I-1140 was from Bachem. Recombinant TF (thromboplastin) was from Dade Innovin (Marburg, Germany). 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phosphoserine (DOPS), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) were obtained from Avanti Polar Lipids. Phospholipids vesicles (20% DOPS, 20% DOPE, and 60% DOPC) were prepared as described (19). Polyclonal anti-protein S and anti-protein C antibodies were obtained from DAKO. Human FXa was obtained from Enzyme Research Laboratories (South Bend, IN). TFPI was kindly provided by T. Lindhout (Cardiovascular Research Institute Maastricht) (40). Full-length TFPI was produced in Escherichia coli, and the truncated variant of TFPI (amino acid residues 1–161) was expressed in Sacharomyces cerevisiae. Purification and analysis of both forms of TFPI has been described (41, 42). The TFPI concentration was determined as described (43). Recombinant FVIIa (NovoSeven) was obtained from Novo-Nordisk (Copenhagen). TFPI-depleted plasma was a kind gift of R. van Oerle (Cardiovascular Research Institute Maastricht). The plasma was depleted from TFPI as described by van’t Veer et al. (44): normal pooled plasma was applied to an antibody column containing rabbit polyclonal antibodies directed against the N-terminal region of TFPI. The remaining TFPI activity (<1%) was determined with an in-house assay based on the chromogenic assay described by Sandset et al. (45).
Measurement of Thrombin Generation.
Thrombin generation was initiated in normal pooled plasma with 1.4 pM TF, 10 μM phospholipid vesicles (20:60:20 DOPS/DOPC/DOPE), and 16 mM CaCl2 (final concentrations) and continuously followed with the fluorogenic substrate I-1140 (Z-Gly-Gly-Arg-7-amino-4-methylcoumarin·HCl) as described (19, 46). Interference by APC activity was excluded in all experiments by addition of inhibitory anti-(activated) protein C antibodies (1.23 μM IgG) sufficient to completely block both activation of endogenous protein C and the effect of 5 nM APC added to plasma. Protein S was inhibited in plasma by the addition of saturating amounts of polyclonal antiserum against protein S (2.73 μM IgG) and preincubation of plasma for 15 min at 37°C before the initiation of thrombin generation as described (19). Where indicated, C4BP was added to plasma to a final concentration of ≈575 nM (≈200 nM endogenous C4BP and 375 nM exogenous C4BP) and incubated for 30 min before the addition of anti-protein C antibodies with or without antibodies against protein S. The ETP (area under the curve) was calculated from thrombin generation curves by means of the calibrated automated thrombogram computer software provided by Synapse BV (Maastricht, The Netherlands) (46).
Inhibition of TF/FVIIa-Catalyzed Activation of FX by TFPI and Protein S.
TF (1 pM) was incubated with 500 pM recombinant FVIIa (NovoSeven) in the presence of 15 μM phospholipids (20:60:20 DOPS/DOPC/DOPE) at 37°C in Hepes-buffered saline (25 mM Hepes/175 mM NaCl, pH 7.5) containing 3 mM CaCl2 and 0.5 mg/ml BSA. FXa generation was started by addition of 160 nM human FX either in the absence or presence of 1 nM TFPI and/or 100 nM protein S (final concentrations). After different time intervals, aliquots taken from the reaction mixture were diluted 10-fold in ice-cold stop buffer (50 mM Tris·HCl/175 mM NaCl, pH 7.9) containing 20 mM EDTA and 0.5 mg/ml ovalbumin, and FXa present in the diluted aliquots was determined with the chromogenic substrate S2765 (Z-d-Arg-Gly-Arg-pNA.2HCl).
Inhibition of FXa by TFPI.
