

Abstract
Cysteine dioxygenase (CDO) catalyzes the oxidation of l-cysteine to cysteine sulfinic acid. Deficiencies in this enzyme have been linked to autoimmune diseases and neurological disorders. The x-ray crystal structure of CDO from Mus musculus was solved to a nominal resolution of 1.75 Å. The sequence is 91% identical to that of a human homolog. The structure reveals that CDO adopts the typical β-barrel fold of the cupin superfamily. The NE2 atoms of His-86, -88, and -140 provide the metal binding site. The structure further revealed a covalent linkage between the side chains of Cys-93 and Tyr-157, the cysteine of which is conserved only in eukaryotic proteins. Metal analysis showed that the recombinant enzyme contained a mixture of iron, nickel, and zinc, with increased iron content associated with increased catalytic activity. Details of the predicted active site are used to present and discuss a plausible mechanism of action for the enzyme.
Keywords: cupin, cysteine metabolism, O2-activation
Mouse cysteine dioxygenase (CDO) catalyzes the initial step in the biochemical pathway used for oxidation of cysteine to sulfate (1), namely the oxidation of l-cysteine to cysteine sulfinic acid as shown in Fig. 1. The enzyme activity has important medical implications because elevated cysteine levels have been associated with Parkinson’s and Alzheimer’s diseases (2). High cysteine-to-sulfate ratios have been observed in patients suffering from systemic lupus erthematosus and rheumatoid arthritis (3, 4). Moreover, the Hallervorden–Spatz syndrome, a neurological disorder associated with iron accumulation, has been linked to a decline in CDO activity (5).
Fig. 1.
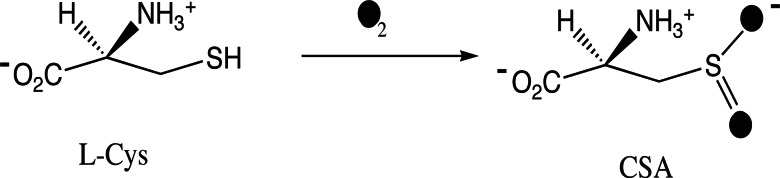
CDO reaction scheme showing that O2 is incorporated into l-cysteine (l-Cys) to yield cysteine sulfinic acid (CSA).
CDO displays significant sequence identity with some members of the cupin superfamily (6), which have a conserved β-barrel fold and share two conserved sequence motifs: G(X)5HXH(X)3,4E(X)6G and G(X)5PXG(X)2H(X)3N (6–8). The two His and Glu residues from the first motif and the His from the second motif coordinate the metal ion in germin, the superfamily archetype (9). The Mus musculus CDO sequence contains the first motif with the exception of the glutamate, which is replaced by cysteine. This substitution is conserved in other eukaryotic CDOs. The second motif is less conserved, and only the His and Asn residues are present in the mouse CDO.
CDO does not require an external reductant (Fig. 1) and incorporates both oxygen atoms from O2 (10), which justifies the dioxygenase classification, but relatively little else is known about the reaction mechanism. The recombinant enzyme from Rattus norvegicus has been purified and characterized by steady-state kinetics (11); the mouse enzyme investigated here has an identical sequence. Reconstitution of the rat apoenzyme with various transition metals confirmed that iron was required for activity, in accord with the earlier conclusions (1). Moreover, the recombinant rat enzyme was active without a second interacting factor, despite previous reports suggesting that additional components were required (12, 13).
Here, we describe the x-ray crystal structure of mouse CDO. The structure reveals a previously undescribed facial triad arrangement of three His ligands at the metal site. In addition, a covalent linkage between Tyr and Cys residues is present, yielding a posttranslational modification analogous to that originally observed in galactose oxidase (14). The modified Tyr and Cys residues are conserved in the eukaryotic CDOs, whereas only the Tyr residue is conserved in the bacterial enzymes. The observed structural features of the active site are used to propose a plausible reaction mechanism for CDO.
Results
Structure Determination.
The time elapsed from the initial selection of CDO as a structure target to deposition, including cloning, purification, crystallization, and structure determination, was 209 days. The structure of CDO was solved to a nominal 1.75-Å resolution. Crystallographic statistics are shown in Table 1. The protein is a monomer in the asymmetric unit and is composed of 200 aa. Terminal residues 1–4 and 191–200 were not modeled because of insufficient electron density. Ramachandran analysis indicated that 166 of the 186 residues modeled were in the most favored regions with the remaining residues in the generously allowed regions. The asymmetric unit also contained 187 ordered water molecules, one ethylene glycol molecule, and a metal ion. The metal was refined as Ni(II). Coordinates and structure factors were deposited in the Protein Data Bank (PDB) with PDB ID code 2atf.
