

Abstract
Myosin-I is the single-headed member of the myosin superfamily that associates with acidic phospholipids through its basic tail domain. Membrane association is essential for proper myosin-I localization and function. However, little is known about the physiological relevance of the direct association of myosin-I with phospholipids or about phospholipid headgroup-binding specificity. To better understand the mechanism of myosin-I–membrane association, we measured effective dissociation constants for the binding of a recombinant myo1c tail construct (which includes three IQ domains and bound calmodulins) to large unilamellar vesicles (LUVs) composed of phosphatidylcholine and various concentrations of phosphatidylserine (PS) or phosphatidylinositol 4,5-bisphosphate (PIP2). We found that the myo1c-tail binds tightly to LUVs containing >60% PS but very weakly to LUVs containing physiological PS concentrations (<40%). The myo1c tail and not the IQ motifs bind tightly to LUVs containing 2% PIP2. Additionally, we found that the myo1c tail binds to soluble inositol-1,4,5-trisphosphate with nearly the same affinity as to PIP2 in LUVs, suggesting that myo1c binds specifically to the headgroup of PIP2. We also show that a GFP-myosin-I-tail chimera expressed in epithelial cells is transiently localized to regions known to be enriched in PIP2. Our results suggest that myo1c does not bind to physiological concentrations of PS but rather binds tightly to PIP2.
Keywords: cell motility, unconventional myosin, lipid signaling
Myosin-Is are widely expressed members of the myosin superfamily that bind to actin filaments and hydrolyze ATP to produce mechanical force (1). Myosin-Is comprise the largest unconventional myosin family in humans (2), where they function in membrane dynamics and mechanical signal transduction (e.g., refs. 3–7). Consistent with these functions, subcellular localization and fractionation experiments show that a large percentage of myosin-I in the cell is associated with lipid membranes (3, 5, 8). This membrane association is primarily electrostatic (9, 10) and is due to the interaction of the basic tail domain of myosin-I with acidic phospholipids (11). Little is known about the relevance of the direct association of myosin-I with phospholipids, and there is little information regarding specificity for lipid headgroups. However, it is clear that myosin-I isoforms are able to bind a variety of anionic phospholipids (10–12).
Myo1c is a widely expressed vertebrate myosin-I isoform that concentrates in perinuclear regions, on ruffling cell membranes, and within stereocilia of hair cells (3, 5, 13, 14). It binds to negatively charged phospholipids, and a single myosin-I binds and clusters phosphatidylserine (PS) molecules upon membrane binding (15). Recent in vivo studies have suggested that myo1c associates with anionic phosphoinositides (12, 16), and that this interaction is regulated by calcium and calmodulin (12). However, it is not clear how myosin-I can specifically associate with phosphoinositides in vivo, given their relatively low abundance on the inner leaflet of the plasma membrane bilayer (1–2%) relative to other negatively charged lipids, such as PS, which makes up ≈20% of the lipid on the inner leaflet of the plasma membrane.
To better understand the biochemical parameters that define the myosin-I–membrane interaction, we performed a quantitative investigation of the steady-state binding of a myo1c-tail construct, consisting of the three calmodulin-binding IQ motifs and the tail domain, to large unilamellar vesicles containing phosphatidylinositol 4,5-bisphosphate (PIP2) or PS at various mol percentages. We report here that myo1c binding to vesicles containing 2% PIP2 is >100-fold tighter than to vesicles containing <40% PS. Additionally, myo1c binds to inositol 1,4,5-trisphosphate (InsP3) with nearly the same affinity as to PIP2, indicating that myo1c binds specifically to the headgroup of PIP2. We also show that a GFP–myosin-I-tail chimera expressed in epithelial cells is transiently localized to regions known to be enriched in PIP2.
Results
Myo1c-Tail Binds PIP2 Tightly.
We determined the effective dissociation constants, expressed in terms of total lipid (Kefflipid) or accessible acidic phospholipid (Keffacidic), for the interaction between myo1c-tail and LUVs of varying phospholipid content (Fig. 1). Low myo1c-tail concentrations were used (40 nM) to ensure that the binding sites on the LUVs were not saturated and that the effective dissociation constants were not affected by the ratio of protein to lipid (15). Binding experiments were performed with LUVs composed of the neutral phospholipid phosphatidylcholine (PC), and the acidic phospholipid PS in mol percentages of 0%, 20%, 40%, 60%, and 80% (Table 1). We found that the myo1c-tail binds weakly to LUVs with ≤40% PS (Kefflipid > 400 μM), whereas LUVs with ≥60% PS exhibit stronger binding (Table 1), thus confirming the finding of Doberstein and Pollard (11) that the affinity of myosin-I depends on the mol percentage of PS.