Conversion of the chromogenic substrate S2222 [Bz-Ile-Glu(γ-OR)-Gly-Arg-pNA.HCl] by FXa was monitored in a Ultra Microplate Reader (Bio-Tek, Burlington, VT). A reaction mixture containing CaCl2, S2222, and phospholipid vesicles (20:60:20 DOPS/DOPC/DOPE) with or without protein S was preincubated for 7 min at 37°C. After FXa was added, the increase in absorbance at 405 nm was followed in time. After ≈5 min, TFPI was added to the reaction mixture, and the reaction was followed until the rate of chromogenic substrate conversion became constant. Final concentrations in all experiments were 0.2 nM human FXa, 500 μM S2222, and 0 or 10 μM phospholipid vesicles in Hepes-buffered saline containing 5 mg/ml BSA and 3 mM CaCl2. The dose-dependent effect of TFPI on FXa inhibition was measured in the absence or presence of 100 nM protein S. Final concentrations were 0–3.9 nM and 0–7.7 nM TFPI with and without phospholipid vesicles, respectively, and 0–12.7 nM TFPI1–161.
Kinetic Analysis.
Progress curves of FXa inhibition by TFPI were fitted to the integrated rate equation (2) for slow binding inhibition, generating values for vo, vs, and kobs: vo and vs are the initial and steady-state velocities of pNA formation, respectively, and kobs is the apparent rate constant for the transition from vo to vs (FXaTFPI to FXaTFPI*, Eq. 1). FXa inhibition at varying TFPI concentrations was measured, and Ki values were calculated from a plot of V/vo versus the concentration of TFPI, in which V is the rate of pNA formation by FXa in the absence of TFPI (22, 47). The x intercept of this line is −Ki(1 + [S]/Km), in which [S] is the concentration chromogenic substrate S2222 in the reaction mixture (0.5 mM). Under the conditions used, the Km value for S2222 conversion by FXa was 1.065 mM. Similarly, Ki* was determined from a plot of V/vs versus the concentration of TFPI. Subsequent application of Eqs. 3 and 4
Acknowledgments
This work was supported in part by Netherlands Organization for Scientific Research Fellowship NWO-VIDI 917.36.372 (to T.M.H.).
Abbreviations
- APC
activated protein C
- TF
tissue factor
- TFPI
TF pathway inhibitor
- FVIIa
factor VIIa
- FX
factor X
- FXa
factor Xa
- ETP
endogenous thrombin potential
- PAR
protease-activated receptor
- C4BP
C4b-binding protein
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPS
1,2-dioleoyl-sn-glycero-3-phosphoserine
- DOPE
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Levi M., Ten Cate H. N. Engl. J. Med. 1999;341:586–592. doi: 10.1056/NEJM199908193410807. [DOI] [PubMed] [Google Scholar]
- 2.Tremoli E., Camera M., Toschi V., Colli S. Atherosclerosis. 1999;144:273–283. doi: 10.1016/s0021-9150(99)00063-5. [DOI] [PubMed] [Google Scholar]
- 3.Riewald M., Ruf W. Proc. Natl. Acad. Sci. USA. 2001;98:7742–7747. doi: 10.1073/pnas.141126698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Camerer E., Huang W., Coughlin S. R. Proc. Natl. Acad. Sci. USA. 2000;97:5255–5260. doi: 10.1073/pnas.97.10.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pawlinski R., Pedersen B., Schabbauer G., Tencati M., Holscher T., Boisvert W., Andrade-Gordon P., Frank R. D., Mackman N. Blood. 2004;103:1342–1347. doi: 10.1182/blood-2003-09-3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belting M., Dorrell M. I., Sandgren S., Aguilar E., Ahamed J., Dorfleutner A., Carmeliet P., Mueller B. M., Friedlander M., Ruf W. Nat. Med. 2004;10:502–509. doi: 10.1038/nm1037. [DOI] [PubMed] [Google Scholar]