Table 1.
Crystal parameters, data collection, and refinement statistics
| Statistic | Peak | Edge | Remote |
|---|---|---|---|
| Space group | P43212 | ||
| Unit cell parameters, Å | a = 57.6, b = 57.6, c = 122.1 | ||
| Wavelength | 0.97915 | 0.96392 | 0.97933 |
| Resolution range | 40.69–1.75 | 40.69–1.75 | 40.69–1.75 |
| Highest resolution shell | 1.79–1.75 | 1.79–1.75 | 1.79–1.75 |
| Measured reflections | 306,248 | 306,149 | 307,785 |
| Unique reflections | 21,529 | 21,553 | 21,554 |
| Completeness, % | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) |
| Rmerge* | 0.118 (0.270) | 0.109 (0.283) | 0.108 (0.257) |
| Redundancy | 14.2 (13.8) | 14.2 (13.4) | 14.3 (14.3) |
| Mean I/σ (I) | 15.89 (5.25) | 14.00 (7.09) | 13.95 (7.63) |
| Resolution range, Å | 1.75–33.86 | ||
| No. reflections, total/test | 20,362/1,101 | ||
| Rcryst† | 0.179 | ||
| Rfree‡ | 0.216 | ||
| Average B factor, Å2 | 19.1 |
Values in parentheses are for the highest-resolution shell.
*Rmerge = ΣhΣi|Ii(h) − 〈I(h)〉|/ΣhΣiIi(h), where Ii(h) is the intensity of an individual measurement of the reflection and 〈I(h)〉 is the mean intensity of the reflection.
†Rcryst = Σh∥Fobs| − |Fcalc∥/Σh|Fobs|, where Fobs and Fcalc are the observed and calculated structure-factor amplitudes, respectively.
‡Rfree was calculated as Rcryst using 5.0% of the randomly selected unique reflections that were omitted from structure refinement.
Protein Fold.
Fig. 2 shows the overall fold of CDO. The protein core consists of the small β-barrel-like structure typical of the cupin superfamily. This barrel is made up of three separate sheets. One side of the barrel consists of a seven-stranded mixed β-sheet containing strands G, D, I, B, A, L, and M. Strands G, D, I, B, and A are antiparallel, as are strands L and M. Strands A and L are parallel. The other half of the barrel is made up of two three-stranded antiparallel β-sheets. The β-sheet made up of β-strands F, E, and H is aligned parallel to the seven-stranded β-sheet, whereas the β-sheet made up of β-strands C, J, and K is rotated ≈90° to the other two sheets and forms a third side of the barrel. The N-terminal region of the protein is primarily helical, and all of the helices are localized along the outer face of the seven-stranded β-sheet. The major axes of helices 1, 2, and 5 are aligned in the same direction as the β-sheet. Helix 3 lies perpendicular to the β-sheet, and helix 4 is a two-residue 310 helix extending from helix 3.
Fig. 2.

The CDO monomer. (Upper) Ribbon illustration showing the CDO fold. The metal atom is shown as a gray sphere, and coordinated water molecules are shown as red spheres. The β-strands are labeled A–M, and the helices are labeled 1–5. Conserved residues are shown as color-coded sticks (blue, His; yellow, Arg; green, Cys; violet, Tyr; orange, Glu). (Lower) Surface illustration showing the entrance to the channel into the active site.
Fig. 2 shows the surface representation and location of a prominent tunnel into the active site, which lies within the β-barrel. The β-sheet made up of β-strands C, J, and K provides the top surface of this tunnel, which continues past the active site and eventually connects to the exterior on the opposite side.
Active Site.
