

Abstract
The ryanodine receptor (RyR)/calcium-release channel on the sarcoplasmic reticulum mediates intracellular calcium release required for striated muscle contraction. RyR2, the predominant isoform in cardiac myocytes, comprises a macromolecular complex that includes calstabin2 (FKBP12.6). Calstabin2, an 11.8-kDa cis-trans peptidyl-prolyl isomerase (apparent molecular mass 12.6 kDa), stabilizes the closed state of the RyR2 channel, but the mechanism by which it achieves this regulation is not fully understood. Protein kinase A (PKA) phosphorylation of RyR2 decreases the affinity of calstabin2 for the RyR2 channel complex. In the present study we identified key aspartic acid residues on calstabin2 that are involved in binding to RyR2 and likely play a role in PKA phosphorylation-induced dissociation of calstabin2 from RyR2. We show that a mutant calstabin2 in which a key negatively charged residue (Asp-37) has been neutralized binds to a mutant RyR2 channel that mimics constitutively PKA-phosphorylated RyR2 (RyR2–S2808D). Furthermore, using wild-type and genetically altered murine models of heart failure induced by myocardial infarction, we show that manipulating the stoichiometry between calstabin2 and RyR2 can restore normal cardiac function in vivo.
Keywords: calcium-release channel, immunophilin, isomerase, myocardial infarction, PKA phosphorylation
Heart failure is a leading cause of morbidity and mortality in the United States (1). Heart failure occurs when the heart cannot adequately pump blood to meet the metabolic needs of the peripheral organs. Deterioration of cardiac function due to heart failure results in activation of the sympathetic nervous system and increased plasma levels of circulating catecholamines (2). The resulting chronic hyperadrenergic state is believed to alter calcium homeostasis in cardiac myocytes (3).
Intracellular calcium release via the ryanodine receptor (RyR) on the sarcoplasmic reticulum (SR) activates muscle contraction. Each RyR is comprised of four RyR monomers, the cytoplasmic domain of which forms a scaffold for protein components of the RyR macromolecular channel complex that are involved in regulating the channel (4).
An important channel modulator associated with RyR is calstabin (FK506-binding protein, FKBP), an ≈12-kDa cis-trans peptidyl-prolyl isomerase. The association of calstabin1 (FKBP12) with RyR1 was first discovered during peptide sequencing of purified RyR, when a peptide sequence was found that corresponded with the immunophilin FKBP12 (calstabin1) (5). Because this 12-kDa protein stabilizes the closed state of the RyR channel (6), this protein and its isoform, FKBP12.6, have been referred to as calstabin1 and calstabin2, respectively (7). Both calstabins contain 108 aa, and their aligned sequences are 88% identical.
Under physiological conditions, calstabin1 (FKBP12) binds selectively to RyR1, and calstabin2 (FKBP12.6) associates predominantly with the RyR2 isoform (8). The calstabins bind to RyRs with a Kd of ≈0.3 μM (9). Each RyR subunit of the channel tetramer binds a single calstabin molecule (10). Protein kinase A (PKA) phosphorylation of RyR2 at Ser-2808 (S2808) reduces the binding affinity of calstabin2 for the channel complex (11). Chronic PKA hyperphosphorylation of RyR2 in heart failure reduces calstabin2 binding to RyR2 and contributes to leaky RyR2 channels that play a role in depleting SR calcium, resulting in decreased contractility in failing hearts (11). Moreover, we reported that decreased affinity for calstabin2 of mutant human RyR2 linked to catecholaminergic polymorphic ventricular tachycardia channels leads to a diastolic SR calcium leak that may trigger fatal cardiac arrhythmias during exercise or stress (12). However, several of these findings have been disputed by others (13–15).
Previous mutagenesis and crystallography studies have identified key residues in the ligand-binding site of calstabin2 bound to the immunosuppressant drug rapamycin (Fig. 1A and ref. 16) and calstabin1 with TGFβR1 (Fig. 1B and ref. 17). The key residues identified from these studies include the negatively charged residue D37 and aromatic residues F36 and F99, which are conserved between calstabin1 and calstabin2. Mutagenesis of these residues resulted in calstabin1 with diminished peptidyl-prolyl isomerase activity (9, 18). Timerman et al. (9) also showed that modulation of RyR1 by calstabin1 was unrelated to peptidyl-prolyl isomerase activity, because they observed normal SR calcium loading rates with RyR1 bound to isomerase-deficient calstabin1 mutants F36Y, W59H, and F99Y (9). The difference in the selectivities of calstabin1 and calstabin2 for RyR1 and RyR2 has been shown to depend on the residues at three other positions, which in calstabin2 are Q31, N32, and F59 (19).
