

Abstract
Mycolic acids and multimethyl-branched fatty acids are found uniquely in the cell envelope of pathogenic mycobacteria. These unusually long fatty acids are essential for the survival, virulence, and antibiotic resistance of Mycobacterium tuberculosis. Acyl-CoA carboxylases (ACCases) commit acyl-CoAs to the biosynthesis of these unique fatty acids. Unlike other organisms such as Escherichia coli or humans that have only one or two ACCases, M. tuberculosis contains six ACCase carboxyltransferase domains, AccD1–6, whose specific roles in the pathogen are not well defined. Previous studies indicate that AccD4, AccD5, and AccD6 are important for cell envelope lipid biosynthesis and that its disruption leads to pathogen death. We have determined the 2.9-Å crystal structure of AccD5, whose sequence, structure, and active site are highly conserved with respect to the carboxyltransferase domain of the Streptomyces coelicolor propionyl-CoA carboxylase. Contrary to the previous proposal that AccD4–5 accept long-chain acyl-CoAs as their substrates, both crystal structure and kinetic assay indicate that AccD5 prefers propionyl-CoA as its substrate and produces methylmalonyl-CoA, the substrate for the biosyntheses of multimethyl-branched fatty acids such as mycocerosic, phthioceranic, hydroxyphthioceranic, mycosanoic, and mycolipenic acids. Extensive in silico screening of National Cancer Institute compounds and the University of California, Irvine, ChemDB database resulted in the identification of one inhibitor with a Ki of 13.1 μM. Our results pave the way toward understanding the biological roles of key ACCases that commit acyl-CoAs to the biosynthesis of cell envelope fatty acids, in addition to providing a target for structure-based development of antituberculosis therapeutics.
Keywords: cell wall lipid, multimethyl-branched fatty acid, mycolic acid, tuberculosis, mycocerosic acid
Mycobacterium tuberculosis causes more human deaths than any other single infectious organism, with an estimated eight million new tuberculosis cases and two million fatalities each year (1, 2). Tuberculosis has two features that render it the deadliest infectious disease to date: its high infectivity (virulence) and its ability to enter latency for subsequent reactivation, a phenomenon that leads to a deadly synergy with AIDS (3, 4). As a result, tuberculosis is also the current leading cause of death for AIDS patients. Given the current backdrop of emerging multidrug-resistant tuberculosis (MDR-TB) (5), tuberculosis treatment is entering a challenging era, where effective control requires the identification of new drugs and novel drug targets (World Health Organization, www.who.int).
The cell envelope of M. tuberculosis contains one of the most sophisticated and exquisite composition of biologically active lipids. Several components are essential for both mycobacterial viability and pathogenicity, and also play an active role in modulating the host immune response (6–8). This complex structure, which is a major contributor to the intrinsic resistance of M. tuberculosis to most commonly used antibiotics, also represents one of the most successful targets of antimycobacterial chemotherapy. For instance, one of the front-line antimycobacterial drugs, isoniazid, targets the biosynthesis of mycolic acid, a major cell wall component that is unique to mycobacteria (9). With the reemergence of tuberculosis infections caused by MDR-TB (9), including resistance to isoniazid, other steps on the pathways involved in the biosynthesis of these unique cell wall lipids should be explored as possible targets for new antimycobacterial drugs.
The most relevant lipids that form part of this cell envelope are the mycolic acids, the long-chain α-alkyl, β-hydroxy fatty acids, and the characteristic methyl-branched long-chain acids and alcohols, such as the mycocerosic and mycolipenic acids and the phthiocerols (Fig. 1A) (9, 10). Many of the genes for fatty acid synthases and polyketide synthases responsible for the biosyntheses of mycolic acid and multimethyl-branched fatty acids have been cloned and characterized (9–14). However, one question remains: What are the enzyme mechanisms that provide the building blocks for these structurally unique fatty acids?
Fig. 1.

Chemical structures of cell wall fatty acids and ACCase mechanism. (A) The chemical structures of mycolic acids and multimethyl-branched fatty acids such as mycocerosic, phthioceranic, hydroxyphthioceranic, mycosanoic, and mycolipenic acids, which form liposugars phthiocerol dimycocerosate (PDIM), sulfated tetraacyl trehalose (SL1), diacyl trehaloses (DAT1), triacyl trehalose (TAT), and pentaacyl trehalose (PAT), respectively. A proposed cell envelope architecture is shown, adopted from ref. 10. (B) Acyl-CoA carboxylase (ACCase) provides the extender units for the biosyntheses of cell envelope fatty acids. The α [biotin carboxylase (BC) and biotin carboxylate carrier protein (BCCP)] and β [carboxyltransferase (CT)] subunits catalyze the first and second steps, respectively. The β subunit is the key domain that determines the ACCase substrate specificity. In M. tuberculosis, there are six β subunits, AccD1–6, whose biological roles are not well defined.