Fig. 1.

Association of the myo1c-tail with LUVs. (A) SYPRO-red stained SDS–polyacrylamide gel of myo1c-tail from the pellets of a sedimentation assay with 0–400 μM LUVs (total lipid) containing 2% PIP2. The last three lanes are myo1c-tail standards used for normalization. (B) Lipid concentration dependence of 40 nM myo1c-tail binding to LUVs composed of PC and (○) 0% PS, (□) 20% PS, (◇) 40% PS, (▴) 60% PS, (▾) 80% PS, and (•) 2% PIP2. Each point is the average of two to six measurements. The solid and dashed curves are the best fits of the 60% PS and 2% PIP2 data to hyperbolae, respectively. The Keff of each data set is listed in Table 1.
Table 1.
Effective dissociation constants for myo1c-tail binding to LUVs
| LUV composition* | Kefflipid, μM† | Keffacidic, μM‡ |
|---|---|---|
| 0% PS | >400 | NA |
| 20% PS | >400 | >40 |
| 40% PS | >400 | >80 |
| 60% PS | 11 ± 2.0 | 3.2 ± 1.0 |
| 80% PS | 4.1 ± 0.70 | 1.7 ± 0.060 |
| 2% PIP2 | 23 ± 5.0 | 0.23 ± 0.050 |
NA, not applicable. 10 mM Hepes, pH 7.0/100 mM NaCl/1 mM EGTA/1 mM DTT/1 μM calmodulin.
*The mol percentages PS and PIP2 are reported with the remaining composed of PC.
†Effective dissociation constants expressed in terms of total phospholipid. Errors are standard errors of the fit.
‡Effective dissociation constants expressed in terms of accessible acidic phospholipid. Errors are standard errors of the fit.
We also measured the affinity of myo1c for LUVs containing the important signaling lipid PIP2, which has a higher negative charge per molecule than PS. Myo1c-tail binds to LUVs composed of PC and 2% PIP2 with Kefflipid = 23 ± 5.0 μM (Keffacidic = 0.23 ± 0.05 μM; Table 1). LUVs composed of 2% PIP2 have approximately the same effective charge as LUVs composed of 6–8% PS (17), yet the affinities of myo1c-tail for LUVs composed of <40% PS are >150-fold weaker (Table 1). Therefore, myo1c-tail has considerable binding specificity for PIP2 over PS.
Myo1c-Tail Binding to PIP2 Is Not Cooperative.
To determine whether there is a cooperative myo1c-tail-binding-dependence on PIP2 density, we measured the binding of myo1c-tail to 30 μM LUVs composed of 0–10% PIP2 while holding the total lipid concentration constant (Fig. 2). A “lag phase” is not present in the binding curve, as seen for proteins that bind multiple PIP2 molecules (18), but rather a hyperbolic dependence of myo1c-tail binding on the mol percent of PIP2 is observed. Additionally, when the data are plotted as myo1c-tail bound versus the accessible PIP2 concentration (i.e., PIP2 available on the outer leaflet of the LUV bilayer), the concentration dependence of binding is nearly identical to that of 60 μM LUVs composed of 0–5% PIP2 and to 2% PIP2 obtained at different total lipid concentrations (Fig. 2 Inset). Therefore, the interaction of the myo1c-tail with PIP2 is not cooperative and likely has a binding stoichiometry of 1:1. This mode of binding is similar to the pleckstrin homology (PH) domain of phospholipase-Cδ (PLCδ-PH), which binds the head group of PIP2 noncooperatively (19).
Fig. 2.

Binding of 40 nM myo1c-tail to 30 μM LUVs (total lipid) composed of 0–10% PIP2. The percent of membrane-bound myo1c-tail is plotted as a function of the percentage of PIP2 in the LUVs rather than total lipid concentration. Each point is the average of three measurements. (Inset) The same data plotted as a function of the accessible PIP2 concentration (○). Data in Inset also include the percent of 40 nM myo1c-tail bound to (▵) 60 μM LUVs containing 0–5% PIP2 and (•) 0–400 μM LUVs containing 2% PIP2 from Fig. 1B. The solid line is the best fit of all of the data to a hyperbola, yielding Keff = 0.31 ± 0.06 μM.
Myo1c-Tail and PLCδ-PH Compete for Binding to PIP2.