Fig. 1.

Location of key calstabin2 residues. (A) Key residues mutated in this study are displayed in wire-frame format of calstabin2 in complex with rapamycin represented by a ribbon diagram (Protein Data Bank ID code 1C9H; ref. 16). (B) Asp residues conserved between calstabin1 and calstabin2 that were mutated in this study are shown on the crystal structure of calstabin1 in complex with TGFβR1. Also labeled are the four residues of TGFβR1 in the GS loop that are phosphorylation sites: T185, S187, S189, and S191 (Protein Data Bank ID code 1B6C; ref. 17). Molecular displays were generated by rasmol.
We previously reported that mutation of calstabin2–D37 to Ser resulted in increased binding to PKA-phosphorylated RyR2 (12). We identified key residues in calstabin2 that are important for modulating RyR2 channel gating by calstabin2. In addition, we used these mutant calstabin2s to test the relevance of isomerase activity with respect to the change in affinity of calstabin2 for PKA-treated RyR2.
To assess the effects of calstabin2 binding to PKA-hyperphosphorylated RyR2 in vivo, we generated two transgenic (Tg) mouse lines overexpressing WT calstabin2 or calstabin2–D37V, a mutant that had the greatest binding affinity for PKA-phosphorylated RyR2 in vitro. Mice engineered for cardiac-specific expression of WT calstabin2 or mutant calstabin2 with enhanced binding to RyR2 exhibited significantly improved cardiac function after myocardial infarction (MI), indicating that binding of calstabin2 to RyR2 alone can improve cardiac function in failing hearts.
Results
Binding of Calstabin2 Mutants to Nonphosphorylated RyR2.
We hypothesized that residues located in or near the catalytic binding site of calstabin2, based on the crystal structure (Fig. 1), may also be involved in the calstabin2–RyR2 interaction. We mutated Asp-37 to Val and to Ser, substituting a nonpolar residue in the first case and a polar but uncharged residue in the second case. Because mutations F36Y and F99Y were previously shown to affect calstabin1–RyR1 binding affinities (9), we generated the corresponding mutations in calstabin2 to study their effects on its binding to RyR2.
WT and mutant 35S-labeled calstabin2 were exchanged with native calstabin2 bound to RyR2 in cardiac SR (CSR) membranes to screen for changes in RyR2 binding of mutant calstabin2 compared with endogenous WT calstabin2. We found that calstabin2 with a Ser or a Val substituted for calstabin2–D37 retained significant binding to cardiac RyR2 (Fig. 2A). Calstabin1 mutants D37V, F36Y, and F99Y were previously found to be isomerase-deficient (9, 20). The same fraction of calstabin2–F36Y bound to RyR2 as did WT calstabin2 (Fig. 2A). However, calstabin2–F99Y exhibited decreased binding to RyR2 (Fig. 2A).
Fig. 2.

Binding of Calstabin2 mutants to native RyR2 and PKA-phosphorylated RyR2. (A) 35S-labeled WT and mutant calstabin2 were expressed and exchanged with native calstabin2 in CSR preparations. Calstabin2 bound to RyR2 was in the pellets and was compared with unbound calstabin2 in the supernatants by using densitometry. (B) Calstabin mutants D37S and D37V exhibit increased binding to PKA-phosphorylated RyR2. As a negative control, duplicate preparations were treated with PKA in the presence of PKA inhibitor to prevent PKA phosphorylation of RyR2 (data not shown). Calstabin2–D37S and calstabin2–D37V showed similar binding to both PKA-phosphorylated and nonphosphorylated RyR2. Calstabin2–D37V and calstabin2–D37S consistently retained significantly higher levels of binding to PKA-phosphorylated RyR2 when compared with WT calstabin2 (P < 0.05). Data are shown as the average ± SD of at least three replicate experiments. Calstabin2 mutants that exhibited significantly more binding to PKA-phosphorylated RyR2 than WT calstabin2 are denoted by an asterisk (P < 0.05).