The first committed step in the biosynthesis of long-chain fatty acids in all animals, plants, and bacteria is catalyzed by acetyl-CoA carboxylase (ACC) (15). This ubiquitous enzyme catalyzes the α-carboxylation of acetyl-CoA to produce malonyl-CoA, which serves as the building blocks for downstream fatty acid biosynthesis (Fig. 1B). As a crucial metabolic enzyme, ACC is also the key regulation point for fatty acid biosynthesis. In actinomycetes, these enzymes usually have broader substrate specificity (15–18), which accounts for their designation as acyl-CoA carboxylases (ACCases) and, in mycobacteria, they are thought to provide the elongation units for the biosynthesis of n-saturated and branched fatty acids and for one of the precursors involved in mycolic acid biosynthesis (19). During the past three decades, much work has been devoted to the regulation, genetics, and biochemistry of ACCases (15–18). As a result of the vigorous research progress, the importance and validity of ACCase for drug discovery is recognized in both eukaryotic and bacterial organisms (15–18).
The ACCase enzymatic reactions proceed stepwise. In M. tuberculosis and Streptomyces coelicolor, ACCase consists of three subunits, the α, β, and ε subunits (20–22). The α subunit is a di-domain protein that consists of biotin carboxylase (BC) and biotin carboxylate carrier protein (BCCP). During the first enzymatic step, BC couples carbonate with biotin to form carboxybiotin, which is attached to BCCP by a lysine residue that alternatively reaches the active sites of the α and β subunits (Fig. 1B). Subsequently, the β subunit (carboxyltransferase, CT) transfers the carboxyl group from biotin to acyl-CoA. The ε subunit, a unique feature of actinomycetes ACCases, is required for the holo complex activity (α–β–ε) (20–22). Because acyl-CoA participates only in the second step, the β subunit has been proposed to control the ACCase substrate specificity in recognizing different acyl-CoAs. This hypothesis has been confirmed recently by our laboratory on the basis of the crystal structure of the S. coelicolor ACCase β subunit (PccB, propionyl-CoA carboxylase β subunit) and relevant mutational studies (22, 23).
Significantly, sequence analysis of the M. tuberculosis genome reveals genes encoding six ACCase β subunits (accD1–6) (19). This abundant number of β subunits is highly unusual, compared with other organisms, which generally have only one or two ACCases (15). Presumably, each β subunit in M. tuberculosis serves a different biological role and provides different extender units for the biosyntheses of different polyketides and fatty acids. Despite the obvious importance of these ACCases in providing building blocks for fatty acid and polyketide biosyntheses, very little is known about the biochemistry and individual physiological roles of AccD1–6. Until recently, the only tuberculosis ACCase characterized was isolated from crude extracts, and it showed, at least in vitro, higher affinity for propionyl-CoA compared with acetyl- or butyryl-CoA (24). Furthermore, the α (AccA3) subunit has been copurified with AccD4 and AccD5, and this tri-domain complex has been hypothesized to provide the extender unit required for mycolic acid biosynthesis (25). In a related Corynebacterium study, long-chain fatty acyl-CoAs (ranging from C16 to C26) have been proposed as the ACCase substrates during mycolic acid biosyntheses (26). These previous results indicate that AccD4, AccD5, and AccD6 are the most likely candidates to provide extender units for cell envelope lipids (9). However, because of the absence of molecular information, the individual substrate specificities of AccD1–6 are not well defined.
Recently, we have successfully reconstituted one ACCase complex from the biotinylated α subunit AccA3, the carboxyltransferase β subunit AccD5, and the ε subunit AccE5 (Rv3281) of M. tuberculosis (20). Kinetics of the holo complex (called AceCC5) indicates that AceCC5 accepts acetyl- and propionyl-CoAs as its substrates, with a 5-fold preference for propionyl-CoA. This finding suggests that the main physiological role of this enzyme may be to generate methylmalonyl-CoA for the biosynthesis of multimethyl-branched fatty acids (examples shown in Fig. 1A) (20). Herein, we report the crystal structure and kinetic analysis of the M. tuberculosis ACCase β subunit. Our structural and functional analyses elucidate the unique substrate specificity of AccD5 and its possible biological role. We also present in silico screening work that leads to the identification of AccD5 inhibitors, which may serve as drug leads for the development of new tuberculosis therapeutics.
Results and Discussion
Overall Structure.
