

Abstract
p53-binding protein 1 (53BP1) participates in the cellular response to DNA double-stranded breaks where it associates with various DNA repair/cell cycle factors including the H2AX histone variant. Mice deficient for 53BP1 (53BP1−/−) are sensitive to ionizing radiation and immunodeficient because of impaired Ig heavy chain class switch recombination. Here we show that, as compared with p53−/− mice, 53BP1−/−/p53−/− animals more rapidly develop tumors, including T cell lymphomas and, at lower frequency, B lineage lymphomas, sarcomas, and teratomas. In addition, T cells from animals deficient for both 53BP1 and p53 (53BP1−/−/p53−/−) display elevated levels of genomic instability relative to T cells deficient for either 53BP1 or p53 alone. In contrast to p53−/− T cell lymphomas, which routinely display aneuploidy but not translocations, 53BP1−/−/p53−/− thymic lymphomas fall into two distinct cytogenetic categories, with many harboring clonal translocations (40%) and the remainder showing aneuploidy (60%). We propose that 53BP1, in the context of p53 deficiency, suppresses T cell lymphomagenesis through its roles in both cell-cycle checkpoints and double-stranded break repair.
Keywords: thymic lymphoma, aneuploidy, translocations
Double-stranded break (DSB) repair is essential for genomic stability and tumor suppression. In mammalian cells, DSBs arise through exogenous means such as ionizing radiation, at stalled replication forks, and in the context of two genetically programmed processes that occur in lymphoid cells (1–3). DSBs are repaired by either of two nonmutually exclusive mechanisms: homologous recombination and nonhomologous end joining. During the development of the immune system, both B and T cells assemble the exons that encode Igs and T cell receptor variable regions by means of V(D)J recombination. In addition, mature B cells can express different Ig heavy chain constant regions through a DSB-mediated process known as Ig heavy chain class switch recombination (CSR). The DSBs that initiate V(D)J recombination are generated by the site-specific RAG endonuclease, whereas those that initiate CSR appear to involve the activation-induced deaminase. However, both V(D)J recombination and CSR are completed by nonhomologous end joining. In contrast to V(D)J recombination, CSR requires components of the DNA DSB repair response, including the histone variant H2AX and p53 binding protein 1 (53BP1) (4–7), potentially to juxtapose broken DSBs for nonhomologous end joining (8). Although 53BP1 is required for joining of S regions during CSR, like H2AX, it does not appear to be absolutely required for V(D)J recombination (6, 7), perhaps because of the fact that RAG also mediates synapsis functions (8).
53BP1 is phosphorylated in response to ionizing radiation, accumulates at or near sites of DNA damage, and participates in the intra S and G2/M checkpoints (reviewed in ref. 9). Accumulation of 53BP1 at sites of irradiation-induced foci depends on the phosphorylation of H2AX (γ-H2AX), a protein that directly interacts with 53BP1 in response to ionizing radiation (10) and the presence of Mdc1, a mediator of the DNA damage response (DDR) (11, 12). ATM phosphorylates 53BP1 (9), but this event is not required for its accumulation at irradiation-induced foci (13). 53BP1 is hyperphosphorylated in response to nocodozole and, for reasons unclear, localizes to the kinetochore during mitosis (14). 53BP1 possesses Tudor motifs that are critical for its localization to irradiation-induced foci (15). A region of 53BP1 upstream of the Tudor domains interacts with DNA and has been shown to facilitate joining events in vitro (16). The Tudor domain directly interacts with methylated lysine residue 79 (K79) of histone H3 (15). Because K79 maps to the histone core, changes in chromatin structure, perhaps as induced by DSBs, have been proposed to be detected by 53BP1, leading to the notion that 53BP1 senses DSBs (15). Thus, 53BP1 may operate both upstream and downstream of ATM activation (15, 17). Both 53BP1 and H2AX appear to influence the joining phase of CSR, leading to the proposal that these DDR regulators may participate in DNA synapsis or joining by “anchoring” the DNA breaks for DSB repair (8). Such a mechanism has been proposed to limit the recombinogenic potential of free, broken DNA ends (8).
The majority (≈75%) of mice defective in p53 (p53−/−) develop thymic lymphoma at an average age of ≈5 months (18–20). These tumors are characterized by aneuploidy and are lacking in clonal translocations (21–24). In contrast, the two independently derived 53BP1−/− mouse lines are only mildly tumor-prone, because up to 8% of these animals die of thymic lymphoma between 4 and 7 months (25, 26). Such low rates of tumorigenesis have been observed in other components of the DDR network, including H2AX (23, 24). Although deficiencies in H2AX lead to increased genomic instability in T cells as measured by DNA breaks and/or translocations, the presence of intact checkpoint mechanisms may prevent tumorigenesis. In the context of p53 deficiency, H2AX−/− mice (H2AX−/−/p53−/−) rapidly develop T cell and, to a lesser extent, B cell lymphomas as well as other tumors (23, 24). In contrast to the well established aneuploid nature of p53−/− thymic tumors, H2AX−/−/p53−/− T cell lymphomas are characterized by clonal translocations. In addition, a limited sampling size of H2AX+/−/p53−/− tumors reveals that these tumors arise from clonal translocation and aneuploid-driven mechanisms (23, 24). Therefore, tumorigenesis in both H2AX−/−/p53−/− and H2AX+/−/p53−/− mice is mechanistically distinct from that observed in p53−/− animals (23, 24).