Binding of Calstabin2 Mutants to PKA-Phosphorylated RyR2.
To evaluate the ability of calstabin2 mutants to bind to PKA-phosphorylated RyR2, RyR2 from CSR membranes were maximally phosphorylated by using exogenous PKA (12). As a negative control, CSR membranes were treated with PKA in the presence of a specific PKA inhibitor (PKI5–24) (data not shown). Calstabin2–D37S and calstabin2–D37V retained the ability to bind to PKA-phosphorylated RyR2 (Fig. 2B). In contrast, binding of WT calstabin2 as well as calstabin2–F36Y and calstabin2–F99Y to PKA-phosphorylated RyR2 was reduced (Fig. 2B).
Binding of Calstabin2 Mutants to RyR2–S2808D Channels.
Calstabin2 binds with much lower affinity to RyR2–S2808D mutant channels that mimic a PKA-phosphorylated RyR2 channel, and RyR2–S2808D mutant channels display gating properties similar to PKA-phosphorylated RyR2 (i.e., increased open probabilities compared with nonphosphorylated RyR2) (12).
We expressed RyR2–S2808D in human embryonic kidney cells, isolated endoplasmic reticulum microsomes, and added calstabin2 separately to control the relative amounts of calstabin2 and RyR2 in the binding experiments. In agreement with the exchange experiments (Fig. 2B), calstabin2–D37V was able to bind RyR2–S2808D, whereas WT and other calstabin2 mutants did not (Fig. 3A).
Fig. 3.

Calstabin2–D37V binds and modulates function of mutant RyR2–S2808D, which mimics chronically PKA-phosphorylated RyR2. (A) Immunoblots showing that calstabin2–D37V bound to RyR2–S2808D (which mimics chronically PKA-phosphorylated RyR2), whereas WT calstabin2 and mutants F36Y and F99Y did not. (B) Single-channel measurements showing that calstabin2–D37V restored normal function to RyR2–S2808D, whereas WT calstabin2, calstabin2–F36Y, and calstabin2–F99Y did not. (C) Bar graphs showing relative reduction in open probability and frequency of openings of RyR2–S2808D channels to which mutant calstabin2 were added as indicated. Calstabin2–D37V significantly reduced open probability and frequency of channel openings compared with WT calstabin2, calstabin2–F36Y, and calstabin2–F99Y.
Single-channel measurements of RyR2–S2808D in the presence of WT calstabin2, calstabin2–F36Y, or calstabin2–F99Y in planar lipid bilayers demonstrated high open probabilities and gating frequencies (Fig. 3 B and C) similar to PKA-phosphorylated RyR2 in the absence of calstabin2. Moreover, these channels exhibited subconductance states, consistent with the lack of binding of calstabin2 to the channel (21). RyR2–S2808D channels incubated with mutant calstabin2–D37V, however, displayed gating properties characterized by significantly lower open probabilities and long closed times, comparable to calstabin2-bound RyR2 (data not shown). Thus, the channel-stabilizing role of calstabin2 with respect to RyR2 channel gating is preserved when residue calstabin2–D37 is mutated to Val. Moreover, the increased channel activity due to PKA phosphorylation of RyR2 may be reversed by enhancing calstabin2 association with the RyR2 complex.
Role of Electrostatic Interactions and Side-Chain Length in the Structural Dynamics Between Calstabin2 and PKA-Phosphorylated RyR.
We next mutated four other Asp residues in calstabin2 (D11G, D41G, D79G, and D100G) to further assess the role of negative charges in calstabin2 that may mediate electrostatic interactions between PKA-phosphorylated RyR2 and calstabin2. These calstabin2 mutants retained the ability to bind to nonphosphorylated RyR2, with the exception of D100G. Binding of calstabin2–D100G to RyR2 may be disrupted because of improper folding of the β-sheet structure of calstabin2 (see Fig. 1). Calstabin2 mutants D11G, D41G, and D79G also bound to PKA-phosphorylated RyR2, suggesting that neutralization of other negative charges in calstabin2 near the hydrophobic binding pocket (Fig. 1) may reduce the electrostatic repulsion between PKA-phosphorylated RyR2–S2808 and calstabin2 (Fig. 4A). However, the fraction of calstabin2–D41G and calstabin2–D79G retained in the PKA-phosphorylated RyR2 membrane pellet was lower than the fraction of calstabin2–D37V (Fig. 4A). Thus, our results suggest that calstabin2–D37 is located near the rim of the binding pocket and may be critical for the decreased binding affinity of calstabin2 to PKA-phosphorylated RyR2.