The AccD5 crystal structure indicates that, unlike the dimeric yeast carboxyltransferase (yeast CT) (16, 27), AccD5 is a 360-kDa hexamer (Fig. 2A), similar to PccB (ACCase CT domain) from S. coelicolor and the 12S domain (a methylmalonyl-CoA transcarboxylase) from Propionibacterium shermanii (23, 28). AccD5 has a sequence identity of 67% and 50% with PccB and 12S domain, respectively. Each monomer of AccD5 consists of two domains (N and C domains), and both domains have a crotonase fold consisting of seven β-strands and α-helices (Fig. 6, which is published as supporting information on the PNAS web site) (29). The ring-shaped hexamer forms two stacks of tightly interacting trimer rings, A–B–C and D–E–F. The six active sites are located at the interfaces of dimer pairs A–D, B–E, and C–F (Fig. 2B), whose interactions are highly conserved between AccB, PccB, AccD4, AccD5, and AccD6 (black ellipses in Fig. 7, which is published as supporting information on the PNAS web site). Although the eukaryotic yeast CT has only 11% sequence identity with AccD5, the structural features at the dimer interface are still highly conserved, demonstrating the importance of dimeric di-domain interactions for the protein stability of CT domains. In addition, the intratrimer interactions A–B–C or D–E–F, important for hexamer formation, are highly conserved between PccB and AccD5, but not conserved between AccD4 and AccD6 (Fig. 2) (23, 28). Indeed, size-exclusion chromatography and native gels showed that AccD5 is a hexamer, whereas AccD4 and AccD6 have different oligomeric states (data not shown). Therefore, the oligomeric architecture of AccD4 and AccD6 may be different from the hexameric ring structures of PccB, 12S, and AccD5 (23, 28).
Fig. 2.
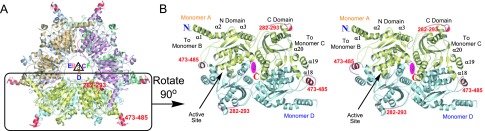
The AccD5 crystal structure. (A) Overall structure of AccD5 as two stacks of trimers. Only minor differences are observed among the six monomers, with an rms deviation of <0.4 Å. The threefold axis of monomers A–B–C and D–E–F is indicated. The loops in red are regions that are different between AccD5 and PccB. The N-domain helices (α1, α2, and α3) of monomer A, B, or C interact extensively with the C-domain helices (α18, α19, and α20) of monomer D, E, or F. (B) Stereoview of the dimeric, di-domain interactions, shown between monomers A (in yellow) and D (in blue) are important for protein stability, enzyme catalysis, and substrate specificity.
Two Different Loops Between PccB and AccD5.
The major differences between PccB and AccD5 lie in two surface loops between residues 282–293 and 473–485 (Fig. 2). These two regions are also the least conserved (Fig. 7). Residues 282–293 fortify A–B–C interaction, but do not change the overall fold or active site geometry of AccD5. The most distinct difference between AccD5 and PccB lies in the 473–485 loop between α17 and α18 that defines the propionyl-CoA pocket entrance, which is important for binding the coenzyme bisphosphate group (23, 28). However, except for the phosphate-binding residues, the CoA-binding motif remains conserved. Because of the importance of the dimeric di-domain interactions for protein stability, enzyme mechanism, and substrate specificity of the CT domains (23, 28), the near-identical overlap between PccB and AccD5 indicates that these important molecular properties should be highly conserved between AccD5 and PccB, which accepts propionyl- and butyryl-CoA as its substrates (22).
The AccD5 Active Site.
PccB is homologous to AccD4, AccD5, and AccD6 (Fig. 7) with sequence identities of 27%, 67%, and 25%, respectively. The key residues of the PccB mechanism involve two pairs of oxyanion-stabilizing residues: Gly-419 and Ala-420 hydrogen-bond with the carbonyl group of biotin, whereas Gly-182 and Gly-183 hydrogen-bond with the carbonyl group of propionyl-CoA (23). These four residues are also conserved in both AccD4 (Gly-410, Ala-411, Gly-182, Gly-183), AccD5 (Gly-434, Ala-435, Gly-193, Gly-194) and AccD6 (Gly-356, Ala-357, Gly-147, Gly-148). Indeed, these four residues are highly conserved in positions when the structures of PccB and AccD5 are overlaid (Fig. 3A), suggesting that the catalytic mechanism is conserved among PccB, AccD4, AccD5, and AccD6 (Fig. 7) (23).
Fig. 3.

The AccD5 active site. (A) Structural overlap between AccD5 and PccB near the active site. Two sets of oxyanion-stabilizing residues are highly conserved, including the NH of G193 and G194 and the NH of G434′ and A435′. The structural similarity strongly suggests a similar enzyme mechanism. (B) Although surfaces outside of the pocket are quite different, the size and shape of the acyl-CoA-binding pockets themselves are very similar between PccB and AccD5, whose pockets are too small to accommodate the 16-carbon palmitoyl-CoA.
Substrate Specificity.
The substrate specificity of AccD1–6 has been an unsolved mystery, whose solutions require detailed analyses of these CT domains. The structural similarity between PccB and AccD5 offers an excellent opportunity to analyze AccD5 in molecular details. Similar to PccB, the tentative biotin and acyl-CoA-binding pockets of AccD5 are located at the conserved dimeric di-domain interface (Figs. 2B and 3). These two pockets are identified by overlapping the crystal structures of apo-AccD5 and substrate-bound PccB. Significantly, all of the biotin-binding residues are conserved (Fig. 7, green triangles) Therefore, the biotin-binding motif is highly conserved among CTs such as 12S, PccB, AccB, and AccD4–6. Further, residues in the acyl-CoA-binding pocket are also highly conserved surrounding the acyl, pantetheine, ribose, and adenine ring, whereas residues that interact with the bisphosphate group such as Q472, R471, and F468 are partially conserved among AccD4–6 and PccB. The high sequence homology strongly suggests that the acyl-CoA-binding pocket size and shape are similar among these four enzymes.