We tested the ability of PLCδ-PH to compete with 100 nM myo1c-tail for binding to LUVs containing 2% PIP2. Myo1c-tail, LUVs, and 0–10 μM PLCδ-PH were sedimented, and pellets were analyzed for the myo1c-tail and PLCδ-PH (Fig. 3C Inset). We found that the myo1c-tail was displaced from the LUVs as the concentration of PLCδ-PH was increased (Fig. 3C). If we assume that the myo1c-tail binds PIP2 with a 1:1 stoichiometry and Keffacidic = 0.23 μM (Table 1), a fit to the competition data yields an effective dissociation constant for PLCδ-PH binding to PIP2 of Keff = 0.30 ± 0.03 μM, which is in close agreement with determined values (20). Therefore, the myo1c-tail and PLCδ-PH compete for the same binding site.
Fig. 3.

The myo1c-tail binds InsP3. (A and B) Gel filtration elution profiles of samples containing 10 μM PLCδ-PH (A) or 10 μM myo1c-tail (B) in the presence of excess 3H-Ins(1,4,5)P3. The concentrations of InsP3 (solid lines) and PLCδ-PH or myo1c-tail (dotted lines) are shown. InsP3 run in the absence of protein in a separate experiment is also shown (dashed line) as a reference elution profile. (C) Binding of 100 nM myo1c-tail to 60 μM LUVs containing 2% PIP2 in the presence of 0–10 μM PLCδ-PH. (Inset) A SYPRO-red-stained SDS–polyacrylamide gel showing a LUV-bound (top) myo1c-tail and (bottom) PLCδ-PH as a function of total PLCδ-PH concentration. (D) Binding of 100 nM myo1c-tail to 60 μM LUVs containing 2% PIP2 in the presence of 0–10 μM InsP3. (Inset) A SYPRO-red-stained SDS–polyacrylamide gel showing a LUV-bound myo1c-tail as a function of total InsP3 concentration. Solid lines are fits to a competition binding equation, yielding Keff = 0.30 ± 0.03 μM for PLCδ-PH and Kd = 0.072 ± 0.12 μM for InsP3. Error bars represent ± 1 standard deviation (n = 6).
Myo1c-Tail Binds to InsP3.
To determine whether the myo1c-tail binds to the headgroup of PIP2 directly, we measured its ability to bind [3H]-labeled InsP3 by gel filtration chromatography. PLCδ-PH, which has been shown to bind InsP3 (20), was used as a positive control. PLCδ-PH (Fig. 3A) and myo1c-tail (Fig. 3B) eluted as single peaks from gel filtration columns. InsP3 eluted as two peaks, one coeluting with the PLCδ-PH (Fig. 3A) or myo1c-tail (Fig. 3B) and the other corresponding to free InsP3. InsP3 eluted as a single nonprotein-bound peak when run on the gel filtration column with GST, a protein that does not bind InsP3 (not shown). These results suggest that myo1c binds to InsP3 directly.
We determined the affinity of InsP3 for the myo1c-tail by competition binding. InsP3 (0–10 μM) was mixed with 100 nM myo1c-tail and LUVs containing 2% PIP2. We found that the myo1c-tail was displaced from LUVs with increasing InsP3 concentrations (Fig. 3D Inset). A fit to the competition data yields a dissociation constant of Kd = 0.072 ± 0.12 μM for the InsP3–myo1c-tail interaction (Fig. 3D). Therefore, the myo1c-tail binds to InsP3 with high affinity, and InsP3 competes with PIP2 for binding to the myo1c-tail.
Calcium and Calmodulin Do Not Affect Myolc-Tail Binding to LUVs.
It has been proposed that one or more of the IQ domains of myo1c binds to lipids directly, and this binding is sensitive to conformational changes in calmodulin upon calcium binding (12, 15, 21). Therefore, we tested the effects of both calmodulin and calcium on the steady-state binding of 40 nM myo1c-tail to LUVs containing 2% PIP2. We found that the binding of the myo1c-tail to the LUVs was not affected by up to 100 μM calmodulin (Fig. 4A). Additionally, we did not detect a significant change in the affinity of myo1c-tail for LUVs containing 2% PIP2 in the presence of 10 μM free calcium (Fig. 4B). Therefore, calmodulin and calcium do not directly affect the binding of myo1c to PIP2 lipid membranes.
Fig. 4.

The effect of calmodulin and calcium on myo1c-binding LUVs. (A) Forty nanomolar myo1c-tail binding to 60 μM LUVs containing (•) 2% PIP2 or (▴) 60% PS LUVs in the presence of 0–100 μM calmodulin. Each point is the average of two measurements. (B) Forty nanomolar myo1c-tail binding to 0–400 μM LUVs containing 2% PIP2 in the (•) absence and (○) presence of 10 μM free calcium. The solid line is the best fit to a rectangular hyperbola. Error bars represent ± 1 standard deviation (n = 4–6). (C) Lipid concentration dependence of 40 nM myo1c–motor–IQ binding to LUVs composed of PC and 20% PS (■, □) or 2% PIP2 (•, ○) in the absence (closed symbols) or presence (open symbols) of 10 μM free calcium.