Fig. 4.

Effects of negative charges and isomerase activity on binding of calstabin2 to RyR2. (A) 35S-labeled WT and mutant calstabin2s with neutralized Asp residues were incubated with PKA-phosphorylated RyR2 (with or without PKA inhibitor as a negative control to inhibit PKA phosphorylation). Pellets containing calstabin2 bound to RyR2 were compared with unbound calstabin2 in the supernatants after size-fractionation by using 15% SDS/PAGE and autoradiography. Neutralization of calstabin2–D37 and calstabin2–D41 enhanced binding to PKA-phosphorylated RyR2, whereas other Asp mutations in calstabin2 exhibited moderately increased or no binding to PKA-phosphorylated RyR2. Densitometry was used to quantify relative binding levels by comparing the fraction of radiolabeled calstabin2 retained in the pellet. Data are shown as the average ± SD of at least three replicate experiments. ∗, P < 0.05. (B) Isomerase activities of calstabin2–D37V, calstabin2–F36Y, and calstabin2–F99Y (diamonds, triangles, and circles, respectively) were significantly decreased compared with WT calstabin2 (squares): kc/Km (s−1mM−1) was 115.4 for WT calstabin2, 16.7 for F36Y, 16.3 for D37V, and 36.2 for F99Y.
Catalytic Efficiencies of Calstabin Mutants Do Not Correlate with Calstabin2 Binding to PKA-Phosphorylated RyR2.
Calstabin2–D37V, calstabin2–F36Y, and calstabin2–F99Y exhibited ≈70–90% reduced rate constants for peptidyl-prolyl isomerization (Fig. 4B), in agreement with previously reported isomerase activities for the corresponding mutations in calstabin1 (9, 20). Because the aforementioned mutants retain the capacity to bind RyR2, we conclude that the binding of calstabin2 to RyR2 does not correlate with the peptidyl-prolyl isomerase activity.
MI Model of Heart Failure Shows Differences in Cardiac Function and Disease Progression for WT Calstabin2 and Calstabin2–D37V Mice.
Calstabin2 was increased 25-fold for WT calstabin2+ mice and 22-fold for calstabin2–D37V+ mice (Fig. 5), and there were no structural differences in the heart in Tg mice at 4 months (data not shown). Cardiac function was assessed by using echocardiography and hemodynamic measurements 6 weeks after MI. Ejection fraction was improved by ≈80% in both Tg mouse strains at 6 weeks after MI, whereas fractional shortening improved by almost 90% (P < 0.01) (Fig. 6A–C). End diastolic volume was ≈50% lower in WT calstabin2+ and calstabin2–D37V+ mice, respectively, suggesting that calstabin2 overexpression in the heart inhibits cardiac dilatation in Tg mice (Fig. 6D). Cardiac contractility was significantly improved in Tg mice: WT mice had an average maximal change in systolic pressure over time (dP/dtmax) of 3,221 ± 178 mmHg per s vs. WT calstabin2+ (4,155 ± 334.3 mmHg per s) and calstabin2–D37V+ (5,536 ± 454 mmHg per s) mice (P < 0.01). There were no significant differences among WT, WT calstabin2+, and calstabin2–D37V+ noninfarcted mice, and they displayed normal contractility at 7,544 ± 112, 8,124 ± 1,288.2, and 7,141 ± 160 mmHg per s, respectively (Fig. 6E). Although some parameters, such as dP/dtmax and end diastolic volume, showed a slight trend toward greater improvement in calstabin2–D37V+ mice when compared with WT calstabin2+ mice, these differences were not significant.
Fig. 5.