On the basis of studies of Corynebacterium glutamicum, a close relative of M. tuberculosis, the ACCase substrates for mycolic acid biosynthesis in M. tuberculosis have been suggested to be C16–C26 acyl-CoAs or acyl carrier proteins (ACPs) (25, 26). It was further proposed that both AccD4 and AccD5 are involved in mycolic acid biosynthesis (25, 26). On the other hand, the substrates for mycocerosic acid biosynthesis have been proven to be propionyl-CoA, which can be condensed to produce the multimethyl-branched structure (Fig. 1A) (10, 24, 30). To distinguish the biological roles of AccD4, AccD5, and AccD6, it is important to determine the substrate specificities of these CT domains. Previously, we identified residue 422 of PccB (corresponding to C437 of AccD5) as an important residue for the substrate specificity of CTs (23): a smaller residue at this position (such as Cys, Asp, or Ala) corresponds to a higher specificity for propionyl-CoA (versus acetyl-CoA), whereas a larger residue (such as Leu or Ile) corresponds to a higher specificity for acetyl-CoA (versus propionyl-CoA) (23). In AccD4, AccD5, and AccD6, this residue is Ala, Cys-437, and Ile, respectively (Fig. 7). Further, the crystal structures of AccD5 and PccB reveal that, although surfaces outside of the AccD5 and PccB substrate pockets are different, the size and shape of the substrate-binding pockets themselves are highly similar between AccD5 and PccB (Fig. 8, which is published as supporting information on the PNAS web site). The acyl-CoA-binding pockets for both AccD5 and PccB have no space to accommodate a long acyl group (such as the 16-carbon palmitoyl-CoA or -ACP, Fig. 3B). In fact, docking simulations of palmitoyl-CoA or palmitoyl-ACP indicate that it is physically impossible to accommodate such a long-chain acyl group in the substrate-binding pocket. Therefore, the structures strongly suggest that, at least for AccD5 (if not for AccD4 and AccD6 as well), the substrate of AccD5 should be a short-chain acyl-CoA such as acetyl- or propionyl-CoA, as opposed to long-chain acyl-CoAs, such as palmitoyl-CoA proposed to be the substrate for AccD4 and AccD5 (25, 26). The structural analysis of the AccD5 active site is consistent with the kinetic assay, which indicates that holo AceCC5 (AccA3–AccD5–AccE5) accepts acetyl- and propionyl-CoAs as its substrate (both with Km values of 220–240 μM), with a 5-fold preference for propionyl-CoA (kcat/Km is 258 and 50 M−1·min−1 for propionyl- and acetyl-CoA, respectively) (20). However, AceCC5 has no detectable activity for palmitoyl-CoA or palmitoyl-ACP (Table 1, which is published as supporting information on the PNAS web site) (20). In previous studies, we have shown that the acyl-CoA specificity is primarily determined by the CT domain (23), namely AccD5 in this case. Therefore, on the basis of structural and kinetic analysis, we conclude that AccD5 is a CT domain that has a high preference to carboxylate propionyl-CoA and produces methylmalonyl-CoA, the basic building block for multimethyl-branched fatty acids such as mycocerosic, phthioceranic, hydroxyphthioceranic, mycosanoic, and mycolipenic acids (Fig. 1A) (10, 14, 30).
Structure-Based Drug Design.
Inhibitors that target cell envelope lipids have already been proven to be frontline tuberculosis therapeutics, such as isoniazid (31, 32). The unusual cell envelope of M. tuberculosis contains many multimethyl-branched fatty acids that are important for antibiotic resistance, pathogen survival, and virulence (10, 14, 30). Therefore, inhibitors aimed at the biosyntheses of these multimethyl-branched fatty acids have high potential to become new antituberculosis therapeutics. AccD5 has been strongly implicated as one of the essential ACCases important for cell envelope lipid biosynthesis (25, 26). In this work, we have shown that AccD5 produces methylmalonyl-CoA that is essential for the biosyntheses of multimethyl-branched fatty acids. Further, a comparison of the AccD5 and yeast CT structures indicates that the substrate-binding pocket is very different between the bacterial and eukaryotic CT domains (27). Consistent with this observation, we found that the activity of AccD5 cannot be inhibited by diclofop and haloxyfop, two inhibitors that specifically inhibit the eukaryotic ACCase CT domains (27). Our result indicates the potential for AccD5 inhibitors that specifically inhibit the pathogen ACCase, but not the human enzyme.