- 7.Carmeliet P., Mackman N., Moons L., Luther T., Gressens P., Van Vlaenderen I., Demunck H., Kasper M., Breier G., Evrard P., et al. Nature. 1996;383:73–75. doi: 10.1038/383073a0. [DOI] [PubMed] [Google Scholar]
- 8.Rosendaal F. R. Thromb. Haemostasis. 1999;82:610–619. [PubMed] [Google Scholar]
- 9.Makris M., Leach M., Beauchamp N. J., Daly M. E., Cooper P. C., Hampton K. K., Bayliss P., Peake I. R., Miller G. J., Preston F. E. Blood. 2000;95:1935–1941. [PubMed] [Google Scholar]
- 10.Dahm A., Van Hylckama Vlieg A., Bendz B., Rosendaal F., Bertina R. M., Sandset P. M. Blood. 2003;101:4387–4392. doi: 10.1182/blood-2002-10-3188. [DOI] [PubMed] [Google Scholar]
- 11.Broze G. J., Jr Annu. Rev. Med. 1995;46:103–112. doi: 10.1146/annurev.med.46.1.103. [DOI] [PubMed] [Google Scholar]
- 12.Girard T. J., Warren L. A., Novotny W. F., Likert K. M., Brown S. G., Miletich J. P., Broze G. J., Jr Nature. 1989;338:518–520. doi: 10.1038/338518a0. [DOI] [PubMed] [Google Scholar]
- 13.Esmon C. T. J. Biol. Chem. 1989;264:4743–4746. [PubMed] [Google Scholar]
- 14.Rosing J., Hoekema L., Nicolaes G. A., Thomassen M. C., Hemker H. C., Varadi K., Schwarz H. P., Tans G. J. Biol. Chem. 1995;270:27852–27858. doi: 10.1074/jbc.270.46.27852. [DOI] [PubMed] [Google Scholar]
- 15.O’Brien L. M., Mastri M., Fay P. J. Blood. 2000;95:1714–1720. [PubMed] [Google Scholar]
- 16.Heeb M. J., Mesters R. M., Tans G., Rosing J., Griffin J. H. J. Biol. Chem. 1993;268:2872–2877. [PubMed] [Google Scholar]
- 17.Heeb M. J., Rosing J., Bakker H. M., Fernandez J. A., Tans G., Griffin J. H. Proc. Natl. Acad. Sci. USA. 1994;91:2728–2732. doi: 10.1073/pnas.91.7.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hackeng T. M., van’t Veer C., Meijers J. C., Bouma B. N. J. Biol. Chem. 1994;269:21051–21058. [PubMed] [Google Scholar]
- 19.Sere K. M., Rosing J., Hackeng T. M. Blood. 2004;104:3624–3630. doi: 10.1182/blood-2004-03-1146. [DOI] [PubMed] [Google Scholar]
- 20.Baugh R. J., Broze G. J., Jr, Krishnaswamy S. J. Biol. Chem. 1998;273:4378–4386. doi: 10.1074/jbc.273.8.4378. [DOI] [PubMed] [Google Scholar]
- 21.Huang Z. F., Wun T. C., Broze G. J., Jr J. Biol. Chem. 1993;268:26950–26955. [PubMed] [Google Scholar]
- 22.Morrison J. F., Walsh C. T. Adv. Enzymol. Relat. Areas Mol. Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 23.Mitchell C. A., Kelemen S. M., Salem H. H. Thromb. Haemostasis. 1988;60:298–304. [PubMed] [Google Scholar]
- 24.Sere K. M., Janssen M. P., Willems G. M., Tans G., Rosing J., Hackeng T. M. Biochemistry. 2001;40:8852–8860. doi: 10.1021/bi002500a. [DOI] [PubMed] [Google Scholar]
- 25.Piro O., Broze G. J., Jr Circulation. 2004;110:3567–3572. doi: 10.1161/01.CIR.0000148778.76917.89. [DOI] [PubMed] [Google Scholar]
- 26.Valentin S., Schousboe I. Thromb. Haemostasis. 1996;75:796–800. [PubMed] [Google Scholar]
- 27.Lockett J. M., Mast A. E. Biochemistry. 2002;41:4989–4997. doi: 10.1021/bi016058n. [DOI] [PubMed] [Google Scholar]