Fig. 3 shows the active site and metal binding environment. The NE2 atoms of His-86, -88, and -140 coordinate the metal in a configuration reminiscent of that given by the 2-His, 1-carboxylate facial triad motif (15). These residues originate from β-strands H and C at the point where the two three-stranded β-sheets meet. His-140 is the only metal ligand that is stabilized by hydrogen-bonding to ND1, in this case, from Glu-104. The metal atom has three additional water or hydroxide ligands that complete a distorted octahedral coordination. There is some heterogeneity in the active site indicating that alternative coordination geometries may exist in the crystal. Figs. 3 and 4 show other residues that are conserved among CDOs. Among these residues are Tyr-58, which has weak π-orbital stacking interactions with Trp-77. The side-chain hydroxyl of Tyr-58 is pointed directly into the active-site cavity. Another conserved Tyr, Tyr-157, has the hydroxyl group located ≈4.2 Å from the metal center. This hydroxyl group is part of a hydrogen-bonding network that includes several other conserved amino acids that line the active site cavity, including His-155, Ser-153, and Trp-77. Specifically, the hydroxyl oxygen of Tyr-157 is 2.7 Å from the NE2 nitrogen of His-155; the ND1 nitrogen of His-155 is 2.7 Å from the side-chain hydroxyl oxygen of Ser-153; and this oxygen is 2.8 Å from the side-chain nitrogen of Trp-77. The residues Tyr-157 (conserved in all CDOs) and Cys-93 (conserved in CDO from eukaryotes only; see Fig. 4) appear to be covalently linked between Tyr-157 CE and Cys-93 SG, because these two atoms lie within 2.2 Å. Many bacterial CDOs have Gly in the position of Cys-93 and so could not have this posttranslational modification. Moreover, Cys-93 replaces the metal-coordinating glutamate found in the 2-His, 1-carboxylate sequence motif of many cupin superfamily members, but it is displaced 4.41 Å away from the metal center in CDO.
Fig. 3.

CDO active site contoured at 1.2σ. The metal is shown as a gray sphere; His-86, -88, and -140 are the metal ligands. Three additional coordination sites are occupied by water (red spheres). Cys-93 and Tyr-157 are covalently linked, and the hydroxyl group of Tyr-157 is 4.4 Å from the metal. Other conserved active-site residues are also shown.
Fig. 4.

Multiple sequence alignment of CDOs. Conserved residues involved in metal binding are highlighted in red; other conserved residues are highlighted in green.
Kinetic Analysis and Metal Incorporation.
An assay of the enzyme used to determine the crystal structure gave kcat of 1.8 min−1 and an apparent KM of 3.4 mM. Inductively coupled plasma-MS analysis of this preparation indicated that nickel, iron, and zinc were present, with ≈0.1 mol of iron present per mol of enzyme. When the enzyme was expressed in medium containing extra iron and purified in buffers lacking EDTA, a kcat of 3.6 min−1 and an apparent KM of 2.1 mM were determined. This latter enzyme preparation had ≈0.25 mol of iron per mol of enzyme.
Discussion
Cupin Superfamily Dioxygenases.
Structures of other cupin-fold dioxygenases are known. These structures include the iron-containing enzymes homogentisate dioxygenase from Homo sapiens (16), 3-hydroxyanthranilate-3,4-dioxygenase from Ralstonia metallidurans (17), quercetin dioxygenase from Bacillus subtilis (18), and the copper-containing quercetin dioxygenase from Aspergillus japonicus (19). The acireductone dioxygenases from Klebsiella pneumoniae and rat have been shown to bind either iron or nickel (20, 21).
The iron of homogenisate 1,2-dioxygenase (PDB ID code 1ey2) has a distorted square pyramidal coordination geometry including two His residues and a glutamate [i.e., the 2-His, 1-carboxylate facial triad (15)]. In the aligned active sites, the CDO residues His-86 and -140 approximate the positions of the His ligands of the facial triad, whereas His-88 approximates the position of the carboxylate ligand. The iron of 3-hydroxyanthranilate-3,4-dioxygenase (PDB ID code 1zvf) has octahedral coordination with two His residues and a bidentate glutamate (17). The iron or copper in the quercetin dioxygenases (PDB ID code 1juh) and the nickel in rat homolog of acireductone dioxygenase (PDB ID code 1vr3) are coordinated by three His residues and a Glu (18, 19, 22). Whereas His-86 aligns well with its structurally comparable His ligand in these other proteins, His-88 and -140 of CDO had adopted unique positions relative to their comparable ligands. Furthermore, the Glu ligand in these other proteins occupies the position of metal-bound water(s) in CDO. No other member of the cupin dioxygenase family contains a Tyr–Cys adduct like that found in CDO.
Enzyme Activity.