To identify inhibitors of AccD5, we have used dock and icm-pro (33, 34) for extensive in silico screening applied to compounds from the National Cancer Institute (NCI) diversity set (1,990 compounds) and the University of California, Irvine, ChemDB database (35), which contains more than four million compounds compiled from commercial vendors and other publicly available sources. The search was restricted to the binding pockets of biotin and propionyl-CoA. Results were scored by ligand-AccD5 binding energy to produce a ranked list of small molecules. To increase the consistency of the docking simulations, multiple (two to five) rounds of in silico screening were performed against each small molecule library, and only the ligands that were repetitively predicted to be the tight-binders by both icm and dock were selected for further experimental screening. The top nine predicted tight-binders are shown in Fig. 4A. Among these nine candidates, only one compound inhibits both the holo complex (AccA3–AccD5–AccE5) as well as the single AccD5 domain (NCI-65828, Fig. 4A), with an IC50 of 10 μM. The holo complex and AccD5 inhibitions were assayed by the pyruvate kinase coupling method (22) and malate dehydrogenase–citrate synthase (MD-CS) assay, respectively. Using the ChemDB similarity search algorithm (35), we found 3,000 chemical analogs in ChemDB that have chemical structures similar to that of NCI-65828, and we conducted the second round of in silico screening. As in the initial screening, only the top 10 ligands that were consistently predicted to be the tight-binders by both icm and dock were selected for further experimental screening. We found that the IC50 values of these chemical analogs of NCI-65828 range from 25 μM to no inhibition (Fig. 4B). We conclude from the two rounds of drug screening effort that (i) the naphthalene ring, two phenyl rings, diazo groups, and sulfate are important for AccD5 binding (Fig. 5A) and (ii) different aromatic substituents on the naphthalene ring result in a large change of IC50 (Fig. 4B). Therefore, subsequent modification of the naphthalene ring substituents should further improve the IC50 of NCI-65828 analogs.
Fig. 4.

The in silico inhibitor leads of AccD5. (A) The lead compounds from the first round of in silico inhibitor screening against the AccD5 active site, in which only NCI-65828 showed extensive enzyme inhibition. (B) The lead compounds from the second round of in silico screening of 3,000 chemical homologs that resemble NCI-65828. The IC50 values of these analogs range from 25 to 300 μM, with >50% lacking inhibitory effect on AccD5.
Fig. 5.

After extensive in silico screening, the ligand NCI-65828 was found to inhibit AccD5 competitively with an experimental Ki of 13.1 μM. (A) Docking of NCI-65828 in the acyl-CoA-binding pocket of AccD5 matches the binding motif of an acyl-CoA, in which the anionic sulfate of NCI-65828 binds the entrance of the CoA pocket and the hydrophobic moiety binds the hydrophobic interior of the CoA pocket. (B) The Lineweaver–Burk plot shows that NCI-65828 is a competitive inhibitor of AccD5.
To further characterize the lead compound, NCI-65828, we conducted a detailed inhibition study of NCI-65828 against AccD5 at different concentrations of the substrate, malonyl-CoA, and the inhibitor. We found that in the absence of an inhibitor, malonyl-CoA binds AccD5 with a Km of 250 μM, similar to the Km of acetyl-CoA or propionyl-CoA from the forward direction (Km = 220 and 240 μM, respectively). The double-reciprocal plot (1/v versus 1/[malonyl-CoA] clearly indicates that NCI-65828 inhibits AccD5 competitively against malonyl-CoA, with a Ki of 13.1 μM (Fig. 5B). Therefore, using extensive in silico effort, we have identified an inhibitor (NCI-65828) that binds AccD5 with an affinity 20-fold higher than its substrates, acetyl- or propionyl-CoA.
Biological Significance.
In summary, two important outcomes are revealed from the above results:
(i) The biological role of AccD5.
Because of the importance of cell envelope lipids to the pathogenesis of M. tuberculosis, 10% of its genome is devoted to fatty acid biosynthesis (10). Because ACCase is the committing enzyme of fatty acid biosynthesis, the pathogen has an unusually high number of ACCase CT domains, AccD1–6 (six, compared with one or two ACCases in humans and Escherichia coli). Despite their importance to pathogenesis, the precise biological roles and substrate specificities of AccD1–6 are not well understood. Here, we have presented the crystal structure and inhibition kinetics of an M. tuberculosis ACCase CT domain, AccD5. Although AccD5 was proposed to form a complex with AccD4 and accept palmitoyl-CoA or palmitoyl-ACP as its substrates (25, 26), both structural and functional work presented herein has shown that AccD5 is structurally and functionally similar to PccB of S. coelicolor, and it accepts propionyl-CoA as its primary substrate to produce methylmalonyl-CoA, the building block for multimethyl-branched fatty acids of the cell envelope, such as mycocerosic, phthioceranic, hydroxyphthioceranic, mycosanoic, and mycolipenic acids that form phthiocerol dimycocerosate (PDIM), sulfated tetraacyl trehalose (SL1), diacyl trehaloses (DAT1), triacyl trehalose (TAT), and pentaacyl trehalose (PAT), respectively (Fig. 1A) (10). PDIM, SL1, DAT1, TAT, and PAT have been shown to be important for pathogen survival and invasion (10).