To further characterize the role of 53BP1 in tumor suppression and genomic stability, we generated 53BP1−/−/p53−/− double mutants. Here, we show that loss of 53BP1 function, in the context of p53 deficiency, leads to rapid T cell lymphomagenesis. In addition, we show that 53BP1 and p53 synergize to suppress genomic instability in T cells. Our data suggest that two separate mechanisms, aneuploidy and clonal translocations, contribute to lymphomagenesis in 53BP1−/−/p53−/− mice, suggesting that both the cell-cycle checkpoint and DSB repair functions of 53BP1 contribute to tumor suppression.
Results
Accelerated Tumorigenesis in 53BP1−/−p53−/− Double-Mutant Mice.
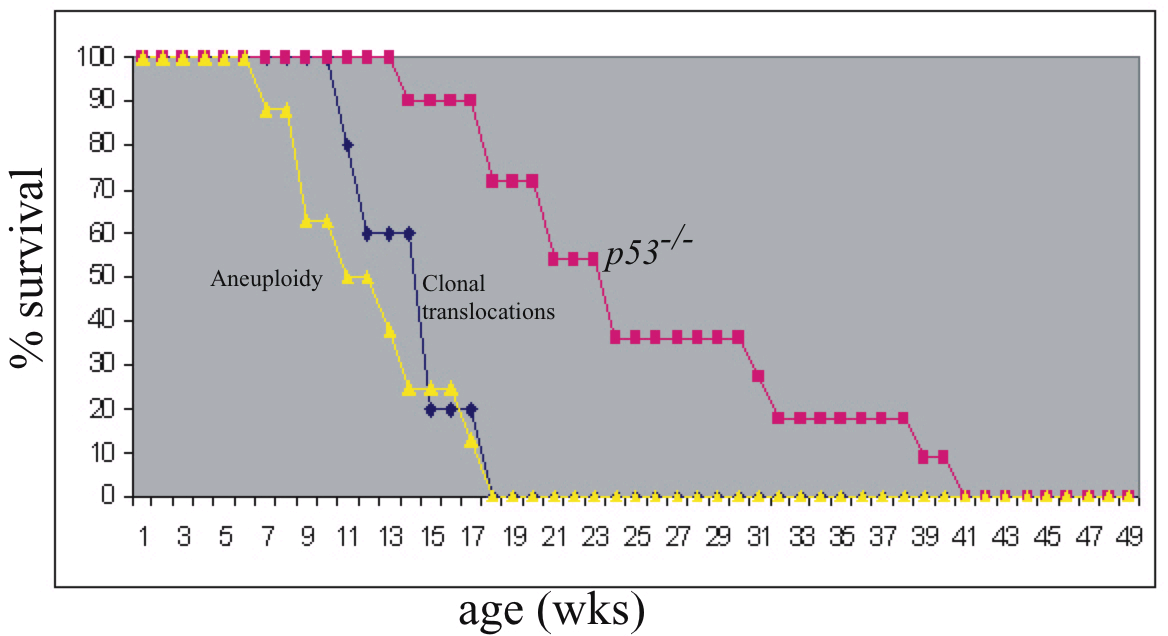
To test for potential cooperation between 53BP1 and p53 in tumor suppression, we bred 53BP1+/− and p53+/− mice to generate cohorts of 53BP1−/−/p53−/− double mutants. We also made cohorts of 53BP1+/+/p53+/+, 53BP1+/−/p53+/+, 53BP1−/−/p53+/+, 53BP1+/−/p53+/−, 53BP1+/+/p53−/−, and 53BP1+/−/p53−/− mice. All genotypes were born at approximately normal Mendelian frequency. However, the 53BP1−/−/p53−/− progeny, because of increased tumorigenesis, had a substantially reduced lifespan compared with cohorts of other genotypes, becoming moribund as early as 7 weeks, with 50% mortality occurring by 12 weeks (Fig. 1A). Of 44 liveborn 53BP1−/−/p53−/− mice, nearly all developed tumors, with most (32/44) succumbing to thymic lymphomas, with an onset time much more rapid than p53−/− tumors, where 50% mortality occurred by 22 weeks (Fig. 1A). 53BP1−/−/p53−/− mice also developed other types of tumors, including B lineage tumors (7/44), sarcomas (2/44), and teratomas (2/44) (Fig. 1B). Although sarcomas and teratomas have been observed in p53-null animals, B lineage tumors are rare in this background (18–20). Thus, 53BP1 deficiency alters both the timing and the tumor spectrum observed in a p53−/− background, indicating that both proteins cooperate to suppress tumorigenesis. Because these 53BP1+/−/p53−/− animals (n = 10) showed a morbidity, primarily due to T cell lymphomas, that was slightly delayed compared with that of p53-deficient animals (Fig. 1B), our 53BP1 allele does not show a haploinsufficient phenotype in this context.
Fig. 1.