Recombinant CDO studied here converted Cys to sulfinic acid in aerobic buffer. The KM-value was comparable with previously determined values, whereas the protein prepared for enzymatic assays had a kcat value ≈10% of that determined for recombinant R. norvegicus CDO (11).
The available results indicate that catalytically active CDO contains iron. Recombinant forms of the enzyme apparently contain substoichiometric amounts of this metal (this work and ref. 11), and in the case of the mouse enzyme, possibly other divalent transition metals. In contrast, the enzyme originally purified from rat (23) had an equimolar stoichiometry between iron and protein. These results suggest that refinement of expression and purification methods beyond semiautomated approaches may lead to improved enzyme preparations.
Conserved Residues.
The conserved residues in CDO include Tyr-58, Arg-60, Trp-77, His-86, His-88, Gly-100, His-140, His-155, and Tyr-157. Tyr-58 is located at the active-site entrance. The hydroxyl group points into the active-site cavity and rests 7.5 Å from the metal center. The position of Tyr-58 is fixed through π-stacking interactions with Trp-77. Arg-60 is the only charged residue in the active site. His-86, -88, and -140 are necessary for coordination of the catalytic metal. The importance of Gly-100 is not immediately obvious, but it is located in a loop between strands D and E. Tyr-157 is involved in a posttranslational modification with Cys-93, and its hydroxyl group is within hydrogen-bonding distance of His-155 ND1. This unique environment should substantially lower the pKa of the Tyr-157 hydroxyl group, which is within 4.2 Å of the metal but clearly not coordinated to the metal center.
Comparison with Other Enzymes.
A Tyr-272–Cys-228 adduct was first observed in galactose oxidase (14). In this enzyme, the modified Tyr-272 is coordinated to the copper center, and the resting state is a spin-coupled pairing of a Tyr–Cys radical and Cu(II) (24). Because the reaction involves 2e− transfer, the participation of the radical cofactor allows reaction without formation of Cu(III).
Nitrile hydratase is a low-spin iron(III)-containing hydrolase that contains ligands provided by a combination of amino and thiol groups from three Cys residues. Two of the thiol groups have been posttranslationally modified to the sulfenic and sulfinic acid oxidation states, respectively (25). The enzyme does not require O2 activation for the catalyzed reaction, and the mechanism leading to these modifications is not known. However, the presence of the sulfinic acid modification suggests a possible coordination geometry for product in CDO.
CDO Mechanism.
Fig. 5 shows a plausible minimal reaction mechanism. We propose that this reaction may follow the paradigm for aromatic ring extradiol dioxygenases (26), including substrate coordination, binding and activation of O2, and then substrate oxidation. Owing to the ability of sulfur to access cation-radical intermediates (27, 28), this reaction may not require the assistance of an additional redox cofactor such as the Tyr-157–Cys-93 adduct.
Fig. 5.

Mechanism for CDO reaction. (A) Resting Fe(II) state. (B) Substrate coordination by sulfur and nitrogen. (C) O2 coordination, forming a ternary Fe(III)-superoxo complex. (D) The bound sulfur acquires partial cation-radical character, which can be stabilized by the adjacent negative charge on Tyr-157. (E) Combination of bound sulfur and Fe(III)-superoxo to give a cyclic peroxo intermediate. (F) O–O bond breakage to form a sulfoxy cation and metal-bound activated oxygen. (G) Transfer of the metal-bound activated oxygen to form product, cysteine sulfinic acid (CSA).
In this mechanism, the resting enzyme (Fig. 5A) is proposed to be Fe(II). The minimum distance between sulfur and nitrogen in substrate is ≈2.8 Å, suggesting a chelating binding mode is possible. Thus, Cys binding (Fig. 5B) may include coordination of either sulfur or O2 trans to His-140. His-140 is unique among the His ligands as ND1 is hydrogen-bonded to Glu-104, which may distinguish this axis from others in terms of bonding and electron transfer effects. The proposed binding orientation also would direct the substrate carboxyl group toward Tyr-58 and Arg-60. Substrate binding might require deprotonation of both the protonated amine cation to the neutral amine group as well as deprotonation of the thiol, possibly facilitated by the participation of His-155–Tyr-157–Cys-93 triad as a general base or bound water or hydroxide molecules. Alternatively, the enzyme may bind the deprotonated states of these functional groups (respective pKa values of ≈9) or the neutral thiol group (29) and thus not require general base catalysis for substrate binding. An alternative chelating mode where sulfur and a carboxylate oxygen from Cys bind the metal is also possible, but this assumption would direct the positively charged amino group into the active site toward Arg-60.