To rule out the possibility that the IQ motifs are sufficient for binding to physiological concentrations of anionic lipids, we tested the ability of a myo1c–motor–IQ construct, consisting of the motor domain and three IQ motifs, to bind LUVs composed of 20% PS or 2% PIP2 in the absence and presence of 10 μM free calcium (Fig. 4C). We did not detect appreciable binding to LUVs at lipid concentrations up to 200 μM. We conclude that the tail domain is responsible for PIP2 binding, and that the IQ motifs do not support membrane association alone.
GFP–Myo1c–Tail Concentrates on Cellular PIP2-Containing Structures.
If the tail domain of myo1c binds tightly to cellular PIP2, it should localize to the plasma membrane and translocate to the cytoplasm upon activation of phospholipase-C (22, 23). We expressed a GFP–myo1c-tail chimera in normal rat kidney epithelial cells and confirmed that it binds to the plasma membrane (Fig. 5), as shown (3, 5). Activation of phospholipase C by the addition of 10 μM ionomycin and 1.2 mM calcium chloride to the growth medium results in the redistribution of GFP–myo1c-tail to the cytoplasm (Fig. 5). GFP–myo1c-tail reassociates with the plasma membrane upon washout of the calcium and ionomycin (Fig. 5; see Movie 1, which is published as supporting information on the PNAS web site). Although this assay is relatively nonspecific, it supports our findings that the myo1c-tail binds to PIP2, and that calcium does not increase the affinity of myo1c for membranes.
Fig. 5.
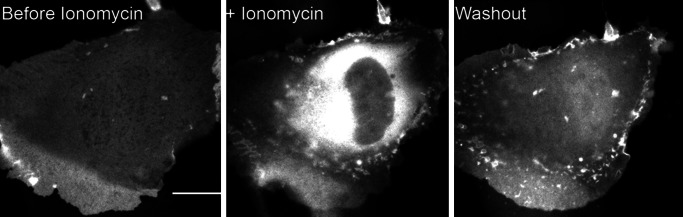
Cellular distribution of GFP–myo1c-tail during influx of calcium. Fluorescence micrographs of a transfected normal rat kidney epithelial cell showing GFP fluorescence before, during, and after incubation with 10 μM ionomycin in medium containing 1.2 mM CaCl2. (Scale bar, 15 μm.) See Movie 1, which is published as supporting information on the PNAS web site, for movie sequence.
In cells undergoing macropinocytosis, we find that the GFP–myo1c-tail transiently concentrates on the membrane during the early phases of membrane internalization (Fig. 6). Once the macropinosome separates from the plasma membrane, the GFP–myo1c-tail dissociates from the membrane. This transient localization closely mirrors the localization of PIP2-specific PH domains during macropinocytosis and phagocytosis (24, 25) and full length GFP–myo1c (3), again supporting the proposal that myo1c primarily binds to cellular PIP2.
Fig. 6.

Cellular distribution of GFP–myo1c-tail during macropinocytosis. Fluorescence micrographs of a transfected NRK cell showing GFP fluorescence around a newly internalized macropinosome (arrow). Note the loss of fluorescence around macropinosome after it is internalized. (The time stamp is min:sec, and the scale bar is 5 μm.) See Movie 2, which is published as supporting information on the PNAS web site, for movie sequence.
Discussion
Mechanism of Membrane Binding.
The binding of myosin-I to membranes is due to electrostatic interactions between the positively charged tail domain and the head groups of acidic phospholipids (11). We have shown previously for LUVs composed of PS and PC that the affinity of myosin-I for LUVs is related to the number of PS molecules bound to the tail (15). Therefore, it is likely that the weak affinity of myo1c-tails for LUVs composed of <40% PS is due to the inability of the myo1c-tail to cluster the lipids needed for stable binding.