Kaplan–Meier survival curve of 53BP1−/−/p53−/− double mutant animals and their cohorts. (A) Survival curve of 53BP1−/−/p53−/− double mutants relative to cohort genotypes, including 53BP1+/−/p53−/− and p53−/− mice. Several genotypes, including 53BP1−/−, survived throughout the duration of this study and, as a result, are represented by a single horizontal line. 53BP1−/−/p53−/− double mutants have a 50% survival rate compared with their p53 mutant cohorts. Sampling size for each genotype is listed. The differences in the timing of morbidity between 53BP1+/−/p53−/− and p53−/− animals was determined to be statistically insignificant (P = 0.476) by using a one-way ANOVA and a t test with 90% confidence. In contrast, P < 0.001 between the survival curves for 53BP1−/−/p53−/− and p53−/− breedings. (B) Pie chart showing the tumor spectrum observed for 53BP1−/−/p53−/− mice (n = 44). T, T lineage lymphomas; B, B lineage lymphomas; S, sarcoma; Te, teratomas; O, other (cause of death undetermined).
Both Aneuploidy and Clonal Translocations Contribute to the Development of 53BP1−/−p53−/− Thymic Lymphomas.
Flow cytometry with anti-CD4 and anti-CD8 antibodies confirmed that the 53BP1−/−/p53−/− tumors were of T cell origin (Fig. 5, which is published as supporting information on the PNAS web site). Most tumors expressed either both CD4 and CD8 (double-positive cells) or were CD8+ CD4− (single-positive cells). Some showed CD8− CD4+ staining. Collectively, this finding suggests that 53BP1−/−/p53−/− tumors had developed past the double-negative stage (Fig. 5).
Thymic lymphomas from p53−/− and H2AX−/−/p53−/− mice arise via distinct mechanisms because the former are routinely aneuploid with no clonal translocations whereas the latter show clonal translocations (21–24). To further elucidate the mechanisms of T cell lymphomagenesis in 53BP1−/−/p53−/− mice, we performed spectral karyotyping (SKY) on metaphase preparations of early-passage thymic tumor cells derived from various 53BP1−/−/p53−/− animals. We analyzed 13 different tumors and found that 8 showed aneuploidy and 5 had clonal translocations with no signs of gross aneuploidy, a feature distinct from p53-derived thymic tumors. In this context, T lineage tumor 3121 (T3121) possessed a clonal, Robertsonian translocation between chromosomes 19 and 5 in 18 of 20 examined metaphases (Fig. 2 and Tables 1 and 2, which are published as supporting information on the PNAS web site). A complete cytogenetic profile for T3074 is shown in Fig. 6, which is published as supporting information on the PNAS web site. We also found that tumor 3074 possessed a clonal t(19;5) translocation in 13 of 15 metaphases but, in contrast to T3121, did not involve fusion of centromeric sequences (Fig. 2). T lineage tumor T3122 had both t(12;4) and t(12;14) translocations (Fig. 2). T2965 possessed a clonal t(14;2) translocation in 8 of 8 examined metaphases (Fig. 2), a translocation that resembles those observed in ATM−/− mice as well as a recently described hypomorphic Nbs1m/m allele (27, 28). In addition, T2965 also exhibited clonal translocations in chromosome 13 [t(9;13)] in 8 of 8 metaphase spreads as well as a t(9;17) translocation in 7 of 8 metaphases examined.
Fig. 2.

53BP1−/−/p53−/− thymic lymphomas display clonal translocations. SKY analysis of four T cell lymphomas displaying clonal translocations was performed as described in Methods. Chromosome number and translocations, if present, are listed for one tumor. See text for details.
In contrast to clonal translocations, 8 of 13 of the 53BP1−/−/p53−/− T cell lymphomas displayed aneuploidy, because these samples had cells with supernumerary chromosomes (Fig. 3). For example, T2961 and 2963 possessed 39–60 and 50–54 chromosomes, respectively, with no evidence of translocations (Fig. 3 and Table 1). T3096 possessed between 40 and 54 chromosomes in addition to three nonclonal translocations in 21 examined metaphases. In addition, T2945 possessed both monosomy and trisomy chromosomes with no evidence of translocations. Thus, despite their much earlier onset relative to p53−/− thymic lymphomas, a subset of 53BP1−/−/p53−/− thymic lymphomas appeared aneuploidy without clonal translocations and, on this basis, appeared “p53-like.” However, in contrast to p53-deficient cells, which frequently exhibit centrosome amplification, we found that 53BP1−/− cells exhibited normal centrosome numbers (unpublished data), a result recently reported by Chen and colleagues (29). In summary, we found that individual 53BP1−/−/p53−/− T cell lymphomas cytogenetically resembled T lineage tumors derived from both H2AX−/−/p53−/− and H2AX+/−/p53−/− (clonal translocations and/or aneuploidy) or p53−/− T lineage tumors (aneuploidy).
Fig. 3.

Supernumerary chromosomes with little evidence of translocations in 53BP1−/−/p53−/− thymic lymphomas. Representative metaphases derived from SKY from a given tumor are shown. n = number of examined metaphase spreads. T2961 possesses 39–60 chromosomes (n = 15). T2963 has 50–54 chromosomes (n = 19). T2945 is essentially euploid (39–42 chromosomes) but has several monosomic chromosomes (6, 10, 17) depending on the cell in addition and trisomy 5 and 11 (n = 8). T3096 was found to have 40–54 chromosomes (n = 20) in addition to nonclonal translocations t(19;14) and t(7;16;4).
Increased Cytogenetic Abnormalities in 53BP1- and p53-Deficient T Cells.