The region of the active site trans to His-86 is hydrophobic and thus represents a satisfactory environment for O2 binding before formation of the ternary complex. Fig. 5C shows that O2 binding to the substrate-activated Fe(II) site would produce a ternary complex including a transient Fe(III)-superoxo substructure analogous to that proposed for the extradiol dioxygenases and other 2-His,1-carboxylate proteins (26, 30). Thiolate Fe(II) complexes are reactive with O2, which could reasonably give rise to the intermediate shown in Fig. 5C (31). One consequence of formation of the Fe(III)-superoxo intermediate (Fig. 5D) would be to increase the cation-radical character of sulfur (27, 28), which would promote recombination with Fe(III)-superoxo to form a cyclic peroxo intermediate (Fig. 5E). The position of sulfur in the substrate complex also may allow the His-155–Tyr-157–Cys-93 triad to stabilize cationic intermediates generated during catalysis. Alternatively, nickel thiolate complexes were hypothesized to be oxidized by nucleophilic attack of the coordinated thiol on O2, leading to a thiadioxirane intermediate (32). However, related work with iron-thiolate complexes did not support formation of the thiadioxirane intermediate (33). In combination with evidence for the reaction of sulfur cation radicals with superoxide (34), we favor the recombination step shown from Fig. 5D to Fig. 5E.
The O–O bond breakage of the intermediate shown in Fig. 5E could proceed by either heterolysis or homolysis; the homolysis option would generate a metal-bound sulfoxy-cation species in close proximity to an activated oxygen atom (Fig. 5F). Recombination of the sulfoxy species and the activated oxygen atom would give the metal-bound product (Fig. 5G). Release of product from the active site and rebinding of water to the metal then would complete the proposed catalytic cycle.
Materials and Methods
Cloning and Expression.
The cdo gene was cloned into expression plasmid pVP16 to allow production of the enzyme in Escherichia coli B834 pRARE2 as an N-terminal fusion to a His-8-maltose binding protein (35). The selenomethionyl-labeled protein was expressed by autoinduction (36); unlabeled protein for enzyme assays was expressed in medium supplemented with 40 μM FeSO4.
Protein Purification.
Protein used for structure determination was purified by using semiautomated procedures (37). In summary, the fusion protein was purified by using immobilized metal affinity chromatography with Ni2+ as the metal, and tobacco etch virus protease was used to release the enzyme from the fusion. The liberated enzyme was separated from the His-8-maltose binding protein by immobilized metal affinity chromatography, further purified by gel filtration chromatography, and concentrated to 10.0 mg/ml in 5 mM 1,3-bis[tris(hydroxymethyl)methylamino]propane (pH 7.0), containing 50 mM NaCl, 0.3 mM Tris-carboxyethylphosphine, and 3.1 mM NaN3. EDTA was omitted from the buffers used to purify enzyme for catalytic studies, and the gel filtration step was not used.
Enzyme Assays.
Reactions containing 20 mM ammonium acetate (pH 7.5) and varying amounts of freshly prepared 100 mM l-cysteine (pH 7.5) were incubated at 37°C for 10 min before the addition of enzyme to a final concentration of 2.2 μM. Aliquots were quenched by flash freezing every 10 min over 60 min and prepared for HPLC by filtration through a Microcon (Millipore, Bedford, MA) 3000 MWCO filter. Cysteine sulfinic acid content was determined by ion-paired reverse-phase chromatography using UV detection at 215 nm in 20 mM sodium acetate (pH 5.0) prepared in 99.4:0.6 (vol/vol) water:methanol and 0.3% (wt/vol) heptafluorobutyric acid at a flow rate of 1 ml/min. Standards were used to confirm the retention times.
Other Methods.
Total metal analysis was by inductively coupled plasma-MS; iron content was determined by dye binding (38). Electrospray ionization (ESI) and MALDI MS performed at the University of Wisconsin–Madison Biotechnology Center were used to confirm the identity of the purified protein (predicted m/z 23,121.5 D; observed m/z 23,132 D by ESI and 22,919 D by MALDI).
Crystallization.