Like the myo1c-tail, the protein myristoylated alanine-rich C kinase substrate (MARCKS) has been shown to bind PS (which has a charge of −1) through electrostatic interactions (17). Because membrane binding of MARCKS requires interaction with multiple PS molecules on the lipid, it was predicted and subsequently demonstrated that it preferentially binds to lipid headgroups with multiple negative charges, i.e., lipids in which the charges are already clustered, like PIP2, which has a charge of −4 at pH 7 (17). To determine whether myo1c binds via a similar mechanism, we measured the binding of myo1c-tail to PIP2 and found that, like MARCKS, myo1c binds PIP2 tightly (Figs. 1B and 2; Table 1). However, unlike proteins with polybasic effectors domains, e.g., MARCKS (17) and N-WASP (18), the interaction appears to be noncooperative and specific for the headgroup of the lipid, because the affinity for InsP3 is nearly identical to the PIP2 affinity (Fig. 3D; Table 1). Thus, the mechanism of myo1c membrane binding is like the specific interaction of PLCδ-PH with PIP2 (19).
The membrane-binding region of myo1c has not been identified. However, it has been predicted that at least part of the binding site is present within the IQ motifs, and that this site is revealed by calcium-dependent changes in the binding of calmodulin (12, 15, 21). We found no effect of the calmodulin or calcium concentrations on lipid binding, as would be predicted if PIP2 competed with calmodulin for a common binding site. Additionally, a construct containing the IQ motifs, but not the tail, did not show tight binding to 20% PS or 2% PIP2. We did find that calcium affects the distribution of LUV size, as determined by dynamic light scattering (D.E.H. and E.M.O., data not shown), and it has been demonstrated that low concentrations of divalent cations catalyze lipid aggregation and fusion (26, 27). Thus, our previous reports of calcium-dependent changes in myo1c-tail binding may have been due to changes in lipid structure (15). Therefore, we propose that calcium does not affect myo1c-tail interactions by direct binding to calmodulin.
Biological Relevance of PIP2 Binding.
Our results suggest that myo1c does not bind tightly to physiological concentrations of PS but rather binds to PIP2. PIP2 is an important second messenger involved in a variety of crucial cellular functions, including the regulation of the actin cytoskeleton (28). PIP2 is highly concentrated in actin-rich structures, where it directly regulates numerous cytoskeletal proteins, including activators of the Arp2/3 complex, capping proteins, and profilin (28). Myosin-I isoforms are also highly enriched in these regions (3, 5, 6, 29–31). Thus, the myosin-I–PIP2 interaction may serve to concentrate myosin-I at regions of new actin polymerization, providing a mechanism for recruitment of motors to the actin-rich structures that drive membrane trafficking and endocytosis, membrane retraction, retrograde flow, and mechano-signal transduction (3, 4, 29, 30, 32). Myosin-I may also use its barbed-end-directed motor activity to keep the fast-growing ends of the actin filament oriented toward the membrane, thus ensuring that the addition of new actin subunits push the membrane forward. These roles are consistent with myosin-I’s ability to bind only the dynamic actin filament population and not tropomyosin-stabilized microfilaments (6). Additionally, myosin-I may regulate cytoskeleton–plasma membrane adhesion by functioning as a link between the cortical actin cytoskeleton and PIP2 (33).
The tight binding of myo1c to InsP3 may be important in the mechanism of myo1c dissociation from the membrane. Hydrolysis of PIP2 by PLC, which yields diacylglycerol and soluble InsP3, is required for the termination of actin polymerization and disassembly of the actin cytoskeleton within some actin-rich structures (34). Because myo1c binds InsP3 and PIP2 with similar affinities, myo1c will not compete exclusively for the remaining pool of PIP2 during PLC activation but will readily bind soluble InsP3, thus facilitating its dissociation from the membrane (Fig. 5).
Materials and Methods
Reagents and Buffers.
All in vitro experiments were performed in HNa100 (10 mM Hepes, pH 7.0/100 mM NaCl/1 mM EGTA/1 mM DTT). Calcium concentrations were adjusted by adding CaCl2 to HNa100 and are reported as free calcium. Unless stated otherwise, all binding experiments were performed with 1 μM free calmodulin. PS, PC, and PIP2 were from Avanti Polar Lipids. Tritated InsP3 [3H-Ins(1,4,5,)P3] was purchased from PerkinElmer.
The mouse myo1c-tail construct (residues 690–1028), which consists of an N-terminal HIS6 tag for purification, three calmodulin-binding IQ motifs, and the tail domain, was expressed and purified (15). The purified myo1c-tail contained three bound calmodulins. The myo1c–motor–IQ construct includes the motor domain and three IQ motifs (residues 1–767). A 15-aa sequence for site-specific biotinylation (35) and a FLAG sequence for purification were inserted at the C terminus. The construct was subcloned into a baculovirus transfer vector, and recombinant baculovirus was generated by using standard procedures and screened by plaque assays. The myo1c–motor–IQ was purified from Sf9 cells that were coinfected with virus containing recombinant myo1c–motor–IQ and calmodulin, as described (36).