(ii) AccD5 as a structure-based drug design target.
For decades, ACCases have been the targets of many herbicides (36). Recently, with the increasing appreciation of its importance in metabolic regulation and the availability of ACCase sequences from different genomes, ACCases have become intensely pursued targets for infectious disease, cancer, and obesity therapeutics (16, 17). The importance of ACCases as drug targets for infectious disease has been validated by bacterial genetic and physiological experiments, which found that ACCase is used almost exclusively as the downstream extender unit provider for fatty acid biosynthesis (37, 38). Here, we have presented the structure-based in silico drug design of AccD5, the CT domain of an M. tuberculosis ACCase. Using the four-million compound ChemDB database developed at the University of California, Irvine, extensive in silico docking followed by enzyme inhibition studies have identified a series of AccD5 inhibitors (Fig. 4), the best of which has a Ki of 13.1 μM and binds AccD5 competitively with 20-fold higher affinity than its substrates (Fig. 5B). In conclusion, the above results help define the biological role of AccD5 and pave the way for the application of ACCase as a potential target for tuberculosis therapeutics.
Materials and Methods
Protein Expression and Purification.
Recombinant AccD5 was expressed in E. coli. Cultures were grown in LB broth with 50 μg·ml−1 kanamycin. At OD600 = 0.6–0.8, the protein expression was induced by 0.1 mM isopropyl β-d-thiogalactoside (IPTG) at 28°C overnight. The cells were harvested by centrifugation (5,000 × g for 15 min), followed by sonication to lyse the cells and centrifugation to remove cell debris (21,000 × g for 45 min). The lysis buffer was 50 mM Tris·HCl/300 mM NaCl/10% glycerol/10 mM imidazole at pH 8.0. The lysate was passed through 10 ml of Qiagen Ni-NTA His-bind Resin, washed twice with 30 ml of 10 mM imidazole and 20 mM imidazole, then eluted with 30 ml each of 40, 60, 80, 100, and 500 mM imidazole. The protein was eluted at 60–100 mM imidazole fractions, buffer-exchanged to 10 mM Hepes, pH 7.0/2 mM DTT by dialysis, and concentrated by Centricon YM-10 to 5 mg/ml.
Coupled Enzyme Assay.
The enzyme assay of holo-AceCC5 complex (AccA3–AccD5–AccE5) follows the rate of ATP hydrolysis by biotin carboxylase spectrophotometrically (22). The production of ADP was coupled to pyruvate kinase and lactate dehydrogenase, and the oxidation of NADH was followed at 340 nm. For the AccD5 inhibition assay, to directly observe the carboxyltransferase reaction, we followed the rate of acetyl-CoA formation from the reverse direction (malonyl-CoA turning into acetyl-CoA) by using the malate dehydrogenase/citrate synthase (MD-CS) assay based on a previously reported protocol that detects the formation of NADH at 340 nm (27). The IC50 of a given inhibitor was determined with this assay at 5–300 μM inhibitor concentrations in the presence of Km concentration (200 μM) of malonyl-CoA, saturation concentration (10 mM) of biocytin, and 10 μM AccD5 in 100 mM Tris (pH 7.0) buffer.
Protein Crystallization.
Crystals of AccD5 were grown in sitting-drop trays at room temperature by vapor diffusion. The protein buffer was 10 mM Hepes, pH 7.0/2 mM DTT. Drops were generated by mixing 1 μl of the purified protein solution with 3 μl of well buffer above 500 μl of the well solution. Two crystallization conditions were found that gave crystals with the same space group and cell dimensions: (i) 1.5 M ammonium sulfate at pH 6.5 (0.1 M Bis-Tris) with 0.1 M sodium chloride, and (ii) 15% polyethylene glycol 4000 at pH 5.6 (0.1 M sodium citrate) with 0.2 M ammonium acetate. The crystals were fragile and readily dissolved in different cryoprotectants at room temperature. Subsequently, the AccD5 crystals were transferred to the 4°C cold room for 24 h, and flash-frozen in liquid nitrogen therein, using 2 M lithium sulfate as the cryoprotectant.
Data Collection.
On the average, one dataset could be collected for every 100 AccD5 crystals screened. Three x-ray diffraction datasets of AccD5 crystals were collected at the Stanford Synchrotron Radiation Laboratory (SSRL) and Advanced Light Source (ALS) to a resolution of 2.9 Å. Diffraction intensities from the three datasets were integrated and reduced by using the program hkl2000 and scaled by using scalepack (Table 2, which is published as supporting information on the PNAS web site) (39).
Molecular Replacement and Refinement.
The initial phase of apo-AccD5 was determined by molecular replacement, using the crystal structure of PccB, and the program cns (40). After rebuilding the structure by using quanta, further refinement was performed by using cns (40). A preliminary round of refinement using torsion angle simulated annealing, followed by energy minimization, positional and individual B-factor refinement reduced Rcrys to 29%. Subsequent rounds of model building and refinement were carried out by using the maximum likelihood approach implemented within cns to an Rcrys of <20% (Rfree < 25%). The water molecules were then added and edited both visually and with an automated water picking program (cns) (Table 2) (40).