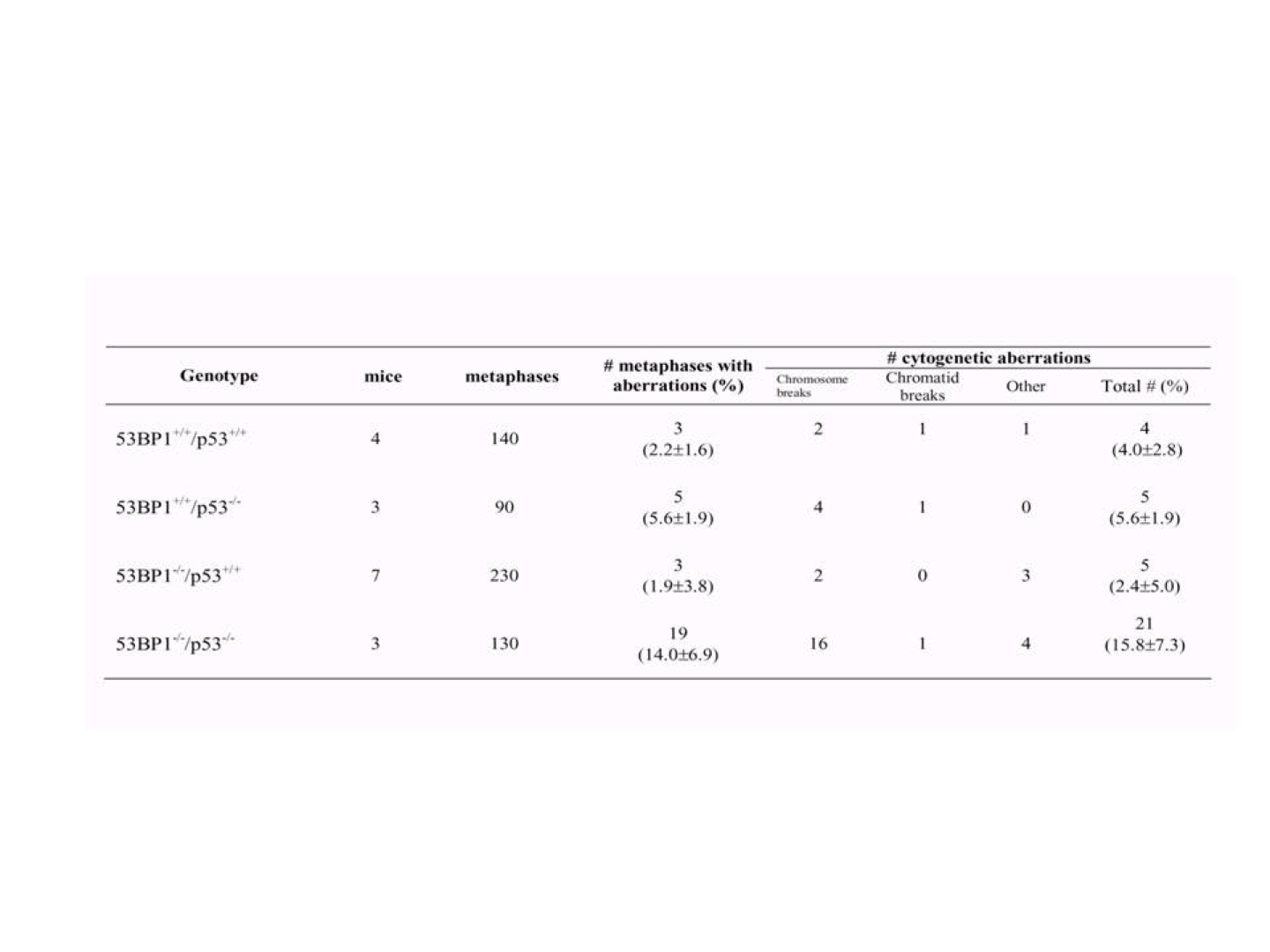
To examine whether 53BP1 and p53 deficiency had synergistic effects on genomic instability, we performed cytogenetic analyses on splenic T cells isolated from wild-type, 53BP1−/−, p53−/−, and 53BP1−/−/p53−/− animals between 4 and 6 weeks of age. Metaphase spreads were prepared from CD43+ spleen cells stimulated with Con A, and cytogenetic aberrations were analyzed by FISH by using a telomere specific probe (PNA) and scored (Fig. 4). We found that 53BP1−/− T cells possessed very low levels of genomic instability, similar to those of T cells isolated from wild type or p53 mutants as measured by chromosome and chromatid breaks as well as other genetic abnormalities (Fig. 4). Thus, 53BP1−/− T cells, unlike H2AX−/− and Nbs1m/m T cells (4, 5, 24, 28), do not possess significant increases in genomic instability. In contrast, 53BP1−/−/p53−/− T cells displayed significant increases (P = 0.047) in aberrant chromosomal structures relative to 53BP1−/− T cells, and this included chromosome breaks, dicentric chromosomes, detached centromeres, and chromatid fusions (Fig. 4). Thus, neither p53 nor 53BP1 deficiency alone resulted in elevated levels of genomic instability in T cells. In fact, such abnormal structures are rarely, if at all, present in p53−/− cells (22, 23). Intriguingly, most of the chromosomal aberrations in 53BP1−/−/p53−/− T cells were derived from chromosome, and not chromatid, breaks (Table 3, which is published as supporting information on the PNAS web site). We conclude that 53BP1 and p53 synergize to suppress genomic instability in T cells.