A bacterial expression plasmid for the PH domain of phospholipase-Cδ (residues 11–140; PLCδ-PH) was a gift from Mark Lemmon (University of Pennsylvania) and was purified as described (37).
Recombinant chicken calmodulin was expressed and purified from bacterial lysates (38) and further purified by FPLC by using a monoQ column (Amersham Pharmacia Biosciences). This final monoQ step was crucial, because we found a bacterial contaminant present in very low abundance in the preparation that effectively competed with the myo1c-tail for binding to anionic lipids.
Lipid Preparation.
LUVs with 100-nm diameter were prepared by extrusion. Lipid components were mixed in the desired ratios in chloroform and dried under a stream of nitrogen. Phospholipid vesicles containing PIP2 were also prepared by first placing the chloroform solution in a 35°C water bath for 5 min and then applying maximum vacuum in a rotovap for 30 min to ensure uniform inclusion of PIP2 and PC (39). Lipids for sedimentation experiments were resuspended in 176 mM sucrose/12 mM Hepes, pH 7.0, to a total concentration of 2 mM, subjected to five cycles of freeze–thaw, and then bath-sonicated 1 min before being passed through 100-nm filters (11 times) by using a miniextruder (Avanti Polar Lipids). LUVs were dialyzed overnight vs. HNa100. Dynamic light-scattering measurements confirmed that the lipids were a preparation of monodisperse 100-nm vesicles. LUVs were stored at 4°C under N2 and discarded after 3 days. PS and PIP2 percentages reported throughout the text are the mol percentages of total PS and PIP2, with the remainder being PC. Lipid concentrations are given as total lipid unless otherwise noted.
Sedimentation Assay.
Myo1c-tail binding to LUVs was determined by sedimentation assays conducted with 200-μl samples in an ultracentrifuge with a TLA-100 rotor (Beckman). Nonspecific binding of myo1c-tail to polycarbonate centrifuge tubes was prevented by incubating the tubes for 1 h in a 50 μM solution of PC in HNa100 and by the addition of 0.25 mg/ml GST to every sample. Sucrose-loaded LUVs were sedimented at 150,000 × g for 30 min at 25°C. The top 160 μl of each sample was removed and analyzed as supernatant. We call the remaining 40 μl the “pellet,” although it contains supernatant. The pellet was resuspended with 10 μl SDS/PAGE sample buffer and boiled for 3 min. Using fluorescently labeled lipids and phosphate assays (40), we confirmed that >95% of all lipid was within this 40 μl after sedimentation. Protein samples were resolved on 12% SDS/PAGE gels that were stained with SYPRO-red (Invitrogen) for quantitation.
Gels were scanned by using a Typhoon 8600 imager, and data were analyzed with metamorph (Universal Imaging, Downington, PA). The integrated intensity of each protein band on the scanned gels was subtracted from the integrated intensity of the region of the gel directly above the protein band to correct for variations in background. Myo1c-tail standards of 40, 120, and 240 nM were run on each gel to ensure that the intensity of the protein bands was within the linear range of the scanner and to translate integrated intensity to mol of bound protein. We report the binding affinity of the myo1c-tail to LUVs as an effective dissociation constant (Keff). Keff is simply the inverse of the partition coefficient (41). The Keff is reported in terms of total lipid concentration (Kefflipid) or as the concentration of acidic phospholipid present on the outer bilayer leaflet of the LUV (Keffacidic). Binding data were fit to hyperbolae by using kaleidagraph (Synergy Software, Reading, PA), and the data from competition experiments were fit as described (42) by using mathcad (Mathsoft, Cambridge, MA).
Gel Filtration.
PLCδ-PH (10 μM) or 10 μM myolc-tail was mixed with excess 3H-Ins(1,4,5,)P3. Samples were applied to a 10 ml Toyo Pearl HW40F desalting column equilibrated with HNa100. Fractions were collected every ≈200 μl. The elution position of protein was determined by absorbance at 280 nm. Scintillation counting was used to detect the 3H-Ins(1,4,5)P3 elution position.
Live Cell Imaging.
A GFP-myo1c-tail construct (residues 690–1028), which contains the three IQ motifs and tail domain, was constructed in pEGFP-C1. Normal rat kidney epithelial cells were cultured and electroporated as described (6). Cells plated on 40-mm glass coverslip were mounted in a temperature-controlled flow chamber (Bioptechs, Butler, PA) and perfused with DMEM with 10% FBS at 37°C as described (6).