Docking Simulation.
icm from Molsoft (San Diego) and dock from the University of California San Francisco (33) were used for all computer simulations and compared to identify AccD5 inhibitors. For icm, the AccD5 Protein Data Bank file was converted to icm objects. Small-molecule ligands were read in SDF format from databases or were created in the icm molecular editor. The AccD5 docking site was identified from the structure overlap between AccD5 and substrate-bound PccB. Compounds from the University of California, Irvine, ChemDB database (35) and the full NCI diversity set (http://dtp.nci.nih.gov/) were docked at thoroughness level 1 or 10 on a Linux server. The in-house dock program was modified to run robustly on a large Linux cluster. Results were sorted by binding energy to produce a prioritized list of small molecules.
Supplementary Material
Acknowledgments
We thank Peter Smith for proofreading the manuscript. Part of this work was supported by Fogarty International Research Collaboration Award R03 TW005778 from the National Institutes of Health (to H.G.), the Agencia Nacional de Promoción Científica y Tecnológica Grant 01-06622 (to H.G.), National Institutes of Health Biomedical Informatics Training Grant LM-07443-01 (to P.B.), and National Science Foundation Major Research Instrumentation Program Grant EIA-0321390 (to P.B.), an International Society for Infectious Diseases (ISID) grant (to G.G.), and a University of California, Irvine, Council on Research, Computing and Library Resources grant (to S.-C.T.). We thank the NCI for providing the compounds used in this study.
Abbreviations
- ACCase
acyl-CoA carboxylase
- ACP
acyl carrier protein
- CT
carboxyltransferase
- NCI
National Cancer Institute
- PccB
propionyl-CoA carboxylase β subunit.
Footnotes
Conflict of interest statement: No conflicts declared.
Data deposition: The atomic coordinates and structure factors of AccD5 have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2A7S).
References
- 1.Ishikawa N. Kekkaku. 2005;80:89–94. [PubMed] [Google Scholar]
- 2.Kunimoto D., Long R. Respir. Care Clin. N. Am. 2005;11:25–34. doi: 10.1016/j.rcc.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 3.Bates I., Fenton C., Gruber J., Lalloo D., Lara A. M., Squire S. B., Theobald S., Thomson R., Tolhurst R. Lancet Infect. Dis. 2004;4:368–375. doi: 10.1016/S1473-3099(04)01047-3. [DOI] [PubMed] [Google Scholar]
- 4.Bates I., Fenton C., Gruber J., Lalloo D., Medina Lara A., Squire S. B., Theobald S., Thomson R., Tolhurst R. Lancet Infect. Dis. 2004;4:267–277. doi: 10.1016/S1473-3099(04)01002-3. [DOI] [PubMed] [Google Scholar]
- 5.Chopra K. Indian J. Pediatr. 1996;63:159–162. doi: 10.1007/BF02845239. [DOI] [PubMed] [Google Scholar]
- 6.Daffe M., Draper P. Adv. Microb. Physiol. 1998;39:131–203. doi: 10.1016/s0065-2911(08)60016-8. [DOI] [PubMed] [Google Scholar]
- 7.Brennan P. J., Nikaido H. Annu. Rev. Biochem. 1995;64:29–63. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- 8.Lee R. E., Brennan P. J., Besra G. S. Curr. Top Microbiol. Immunol. 1996;215:1–27. doi: 10.1007/978-3-642-80166-2_1. [DOI] [PubMed] [Google Scholar]
- 9.Takayama K., Wang C., Besra G. S. Clin. Microbiol. Rev. 2005;18:81–101. doi: 10.1128/CMR.18.1.81-101.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minnikin D. E., Kremer L., Dover L. G., Besra G. S. Chem. Biol. 2002;9:545–553. doi: 10.1016/s1074-5521(02)00142-4. [DOI] [PubMed] [Google Scholar]
- 11.Fernandes N. D., Kolattukudy P. E. Gene. 1996;170:95–99. doi: 10.1016/0378-1119(95)00842-x. [DOI] [PubMed] [Google Scholar]
- 12.Mathur M., Kolattukudy P. E. J. Biol. Chem. 1992;267:19388–19395. [PubMed] [Google Scholar]
- 13.Quadri L. E., Sello J., Keating T. A., Weinreb P. H., Walsh C. T. Chem. Biol. 1998;5:631–645. doi: 10.1016/s1074-5521(98)90291-5. [DOI] [PubMed] [Google Scholar]
- 14.Trivedi O. A., Arora P., Vats A., Ansari M. Z., Tickoo R., Sridharan V., Mohanty D., Gokhale R. S. Mol. Cell. 2005;17:631–643. doi: 10.1016/j.molcel.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 15.Cronan J. E., Jr., Waldrop G. L. Prog. Lipid Res. 2002;41:407–435. doi: 10.1016/s0163-7827(02)00007-3. [DOI] [PubMed] [Google Scholar]