Fig. 4.

Increased genomic instability in 53BP1−/−/p53−/− thymocytes. (A) Quantification of structural chromosomal aberrations in Con A-stimulated T cells from wild-type (n = 4), p53−/− (n = 3), 53BP1−/− (n = 7), and 53BP1−/−/p53−/− (n = 3) mice. Of the three p53 mutants analyzed, two were derived from Donehower and colleagues (18, 19), and the third was from Jacks and colleagues (20). Metaphase spreads were hybridized with a telomere-specific Cy3-labeled PNA probe (red) to visualize the four ends of each chromosome and counterstained with DAPI (blue). At least 30 metaphases per culture were analyzed. Aberrations are represented as follows: % aberrations per metaphase = (total number of aberrations/total number of metaphases) × 100. (B) Partial metaphase spreads of 53BP1−/−/p53−/− T cells showing three chromosomal breaks (yellow arrows) and a dicentric resulting from rejoining of two broken chromosomes. A normal chromosome with four telomere signals is shown on the left for comparison.
Discussion
Recent studies with human tumors have shown that the loss of 53BP1 expression correlates with cancer progression (30). We found that impaired murine 53BP1 function significantly alters cancer susceptibility in the context of p53 deficiency by accelerating the rate of tumorigenesis and altering the tumor spectrum. Our data indicate that two distinct cytogenetic classes of tumors are found in 53BP1−/−/p53−/− thymic lymphomas, and, based on this finding, we propose that tumorigenesis in 53BP1−/−/p53−/− T cells proceeds through either of two separate mechanisms: aneuploidy and clonal translocations. Thus, on one hand, 53BP1−/−/p53−/− thymic lymphomas resemble H2AX+/−/p53−/− and H2AX−/−/p53−/−T lineage tumors, and, on the other hand, a different, aneuploid subset resembles p53−/− thymic lymphomas, although their timing of onset is much earlier. Although there is no a priori reason why both mechanisms cannot operate within the same 53BP1−/−/p53−/− tumor, we found little evidence for this occurrence. Thus, 53BP1 may have two distinct functions, both of which synergize with p53 to suppress tumorigenesis. In the first, 53BP1 and p53 synergize to suppress oncogenic translocations, a function that likely reflects the role of 53BP1 in DSB repair, perhaps as an “anchor” of DSBs that prevents aberrant recombination/translocation events (8). This event may be expected to operate in concert with H2AX, Nbs1, and Mdc1, given that H2AX and Mdc1 are required for 53BP1 foci formation (11, 12) and that H2AX and Nbs1 synergize with p53 to suppress lymphomagenesis (23, 24, 28). Interestingly, although Mre11ATLD hypomorphic animals display elevated levels of genomic instability as measured by breakage and rearrangement (36), the allele is insufficient to cause tumorigenesis. Moreover, in the context of p53 deficiency, Mre11ATLD/p53−/− double mutants (and Nbsm/m mutants) show only a modest effect on the latency of lymphomagenesis (28, 31), illustrating that DDR regulators such as H2AX, 53BP1, and Mre11 have distinct effects on suppressing genomic instability and tumorigenesis. One possibility, as discussed in ref. 31, is that, because Mre11 and Nbs1 function primarily in checkpoints (i.e., S and G2/M) that are believed to operate largely independent of p53 function, there would be little enhancement of p53-driven lymphomagenesis. In this capacity, 53BP1−/− cells may expedite lymphomagenesis in a p53-deficient background through common functions with p53. How this event would operate is unknown, but defects in the putative kinetochore function of 53BP1 could be synergistic with p53 deficiency, resulting in an increased rate of mitotic failure. In addition, because 53BP1 is required for recruiting both BLM and p53 to stalled replication forks (32), defects in this H2AX-independent mechanism could contribute to increased genomic instability and tumorigenesis. In this light, it is interesting to note that 53BP1, like p53 and BLM, displays a “hyper-rec” phenotype (33).
The T cells examined from 53BP1−/−/p53−/− thymic lymphomas developed into and through the DP stage and appear to have properly executed V(D)J recombination. Nevertheless, a subset of our 53BP1−/−/p53−/− thymic lymphomas (i.e., T2965 and T3122) possessed clonal translocations in chromosomes that harbor antigen receptor loci, raising the possibility that aberrant DNA repair during V(D)J recombination may be causal for tumorigenesis. Indeed, Chen and colleagues (29) recently showed the involvement of the T cell receptor α locus of chromosome 14 in a 53BP1−/−/p53−/− thymic lymphoma. Therefore, although 53BP1 appears dispensable for V(D)J recombination as based on several criteria (6, 7), subtle defects below the levels of assay detection may be selected for during T cell clonal expansion. Additionally, the clonal translocations found in these tumors may have occurred in association with the rapid cellular proliferation (i.e., DNA replication) observed during thymocyte expansion as previously proposed for H2AX−/−/p53−/− tumors (23, 24). Such rapid clonal expansion of 53BP1−/−/p53−/− T cells would generate numerous translocation donors and acceptors in S-phase, perhaps through aberrant resolution of stalled replication forks, a structure known to recruit 53BP1 (32).