Crystals were grown by hanging-drop vapor-diffusion. The reservoir contained 20% methoxypolyethylene glycol 5000, 160 mM CaCl2, and 100 mM 2-morpholinoethanesulfonic acid (pH 6.5). Hanging drops consisted of 2 μl of protein solution mixed with 2 μl of reservoir solution. Diffraction-quality crystals grew within a week at 25°C. Protein crystals were soaked in reservoir solutions containing increasing amounts of ethylene glycol up to 20% (vol/vol) and were flash-cooled in a stream of cryogenic nitrogen.
Diffraction Analysis.
Diffraction data were collected at General Medicine and Cancer Institutes Collaborative Access Team (GM/CA-CAT) Sector 23 at Argonne National Laboratories at wavelengths of 0.97915, 0.97933, and 0.96392 Å at 100 K. The diffraction images were integrated and scaled by using hkl2000 (39). The selenium substructure of the crystal was determined by using hyss from phenix (40, 41), and the selenium positions were input into autosharp (42) to determine phases using multiwavelength anomalous diffraction. Auxiliary programs used by autosharp were from the ccp4 suite (43). Density modification was carried out with solomon (44). arp/warp was used to build the initial model (45). The model was completed with coot (46). The structure was refined with refmac (43, 47). Figures were made with pymol (DeLano Scientific, San Carlos, CA).
Acknowledgments
We thank Craig S. Newman, Zhaohui Sun, Russell L. Wrobel, Eric Steffan, Zachary Eggers, Megan Riters, Ronnie O. Frederick, John Kunert, Hassan Sreenath, Brendan T. Burns, Kory D. Seder, Holalkere V. Geetha, Frank C. Vojtik, Won Bae Jeon, Euiyoung Bae, Byung Woo Han, Jason M. Ellefson, Andrew C. Olson, Janet E. McCombs, Janelle T. Warick, Bryan Ramirez, Gary Wesenberg, Zsolt Zolnai, Peter T. Lee, Mike Runnels, John Cao, Jianhua Zhang, John G. Primm, Donna M. Troestler, Michael R. Sussman, John L. Markley, and other members of the Center for Eukaryotic Structural Genomics staff. We thank Ward Smith for assistance at General Medicine and Cancer Institutes Collaborative Access Team (GM/CA-CAT). This work was supported by National Institutes of Health Protein Structure Initiative Grants P50 GM 64598 and U54 GM 074901 (to John L. Markley, Principal Investigator and G.N.P. and B.G.F., Coinvestigators); National Institute of General Medical Sciences Grant GM-50853; National Science Foundation Grant MCB 0316232 (to B.G.F.); and National Library of Medicine Training Grant T15 LM007359 (to J.G.M). The Advanced Photon Source is supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science Contract W-31-109-ENG-38. GM/CA CAT is supported by National Cancer Institute Grant Y1-CO-1020 and National Institute of General Medical Science Grant Y1-GM-1104.
Abbreviations
- CDO
cysteine dioxygenase
- PDB
Protein Data Bank.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2atf).
References
- 1.Sorbo B., Ewetz L. Biochem. Biophys. Res. Commun. 1965;18:359–363. doi: 10.1016/0006-291x(65)90714-x. [DOI] [PubMed] [Google Scholar]
- 2.Heafield M. T., Fearn S., Steventon G. B., Waring R. H., Williams A. C., Sturman S. G. Neurosci. Lett. 1990;110:216–220. doi: 10.1016/0304-3940(90)90814-p. [DOI] [PubMed] [Google Scholar]
- 3.Gordon C., Bradley H., Waring R. H., Emery P. Lancet. 1992;339:25–26. doi: 10.1016/0140-6736(92)90144-r. [DOI] [PubMed] [Google Scholar]