Supplementary Material
Acknowledgments
We are especially grateful to Nanyun Tang and Tianming Lin (University of Pennsylvania) for providing expression plasmids, virus, and protein. We thank Dr. Mark Lemmon for the PLCδ-PH plasmid and for helpful discussions and Dr. Paul Janmey’s laboratory for assistance in dynamic light scattering and for advice. This work was supported by the National Institutes of Health (GM57247) and an Established Investigator Award (to E.M.O.).
Abbreviations
- InsP3
inositol 1,4,5-trisphosphate
- LUV
large unilamellar vesicle
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PH
pleckstrin homology
- PLCδ-PH
PH domain of phospholipase-Cδ
- PC
phosphatidylcholine
- PS
phosphatidylserine.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Sokac A. M., Bement W. M. Int. Rev. Cytol. 2000;200:197–304. doi: 10.1016/s0074-7696(00)00005-x. [DOI] [PubMed] [Google Scholar]
- 2.Berg J. S., Powell B. C., Cheney R. E. Mol. Biol. Cell. 2001;12:780–794. doi: 10.1091/mbc.12.4.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bose A., Robida S., Furcinitti P. S., Chawla A., Fogarty K., Corvera S., Czech M. P. Mol. Cell. Biol. 2004;24:5447–5458. doi: 10.1128/MCB.24.12.5447-5458.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holt J. R., Gillespie S. K., Provance D. W., Shah K., Shokat K. M., Corey D. P., Mercer J. A., Gillespie P. G. Cell. 2002;108:371–381. doi: 10.1016/s0092-8674(02)00629-3. [DOI] [PubMed] [Google Scholar]
- 5.Ruppert C., Godel J., Muller R. T., Kroschewski R., Reinhard J., Bahler M. J. Cell. Sci. 1995;108:3775–3786. doi: 10.1242/jcs.108.12.3775. [DOI] [PubMed] [Google Scholar]
- 6.Tang N., Ostap E. M. Curr. Biol. 2001;11:1131–1135. doi: 10.1016/s0960-9822(01)00320-7. [DOI] [PubMed] [Google Scholar]
- 7.Tyska M. J., Mackey A. T., Huang J. D., Copeland N. G., Jenkins N. A., Mooseker M. S. Mol. Biol. Cell. 2005;16:2443–2457. doi: 10.1091/mbc.E04-12-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balish M. F., Moeller E. F., III, Coluccio L. M. Arch. Biochem. Biophys. 1999;370:285–293. doi: 10.1006/abbi.1999.1409. [DOI] [PubMed] [Google Scholar]
- 9.Adams R. J., Pollard T. D. Nature. 1989;340:565–568. doi: 10.1038/340565a0. [DOI] [PubMed] [Google Scholar]
- 10.Hayden S. M., Wolenski J. S., Mooseker M. S. J. Cell Biol. 1990;111:443–451. doi: 10.1083/jcb.111.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doberstein S. K., Pollard T. D. J. Cell Biol. 1992;117:1241–1249. doi: 10.1083/jcb.117.6.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirono M., Denis C. S., Richardson G. P., Gillespie P. G. Neuron. 2004;44:309–320. doi: 10.1016/j.neuron.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 13.Hasson T., Gillespie P. G., Garcia J. A., MacDonald R. B., Zhao Y., Yee A. G., Mooseker M. S., Corey D. P. J. Cell Biol. 1997;137:1287–1307. doi: 10.1083/jcb.137.6.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wagner W., Hammer J. A., III J. Cell Biol. 2003;163:1193–1196. doi: 10.1083/jcb.200311077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang N., Lin T., Ostap E. M. J. Biol. Chem. 2002;277:42763–42768. doi: 10.1074/jbc.M206388200. [DOI] [PubMed] [Google Scholar]
- 16.Huang S., Liftshtz L., Patki-Kamath V., Tuft R., Fogarty K., Czech M. P. Mol. Cell. Biol. 2004;24:9102–9123. doi: 10.1128/MCB.24.20.9102-9123.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McLaughlin S., Wang J., Gambhir A., Murray D. Annu. Rev. Biophys. Biomol. Struct. 2002;31:151–175. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 18.Papayannopoulos V., Co C., Prehoda K. E., Snapper S., Taunton J., Lim W. A. Mol. Cell. 2005;17:181–191. doi: 10.1016/j.molcel.2004.11.054. [DOI] [PubMed] [Google Scholar]