A subset of 53BP1−/−/p53−/− T cell lymphomas also develop through aneuploidy, and, in this respect, they appear p53-like although their timing of onset is significantly much faster than p53 tumorigenesis. Because 53BP1deficiency does not appear to significantly alter centrosome number (unpublished data and ref. 29), 53BP1 appears to suppress aneuploidy through a mechanism distinct from p53. Although there are two distinct classes of tumors in these mice, we found no differences in phenotype and, at most, only a slight increase in the timing of onset in anueploid-driven tumorigenesis relative to those identified with translocations (Fig. 7, which is published as supporting information on the PNAS web site). Because 53BP1 participates in cell-cycle checkpoints, localizes to the kinetochore in an H2AX-independent manner, and is hyperphosphorylated in response to nocodozole, deficiencies in any or all of these functions may compromise its ability to function in suppressing aneuploidy. How any putative kinetochore or checkpoint defects in 53BP1 mutants contribute to T cell lymphomagenesis in the context of p53 deficiency remains an open question, although some clues may be emerging. 53BP1 has been reported to interact with Cdc27, a component of the anaphase-promoting complex (34). Intriguingly, the Artemis-related protein Snm1 has also been reported to associate with 53BP1 and coimmunoprecipitates with Cdc27 (31, 35). It is thus possible that 53BP1 influences anaphase-promoting complex function, a result that would conceivably alter cell-cycle control in both mitosis and interphase.
Although 53BP1−/−/p53−/− mice died of lymphomas with similar kinetics to H2AX−/−/p53−/− animals, there are some important differences that differentiate 53BP1 deficiency from H2AX deficiency, indicating that the proteins have overlapping, as well as distinct, functions within DDR pathways. In this regard, 53BP1−/−/p53−/− thymic lymphomas are cytogenetically akin to haploinsufficient H2AX+/−/p53−/− and H2AX−/−/p53−/− thymic lymphomas (23, 24). In addition, 53BP1−/− T cells, unlike those from H2AX-deficient animals, display little detectable genomic instability. Rather, 53BP1−/− T cells have significantly elevated levels of detectable DNA breaks (primarily of chromosome, as opposed to chromatid, origin) and other forms of genomic instability only in the context of p53 deficiency. This level of genomic instability is in contrast to H2AX−/− T cells as well as Nbs1m/m T cells, both of which possess significantly elevated levels of chromosomal aberrations that are not augmented by an additional mutation in p53 (23, 24, 28). Although we cannot rule out genetic background effects, this occurrence is perhaps due to the observation that 53BP1−/− T cells, but not H2AX−/− T cells, increase their apoptotic response in the presence of DNA damage (7). Thus, for any given population of 53BP1−/− T cells, those that exhibit increased genomic stability through abrogated DSB repair could be targeted for apoptosis, a result that would enrich viable cells for those that possess fewer overall chromosomal aberrations. Despite the elevated levels of genomic instability in H2AX−/− T cells, H2AX-deficient mice show little predisposition to develop cancer, indicating that genomic instability per se is not sufficient for tumorigenesis, a scenario that has been observed for alleles of BRCA2 and Mre11 (31, 36).
As this manuscript was being prepared, Chen and colleagues (29) used an independently derived 53BP1−/− strain and reported a synergistic effect between 53BP1 and p53 with respect to lymphomagenesis. Although the timing, tumor spectrum, and cytogenetics appear similar between the two studies, there is one noteworthy difference. Although Chen and colleagues (29) observed a haploinsufficient phenotype for tumorigenesis in 53BP1+/−/p53−/− mice (n = 37), we thus far found no evidence for this phenotype in our more limited studies (n = 10). Although our 53BP1−/− allele encodes for a truncated protein (25), it is missing >700 C-terminal amino acids that include critical elements such as the tandem Tudor and BRCT repeats. In addition, this protein fails to localize to the nucleus, the site where 53BP1 most likely executes its essential functions (14, 25). Importantly, because both independently derived 53BP1 alleles are sensitive to ionizing radiation and severely impaired in CSR (6, 7, 25, 26), each one likely represents the null phenotype. Given this observation, possible explanations for the observed differences between the haploinsufficient phenotypes in each 53BP1−/−/p53−/− double mutant may include differences in genetic background, the p53 targeted mutation used, our limited sample size, and/or the breeding facilities.
Methods
Generation of 53BP1−/−/p53−/− Mice.
p53+/− mice (20) were obtained from G. Lozano (University of Texas M.D. Anderson Cancer Center) and bred with 53BP1+/− mice (25) to generate heterozygous offspring (53BP1+/−/p53+/−). These mice were bred to generate wild-type mice (53BP1+/+p53+/+) and animals with mutations in 53BP1 and p53 (53BP1−/−/p53−/−). Genomic DNA from tail snips was used for genotyping. A PCR assay with three primers was used to establish the genotype at the 53BP1 locus, where two of the primers (53BP1-F and 53BP1-R) recognize wild-type sequences, and the third (53BP1-NEOR) is derived from the neomycin insertion. We determined the genotype for p53 animals with a similar strategy. PCR primer sequences and reaction conditions will be made available upon request.
T Cell Isolation from 53BP1−/−/p53−/− Thymic Lymphomas and SKY.