- 28.Lindhout T., Willems G., Blezer R., Hemker H. C. Biochem. J. 1994;297:131–136. doi: 10.1042/bj2970131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wesselschmidt R., Likert K., Girard T., Wun T. C., Broze G. J., Jr Blood. 1992;79:2004–2010. [PubMed] [Google Scholar]
- 30.Huang Z. F., Higuchi D., Lasky N., Broze G. J., Jr Blood. 1997;90:944–951. [PubMed] [Google Scholar]
- 31.Mahasandana C., Suvatte V., Marlar R. A., Manco-Johnson M. J., Jacobson L. J., Hathaway W. E. Lancet. 1990;335:61–62. doi: 10.1016/0140-6736(90)90201-f. [DOI] [PubMed] [Google Scholar]
- 32.Hansen J. B., Huseby K. R., Huseby N. E., Ezban M., Nordoy A. Thromb. Res. 1997;85:413–425. doi: 10.1016/s0049-3848(97)00029-7. [DOI] [PubMed] [Google Scholar]
- 33.Hockin M. F., Jones K. C., Everse S. J., Mann K. G. J. Biol. Chem. 2002;277:18322–18333. doi: 10.1074/jbc.M201173200. [DOI] [PubMed] [Google Scholar]
- 34.Koyama T., Nishida K., Ohdama S., Sawada M., Murakami N., Hirosawa S., Kuriyama R., Matsuzawa K., Hasegawa R., Aoki N. Br. J. Haematol. 1994;87:343–347. doi: 10.1111/j.1365-2141.1994.tb04919.x. [DOI] [PubMed] [Google Scholar]
- 35.Mueller B. M., Reisfeld R. A., Edgington T. S., Ruf W. Proc. Natl. Acad. Sci. USA. 1992;89:11832–11836. doi: 10.1073/pnas.89.24.11832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riewald M., Kravchenko V. V., Petrovan R. J., O’Brien P. J., Brass L. F., Ulevitch R. J., Ruf W. Blood. 2001;97:3109–3116. doi: 10.1182/blood.v97.10.3109. [DOI] [PubMed] [Google Scholar]
- 37.Hjortoe G. M., Petersen L. C., Albrektsen T., Sorensen B. B., Norby P. L., Mandal S. K., Pendurthi U. R., Rao L. V. Blood. 2004;103:3029–3037. doi: 10.1182/blood-2003-10-3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahamed J., Ruf W. J. Biol. Chem. 2004;279:23038–23044. doi: 10.1074/jbc.M401376200. [DOI] [PubMed] [Google Scholar]
- 39.Ahamed J., Belting M., Ruf W. Blood. 2005;105:2384–2391. doi: 10.1182/blood-2004-09-3422. [DOI] [PubMed] [Google Scholar]
- 40.Salemink I., Franssen J., Willems G. M., Hemker H. C., Lindhout T. J. Biol. Chem. 1999;274:28225–28232. doi: 10.1074/jbc.274.40.28225. [DOI] [PubMed] [Google Scholar]
- 41.Diaz-Collier J. A., Palmier M. O., Kretzmer K. K., Bishop B. F., Combs R. G., Obukowicz M. G., Frazier R. B., Bild G. S., Joy W. D., Hill S. R., et al. Thromb. Haemostasis. 1994;71:339–346. [PubMed] [Google Scholar]
- 42.Petersen J. G., Meyn G., Rasmussen J. S., Petersen J., Bjorn S. E., Jonassen I., Christiansen L., Nordfang O. J. Biol. Chem. 1993;268:13344–13351. [PubMed] [Google Scholar]
- 43.Girard T. J., Broze G. J., Jr Methods Enzymol. 1993;222:195–209. doi: 10.1016/0076-6879(93)22014-7. [DOI] [PubMed] [Google Scholar]
- 44.van’t Veer C., Hackeng T. M., Delahaye C., Sixma J. J., Bouma B. N. Blood. 1994;84:1132–1142. [PubMed] [Google Scholar]
- 45.Sandset P. M., Larsen M. L., Abildgaard U., Lindahl A. K., Odegaard O. R. Blood Coagul. Fibrinolysis. 1991;2:425–433. [PubMed] [Google Scholar]
- 46.Hemker H. C., Giesen P., Al Dieri R., Regnault V., de Smedt E., Wagenvoord R., Lecompte T., Beguin S. Pathophysiol. Haemostasis Thromb. 2003;33:4–15. doi: 10.1159/000071636. [DOI] [PubMed] [Google Scholar]
- 47.Longstaff C., Gaffney P. J. Biochemistry. 1991;30:979–986. doi: 10.1021/bi00218a014. [DOI] [PubMed] [Google Scholar]
- 48.Morrison J. F. Trends Biochem. Sci. 1982;7:102–105. [Google Scholar]