- 19.Lemmon M. A., Ferguson K. M. Biochem. Soc. Trans. 2001;29:377–384. doi: 10.1042/bst0290377. [DOI] [PubMed] [Google Scholar]
- 20.Lemmon M. A., Ferguson K. M., O’Brien R., Sigler P. B., Schlessinger J. Proc. Natl. Acad. Sci. USA. 1995;92:10472–10476. doi: 10.1073/pnas.92.23.10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swanljung-Collins H., Collins J. H. J. Biol. Chem. 1992;267:3445–3454. [PubMed] [Google Scholar]
- 22.Botelho R. J., Teruel M., Dierckman R., Anderson R., Wells A., York J. D., Meyer T., Grinstein S. J. Cell Biol. 2000;151:1353–1368. doi: 10.1083/jcb.151.7.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varnai P., Balla T. J. Cell Biol. 1998;143:501–510. doi: 10.1083/jcb.143.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cullen P. J., Cozier G. E., Banting G., Mellor H. Curr. Biol. 2001;11:R882–R893. doi: 10.1016/s0960-9822(01)00523-1. [DOI] [PubMed] [Google Scholar]
- 25.Simonsen A., Wurmser A. E., Emr S. D., Stenmark H. Curr. Opin. Cell. Biol. 2001;13:485–492. doi: 10.1016/s0955-0674(00)00240-4. [DOI] [PubMed] [Google Scholar]
- 26.Wilschut J., Duzgunes N., Hoekstra D., Papahadjopoulos D. Biochemistry. 1985;24:8–14. doi: 10.1021/bi00322a002. [DOI] [PubMed] [Google Scholar]
- 27.Portis A., Newton C., Pangborn W., Papahadjopoulos D. Biochemistry. 1979;18:780–790. doi: 10.1021/bi00572a007. [DOI] [PubMed] [Google Scholar]
- 28.Yin H. L., Janmey P. A. Annu. Rev. Physiol. 2003;65:761–789. doi: 10.1146/annurev.physiol.65.092101.142517. [DOI] [PubMed] [Google Scholar]
- 29.Jung G., Wu X., Hammer J. A., III J. Cell Biol. 1996;133:305–323. doi: 10.1083/jcb.133.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Novak K. D., Peterson M. D., Reedy M. C., Titus M. A. J. Cell Biol. 1995;131:1205–1221. doi: 10.1083/jcb.131.5.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ostap E. M., Maupin P., Doberstein S. K., Baines I. C., Korn E. D., Pollard T. D. Cell. Motil. Cytoskeleton. 2003;54:29–40. doi: 10.1002/cm.10081. [DOI] [PubMed] [Google Scholar]
- 32.Diefenbach T. J., Latham V. M., Yimlamai D., Liu C. A., Herman I. M., Jay D. G. J. Cell Biol. 2002;158:1207–1217. doi: 10.1083/jcb.200202028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raucher D., Stauffer T., Chen W., Shen K., Guo S., York J. D., Sheetz M. P., Meyer T. Cell. 2000;100:221–228. doi: 10.1016/s0092-8674(00)81560-3. [DOI] [PubMed] [Google Scholar]
- 34.Simmen R. C., Tanaka T., Ts’ui K. F., Putkey J. A., Scott M. J., Lai E. C., Means A. R. J. Biol. Chem. 1985;260:907–912. [PubMed] [Google Scholar]
- 35.Schatz P. J. Biotechnology. 1993;11:1138–1143. doi: 10.1038/nbt1093-1138. [DOI] [PubMed] [Google Scholar]
- 36.El Mezgueldi M., Tang N., Rosenfeld S. S., Ostap E. M. J. Biol. Chem. 2002;277:21514–21521. doi: 10.1074/jbc.M200713200. [DOI] [PubMed] [Google Scholar]
- 37.Ferguson K. M., Lemmon M. A., Schlessinger J., Sigler P. B. Cell. 1994;79:199–209. doi: 10.1016/0092-8674(94)90190-2. [DOI] [PubMed] [Google Scholar]
- 38.Putkey J. A., Slaughter G. R., Means A. R. J. Biol. Chem. 1985;260:4704–4712. [PubMed] [Google Scholar]
- 39.Gambhir A., Hangyas-Mihalyne G., Zaitseva I., Cafiso D. S., Wang J., Murray D., Pentyala S. N., Smith S. O., McLaughlin S. Biophys. J. 2004;86:2188–2207. doi: 10.1016/S0006-3495(04)74278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fiske C. H., Subbarow Y. J. Biol. Chem. 1925;66:374–389. [Google Scholar]
- 41.Peitzsch R. M., McLaughlin S. Biochemistry. 1993;32:10436–10443. doi: 10.1021/bi00090a020. [DOI] [PubMed] [Google Scholar]
- 42.Kubala M., Plasek J., Amler E. Physiol. Res. 2004;53:109–113. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.