Thymic lymphomas were placed in a 0.70-μm cell strainer in 5 ml of RPMI medium 1640 (GIBCO) with nonessential amino acids, 2-mercaptoethanol, sodium pyruvate, and antibiotics. Isolated cells were pelleted and resuspended in RBC lysis buffer and transferred to RPMI medium 1640. The cells were processed for flow cytometry with antibodies against CD4 and CD8 (Pharmingen) or seeded in RPMI medium 1640 at 2 × 106 cells per ml, treated with KaryoMAX colcemid solution (GIBCO) for 2–4 h at 37°C, and processed for SKY as described in ref. 23.
PNA FISH of TTAGGG Sequences.
CD43+ T lymphocytes were purified from splenocyte cell suspensions by using LS columns (Beads, Miltenyi Biotec), incubated in colcemid (KaryoMAX), swollen in prewarmed 30 mM sodium citrate for 25 min at 37°C, fixed in methanol/acetic acid (3/1), and air-dried on slides overnight. After pepsin digestion, slides were denatured at 80°C for 3 min, hybridized with a Cy3-labeled PNA telomeric probe (Cy3-(TTAGGG)3 in 70% formamide at room temperature for 2 h, washed, dehydrated, and mounted in Vectashield with DAPI. Metaphase images were captured by using a Nikon Eclipse microscope equipped with a charge-coupled device camera and a ×63 objective lens.
Supplementary Material
Acknowledgments
We thank Gigi Lozano for providing murine p53 mutants and Sandy Chang, L. J. Medeiros, and Ben Zhu for advice; Drs. John Petrini, Ron Depinho, and Yang Xu for the critical reading of the manuscript; and Raffaella Righetti for assistance with the statistical analysis. This work was supported by National Institutes of Health Grants GM65812 (to P.B.C.) and CA92625 and CA109901 (to F.W.A.). C.H.B. is a Pew Scholar in the Biomedical Sciences and a Lymphoma Research Foundation Fellow. S.F. was supported by a Long-Term Fellowship of the European Molecular Biology Foundation. F.W.A. is an investigator of the Howard Hughes Medical Institute.
Abbreviations
- DSB
double-stranded break
- DDR
DNA damage response
- CSR
class switch recombination
- SKY
spectral karyotyping.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Bassing C. H., Swat W., Alt F. W. Cell. 2002;109(Suppl.):S45–S55. doi: 10.1016/s0092-8674(02)00675-x. [DOI] [PubMed] [Google Scholar]
- 2.Li Z., Woo C. J., Iglesias-Ussel M. D., Ronai D., Scharff M. D. Genes Dev. 2004;18:1–11. doi: 10.1101/gad.1161904. [DOI] [PubMed] [Google Scholar]
- 3.Lombard D. B., Chua K. F., Mostoslavsky R., Franco S., Gostissa M., Alt F. W. Cell. 2005;120:497–512. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 4.Bassing C. H., Chua K. F., Sekiguchi J., Suh H., Whitlow S. R., Fleming J. C., Monroe B. C., Ciccone D. N., Yan C., Vlasakova K., et al. Proc. Natl. Acad. Sci. USA. 2002;99:8173–8178. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Celeste A., Petersen S., Romanienko P. J., Fernandez-Capetillo O., Chen H. T., Sedelnikova O. A., Reina-San-Martin B., Coppola V., Meffre E., Difilippantonio M. J., et al. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manis J. P., Morales J. C., Xia Z., Kutok J. L., Alt F. W., Carpenter P. B. Nat. Immunol. 2004;5:481–487. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- 7.Ward I. M., Reina-San-Martin B., Olaru A., Minn K., Tamada K., Lau J. S., Cascalho M., Chen L., Nussenzweig A., Livak F., et al. J. Cell Biol. 2004;165:459–464. doi: 10.1083/jcb.200403021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bassing C., Alt F. W. Cell Cycle. 2004;2:149–153. doi: 10.4161/cc.3.2.689. [DOI] [PubMed] [Google Scholar]
- 9.Mochan T. A., Venere M., DiTullio R. A., Jr., Halazonetis T. D. DNA Repair. 2004;3:945–952. doi: 10.1016/j.dnarep.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 10.Ward I. M., Minn K., Jorda K., Chen J. J. Biol. Chem. 2003;278:19579–19582. doi: 10.1074/jbc.C300117200. [DOI] [PubMed] [Google Scholar]
- 11.Stewart G. S., Wang B., Bignell C. R., Taylor A. M., Elledge S. J. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 12.Bekker-Jensen S., Lukas C., Melander F., Bartek J., Lukas J. J. Cell Biol. 2005;170:201–211. doi: 10.1083/jcb.200503043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernandez-Capetillo O., Chen H.-T., Celeste A., Ward I., Romanienko P. J., Morales J. C., Naka K., Xia Z., Camerini-Otero R. D., Motoyama N., et al. Nat. Cell Biol. 2002;12:993–997. doi: 10.1038/ncb884. [DOI] [PubMed] [Google Scholar]
- 14.Jullien D., Vagnarelli P., Earnshaw W. C., Adachi Y. J. Cell Sci. 2002;115:71–79. doi: 10.1242/jcs.115.1.71. [DOI] [PubMed] [Google Scholar]