- 4.Emery P., Bradley H., Arthur V., Tunn E., Waring R. Br. J. Rheumatol. 1992;31:449–451. doi: 10.1093/rheumatology/31.7.449. [DOI] [PubMed] [Google Scholar]
- 5.Perry T. L., Norman M. G., Yong V. W., Whiting S., Crichton J. U., Hansen S., Kish S. J. Ann. Neurol. 1985;18:482–489. doi: 10.1002/ana.410180411. [DOI] [PubMed] [Google Scholar]
- 6.Gough J., Karplus K., Hughey R., Chothia C. J. Mol. Biol. 2001;313:903–919. doi: 10.1006/jmbi.2001.5080. [DOI] [PubMed] [Google Scholar]
- 7.Dunwell J. M. Microb. Comp. Genomics. 1998;3:141–148. doi: 10.1089/omi.1.1998.3.141. [DOI] [PubMed] [Google Scholar]
- 8.Dunwell J. M., Purvis A., Khuri S. Phytochemistry. 2004;65:7–17. doi: 10.1016/j.phytochem.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 9.Woo E. J., Dunwell J. M., Goodenough P. W., Marvier A. C., Pickersgill R. W. Nat. Struct. Biol. 2000;7:1036–1040. doi: 10.1038/80954. [DOI] [PubMed] [Google Scholar]
- 10.Lombardini J. B., Singer T. P., Boyer P. D. J. Biol. Chem. 1969;244:1172–1175. [PubMed] [Google Scholar]
- 11.Chai S. C., Jerkins A. A., Banik J. J., Shalev I., Pinkham J. L., Uden P. C., Maroney M. J. J. Biol. Chem. 2005;280:9865–9869. doi: 10.1074/jbc.M413733200. [DOI] [PubMed] [Google Scholar]
- 12.Sakakibara S., Yamaguchi K., Ueda I., Sakamoto Y. Biochem. Biophys. Res. Commun. 1973;52:1093–1099. doi: 10.1016/0006-291x(73)91050-4. [DOI] [PubMed] [Google Scholar]
- 13.Sakakibara S., Yamaguchi K., Hosokawa Y., Kohashi N., Ueda I. Biochim. Biophys. Acta. 1976;422:273–279. doi: 10.1016/0005-2744(76)90138-8. [DOI] [PubMed] [Google Scholar]
- 14.Ito N., Phillips S. E., Stevens C., Ogel Z. B., McPherson M. J., Keen J. N., Yadav K. D., Knowles P. F. Nature. 1991;350:87–90. doi: 10.1038/350087a0. [DOI] [PubMed] [Google Scholar]
- 15.Koehntop K. D., Emerson J. P., Que L., Jr J. Biol. Inorg. Chem. 2005;10:87–93. doi: 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- 16.Titus G. P., Mueller H. A., Burgner J., Rodriguez De Cordoba S., Penalva M. A., Timm D. E. Nat. Struct. Biol. 2000;7:542–546. doi: 10.1038/76756. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y., Colabroy K. L., Begley T. P., Ealick S. E. Biochemistry. 2005;44:7632–7643. doi: 10.1021/bi047353l. [DOI] [PubMed] [Google Scholar]
- 18.Gopal B., Madan L. L., Betz S. F., Kossiakoff A. A. Biochemistry. 2005;44:193–201. doi: 10.1021/bi0484421. [DOI] [PubMed] [Google Scholar]
- 19.Fusetti F., Schroter K. H., Steiner R. A., van Noort P. I., Pijning T., Rozeboom H. J., Kalk K. H., Egmond M. R., Dijkstra B. W. Structure (London) 2002;10:259–268. doi: 10.1016/s0969-2126(02)00704-9. [DOI] [PubMed] [Google Scholar]
- 20.Dai Y., Wensink P. C., Abeles R. H. J. Biol. Chem. 1999;274:1193–1195. doi: 10.1074/jbc.274.3.1193. [DOI] [PubMed] [Google Scholar]
- 21.Pochapsky T. C., Pochapsky S. S., Ju T., Mo H., Al-Mjeni F., Maroney M. J. Nat. Struct. Biol. 2002;9:966–972. doi: 10.1038/nsb863. [DOI] [PubMed] [Google Scholar]
- 22.Steiner R. A., Kalk K. H., Dijkstra B. W. Proc. Natl. Acad. Sci. USA. 2002;99:16625–16630. doi: 10.1073/pnas.262506299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Griffith O. W. Methods Enzymol. 1987;143:366–376. doi: 10.1016/0076-6879(87)43065-6. [DOI] [PubMed] [Google Scholar]
- 24.Whittaker J. W. Arch. Biochem. Biophys. 2005;433:227–239. doi: 10.1016/j.abb.2004.08.034. [DOI] [PubMed] [Google Scholar]
- 25.Nagashima S., Nakasako M., Dohmae N., Tsujimura M., Takio K., Odaka M., Yohda M., Kamiya N., Endo I. Nat. Struct. Biol. 1998;5:347–351. doi: 10.1038/nsb0598-347. [DOI] [PubMed] [Google Scholar]