- 16.Tong L. Cell Mol. Life Sci. 2005;62:1784–1803. doi: 10.1007/s00018-005-5121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heath R. J., White S. W., Rock C. O. Appl. Microbiol. Biotechnol. 2002;58:695–703. doi: 10.1007/s00253-001-0918-z. [DOI] [PubMed] [Google Scholar]
- 18.Kim K. H. Annu. Rev. Nutr. 1997;17:77–99. doi: 10.1146/annurev.nutr.17.1.77. [DOI] [PubMed] [Google Scholar]
- 19.Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E., III, et al. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 20.Gago G., Kurth D., Diacovich L., Tsai S. C., Gramajo H. J. Bacteriol. 2006;188:477–486. doi: 10.1128/JB.188.2.477-486.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez E., Banchio C., Diacovich L., Bibb M. J., Gramajo H. Appl. Environ. Microbiol. 2001;67:4166–4176. doi: 10.1128/AEM.67.9.4166-4176.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diacovich L., Peiru S., Kurth D., Rodriguez E., Podesta F., Khosla C., Gramajo H. J. Biol. Chem. 2002;277:31228–31236. doi: 10.1074/jbc.M203263200. [DOI] [PubMed] [Google Scholar]
- 23.Diacovich L., Mitchell D. L., Pham H., Gago G., Melgar M. M., Khosla C., Gramajo H., Tsai S. C. Biochemistry. 2004;43:14027–14036. doi: 10.1021/bi049065v. [DOI] [PubMed] [Google Scholar]
- 24.Rainwater D. L., Kolattukudy P. E. J. Bacteriol. 1982;151:905–911. doi: 10.1128/jb.151.2.905-911.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Portevin D., de Sousa-D’Auria C., Montrozier H., Houssin C., Stella A., Laneelle M. A., Bardou F., Guilhot C., Daffe M. J. Biol. Chem. 2005;280:8862–8874. doi: 10.1074/jbc.M408578200. [DOI] [PubMed] [Google Scholar]
- 26.Gande R., Gibson K. J., Brown A. K., Krumbach K., Dover L. G., Sahm H., Shioyama S., Oikawa T., Besra G. S., Eggeling L. J. Biol. Chem. 2004;279:44847–44857. doi: 10.1074/jbc.M408648200. [DOI] [PubMed] [Google Scholar]
- 27.Zhang H., Yang Z., Shen Y., Tong L. Science. 2003;299:2064–2067. doi: 10.1126/science.1081366. [DOI] [PubMed] [Google Scholar]
- 28.Hall P. R., Wang Y. F., Rivera-Hainaj R. E., Zheng X., Pustai-Carey M., Carey P. R., Yee V. C. EMBO J. 2003;22:2334–2347. doi: 10.1093/emboj/cdg244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holden H. M., Benning M. M., Haller T., Gerlt J. A. Acc. Chem. Res. 2001;34:145–157. doi: 10.1021/ar000053l. [DOI] [PubMed] [Google Scholar]
- 30.Rainwater D. L., Kolattukudy P. E. J. Biol. Chem. 1983;258:2979–2985. [PubMed] [Google Scholar]
- 31.Dessen A., Quemard A., Blanchard J. S., Jacobs W. R., Jr., Sacchettini J. C. Science. 1995;267:1638–1641. doi: 10.1126/science.7886450. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y. Annu. Rev. Pharmacol. Toxicol. 2005;45:529–564. doi: 10.1146/annurev.pharmtox.45.120403.100120. [DOI] [PubMed] [Google Scholar]
- 33.Ewing T. J., Makino S., Skillman A. G., Kuntz I. D. J. Comput. Aided Mol. Des. 2001;15:411–428. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]
- 34.Bursulaya B. D., Totrov M., Abagyan R., Brooks C. L., III J. Comput. Aided Mol. Des. 2003;17:755–763. doi: 10.1023/b:jcam.0000017496.76572.6f. [DOI] [PubMed] [Google Scholar]
- 35.Chen J., Swamidass S. J., Dou Y., Bruand J., Baldi P. Bioinformatics. 2005;21:4133–4139. doi: 10.1093/bioinformatics/bti683. [DOI] [PubMed] [Google Scholar]
- 36.Gronwald J. W. Biochem. Soc. Trans. 1994;22:616–621. doi: 10.1042/bst0220616. [DOI] [PubMed] [Google Scholar]
- 37.Davis M. S., Cronan J. E., Jr J. Bacteriol. 2001;183:1499–1503. doi: 10.1128/JB.183.4.1499-1503.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davis M. S., Solbiati J., Cronan J. E., Jr J. Biol. Chem. 2000;275:28593–28598. doi: 10.1074/jbc.M004756200. [DOI] [PubMed] [Google Scholar]
- 39.Otwinowski Z., Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 40.Brunger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., et al. Acta Crystallogr. D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.