- 15.Huyen Y., Zgheib O., Ditullio R. A., Jr., Gorgoulis V. G., Zacharatos P., Petty T. J., Sheston E. A., Mellert H. S., Stavridi E. S., Halazonetis T. D. Nature. 2004;432:406–411. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- 16.Iwabuchi K., Piku Basu B., Kysela B., Kurihara T., Shibata M., Guan D., Cao Y., Tomio H., Doherty J. J. Biol. Chem. 2003;278:36487–36495. doi: 10.1074/jbc.M304066200. [DOI] [PubMed] [Google Scholar]
- 17.Mochan T. A., Venere M., DiTullio R. A., Jr., Halazonetis T. D. Cancer Res. 2003;63:8586–8591. [PubMed] [Google Scholar]
- 18.Donehower L. A., Harvey M., Slagle B. L., McArthur M. J., Montgomery C. A., Jr., Butel J. S., Bradley A. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 19.Donehower L. A., Harvey M., Vogel H., McArthur M. J., Montgomery C. A., Park S. H., Thompson T., Ford R. J., Bradley A. Mol. Carcinog. 1995;14:16–22. doi: 10.1002/mc.2940140105. [DOI] [PubMed] [Google Scholar]
- 20.Jacks T., Remington L., Williams B. O., Schmitt E. M., Halachmi S., Bronson R. T., Weinberg R. A. Curr. Biol. 1994;1:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 21.Rooney S., Sekiguchi J., Whitlow S., Eckersdorff M., Manis J. P., Lee C., Ferguson D. O., Alt F. W. Proc. Natl. Acad. Sci. USA. 2004;101:2410–2415. doi: 10.1073/pnas.0308757101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liao M. J., Zhang X. X., Hill R., Gao J., Qumsiyeh M. B., Nichols W., Van Dyke T. Mol. Cell. Biol. 1998;18:3495–3501. doi: 10.1128/mcb.18.6.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bassing C. H., Suh H., Ferguson D. O., Chua K., Manis J., Eckersdorff M., Gleason M., Bronson R., Lee C., Alt F. W. Cell. 2003;114:359–370. doi: 10.1016/s0092-8674(03)00566-x. [DOI] [PubMed] [Google Scholar]
- 24.Celeste A., Difilippantonio S., Difilippantonio M. J., Fernandez-Capetillo O., Pilch D. R., Sedelnikova O. A., Eckhaus M., Ried T., Bonner W. M., Nussenzweig A. Cell. 2003;114:371–383. doi: 10.1016/s0092-8674(03)00567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morales J. C., Xia Z., Lu T., Aldrich M. B., Wang B., Rosales C., Kellems R. E., Hittelman W. N., Elledge S. J., Carpenter P. B. J. Biol. Chem. 2003;278:14971–14977. doi: 10.1074/jbc.M212484200. [DOI] [PubMed] [Google Scholar]
- 26.Ward I. M., Minn K., Van Duersen J., Chen J. Mol. Cell. Biol. 2003;23:2556–2563. doi: 10.1128/MCB.23.7.2556-2563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liyanage M., Coleman A., du Manoir S., Veldman T., McCormack S., Dickson R. B., Barlow C., Wynshaw-Boris A., Janz S., Wienberg J., et al. Nat. Genet. 1996;14:312–315. doi: 10.1038/ng1196-312. [DOI] [PubMed] [Google Scholar]
- 28.Kang J., Ferguson D., Song H., Bassing C., Eckersdorff M., Alt F. W., Xu Y. Mol. Cell. Biol. 2005;25:661–670. doi: 10.1128/MCB.25.2.661-670.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ward I. M., Difilippantonio S., Minn K., Mueller M. D., Molina J. R., Yu X., Frisk C. S., Ried T., Nussenzweig A., Chen J. Mol. Cell. Biol. 2005;25:10079–10086. doi: 10.1128/MCB.25.22.10079-10086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gorgoulis V. G., Vassiliou L. V., Karakaidos P., Zacharatos P., Kotsinas A., Liloglou T., Venere M., Ditullio R. A., Jr., Kastrinakis N. G., Levy B., et al. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 31.Cheung A. M., Hande M. P., Jalali F., Tsao M. S., Skinnider B., Hirao A., McPherson J. P., Karaskova J., Suzuki A., Wakeham A., et al. Cancer Res. 2002;62:6194–6204. [PubMed] [Google Scholar]
- 32.Sengupta S., Robles A. I., Linke S. P., Sinogeeva N. I., Zhang R., Pedeux R., Ward I. M., Celeste A., Nussenzweig A., Chen J., et al. J. Cell Biol. 2004;166:801–813. doi: 10.1083/jcb.200405128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adams M. M., Wang. B., Xia Z., Morales J. C., Lu X., Donehower L. A., Bochar D. A., Elledge S. J., Carpenter P. B. Cell Cycle. 2005;4:1854–1861. doi: 10.4161/cc.4.12.2282. [DOI] [PubMed] [Google Scholar]
- 34.Akhter S., Richie C. T., Deng J. M., Brey E., Zhang X., Patrick C., Jr., Behringer R. R., Legerski R. J. Mol. Cell. Biol. 2004;23:10448–10455. doi: 10.1128/MCB.24.23.10448-10455.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richie C. T., Peterson C., Lu T., Hittelman W. N., Carpenter P. B., Legerski R. J. Mol. Cell. Biol. 2002;24:8635–8647. doi: 10.1128/MCB.22.24.8635-8647.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Theunissen J. W., Kaplan M. I., Hunt P. A., Williams B. R., Ferguson D. O., Alt F. W., Petrini J. H. Mol. Cell. 2003;12:1511–1523. doi: 10.1016/s1097-2765(03)00455-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}