- 26.Que L., Jr., Ho R. Y. Chem. Rev. 1996;96:2607–2624. doi: 10.1021/cr960039f. [DOI] [PubMed] [Google Scholar]
- 27.Hong J., Schoneich C. Free Radical Biol. Med. 2001;31:1432–1441. doi: 10.1016/s0891-5849(01)00722-5. [DOI] [PubMed] [Google Scholar]
- 28.Montellano P. R. O. Annu. Rev. Pharmacol. Toxicol. 1992;32:89–107. doi: 10.1146/annurev.pa.32.040192.000513. [DOI] [PubMed] [Google Scholar]
- 29.Perera R., Sono M., Sigman J. A., Pfister T. D., Lu Y., Dawson J. H. Proc. Natl. Acad. Sci. USA. 2003;100:3641–3646. doi: 10.1073/pnas.0737142100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bassan A., Borowski T., Siegbahn P. E. Dalton Trans. 2004:3153–3162. doi: 10.1039/B408340G. [DOI] [PubMed] [Google Scholar]
- 31.Theisen R. M., Shearer J., Kaminsky W., Kovacs J. A. Inorg. Chem. 2004;43:7682–7690. doi: 10.1021/ic0491884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mirza S., Pressler M., Kumar M., Day R., Maroney M. J. Inorg. Chem. 1993;32:977–987. [Google Scholar]
- 33.Musie G., Lai C. H., Reibenspies J. H., Sumner L. W., Darensbourg M. Y. Inorg. Chem. 1998;37:4086–4093. doi: 10.1021/ic980475f. [DOI] [PubMed] [Google Scholar]
- 34.Miller B. L., Williams T. D., Schoneich C. J. Am. Chem. Soc. 1996;118:11014–11025. [Google Scholar]
- 35.Thao S., Zhao Q., Kimball T., Steffen E., Blommel P. G., Riters M., Newman C. S., Fox B. G., Wrobel R. L. J. Struct. Funct. Genomics. 2004;5:267–276. doi: 10.1007/s10969-004-7148-4. [DOI] [PubMed] [Google Scholar]
- 36.Sreenath H. K., Bingman C. A., Buchan B. W., Seder K. D., Burns B. T., Geetha H. V., Jeon W. B., Vojtik F. C., Aceti D. J., Frederick R. O., et al. Protein Expression Purif. 2005;40:256–267. doi: 10.1016/j.pep.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 37.Jeon W. B., Aceti D. J., Bingman C. A., Vojtik F. C., Olson A. C., Ellefson J. M., McCombs J. E., Sreenath H. K., Blommel P. G., Seder K. D., et al. J. Struct. Funct. Genomics. 2005;6:143–147. doi: 10.1007/s10969-005-1908-7. [DOI] [PubMed] [Google Scholar]
- 38.Fischer D. S., Price D. C. Clin. Chem. 1964;10:21–30. [PubMed] [Google Scholar]
- 39.Otwinowski Z., Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 40.Weeks C. M., Adams P. D., Berendzen J., Brunger A. T., Dodson E. J., Grosse-Kunstleve R. W., Schneider T. R., Sheldrick G. M., Terwilliger T. C., Turkenburg M. G., Uson I. Methods Enzymol. 2003;374:37–83. doi: 10.1016/S0076-6879(03)74003-8. [DOI] [PubMed] [Google Scholar]
- 41.Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. Acta. Crystallogr. D. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 42.Bricogne G., Vonrhein C., Flensburg C., Schiltz M., Paciorek W. Acta. Crystallogr. D. 2003;59:2023–2030. doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- 43.Collaborative Computational Project, No. 4. Acta. Crystallogr. D. 1994;50:760–763. [Google Scholar]
- 44.Abrahams J. P., Leslie A. G. Acta. Crystallogr. D. 1996;52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- 45.Blanc E., Roversi P., Vonrhein C., Flensburg C., Lea S. M., Bricogne G. Acta Crystallogr. D. 2004;60:2210–2221. doi: 10.1107/S0907444904016427. [DOI] [PubMed] [Google Scholar]
- 46.Emsley P., Cowtan K. Acta. Crystallogr. D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 47.Murshudov G. N., Vagin A. A., Dodson E. J. Acta. Crystallogr. D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]