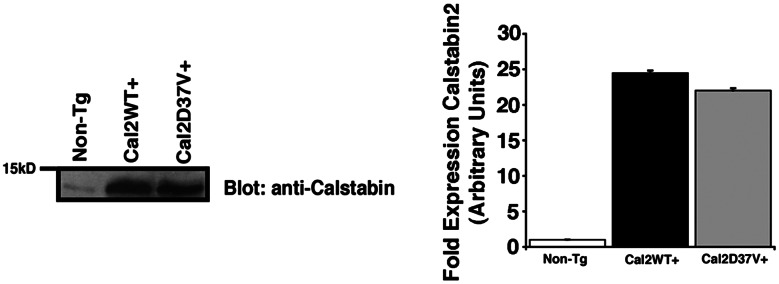
Murine models of cardiac-specific calstabin2 expression. Immunoblots show that WT expressers had ≈25-fold more calstabin2 than native mouse heart, and calstabin2–D37V+ Tg mice expressed ≈22-fold more calstabin2–D37V. The bar graph shows quantification of the immunoblots by using densitometry.
Fig. 6.

Evaluation of mouse cardiac function 6 weeks after MI. (A and B) Ejection fraction (A) and fractional shortening (B) assessed by M-mode echocardiography exhibited improved cardiac function in WT calstabin2+ and calstabin2–D37V+ mice compared with non-Tg mice after MI. (C) Hemodynamic measurements showed significant preservation of cardiac function in Tg mice compared with WT. (D) End diastolic volume was decreased in Tg mice as measured by pressure–volume loop analysis consistent with reduced dilatation after MI. (E) Maximum dP/dt was increased in Tg mice consistent with improved cardiac contractility after MI. Black bars represent infarcted animals, and white bars represent noninfarcted, sham-operated mice. ∗, P < 0.05 (significance compared with non-Tg group).
Increased Calstabin2 Binding to RyR2 Occurs in WT Calstabin2+ and Calstabin2–D37V+ Mice After MI-Induced Heart Failure.
There was decreased binding of calstabin2 to RyR2 and PKA hyperphosphorylation of the channel in WT mice 6 weeks after MI (Fig. 7A). We observed significantly greater calstabin2 binding to PKA-hyperphosphorylated RyR2 in calstabin2D37V+ mice. In addition, we also observed significant binding of WT calstabin2 to PKA-hyperphosphorylated RyR2 in the Tg animals, most likely because of the high levels (≈25-fold overexpression) of WT calstabin2 (Fig. 7B shows that, at high concentrations, WT calstabin2 was able to bind to PKA-treated RyR2 in vitro), which exceeded the Kd for calstabin2 binding to RyR2 (≈300 nM). Levels of PKA phosphorylation of RyR2 were slightly lower in WT calstabin2+ and calstabin2–D37V+ mice, consistent with the global improvements in cardiac function that are known to reduce chronic PKA hyperphosphorylation of RyR2–S2808 (Fig. 7A).
Fig. 7.

Increased expression of calstabin2 in the heart restores calstabin2 binding to PKA-phosphorylated RyR2. (A) Immunoprecipitated RyR2 complexes from heart lysates showed that WT calstabin2+ and calstabin2–D37V+ mice retained calstabin2 binding to the RyR2 complex despite PKA phosphorylation of RyR2. Densitometry was used to quantify the amount of PKA phosphorylation of RyR2–S2808 and the amount of calstabin2 bound to the RyR2 complex relative to non-Tg, noninfarcted mice. (B) Concentration curve of WT calstabin2 vs. calstabin2–D37V binding to PKA-phosphorylated RyR2 showed that, at physiologic levels of calstabin2, calstabin2–D37V exhibited increased binding to PKA-phosphorylated RyR2 compared with WT calstabin2. At high nonphysiological concentrations of WT calstabin2, binding to PKA-phosphorylated RyR2 was observed.
Discussion
In the present study we identified key negatively charged residues in calstabin2 that play a role in the binding of calstabin2 to PKA-hyperphosphorylated RyR2. One possibility is that electrostatic forces between these two proteins could contribute to the changes in binding of calstabin2 upon PKA phosphorylation of RyR2, and neutralization of charged residues on calstabin2 at the interface of these two proteins can allow binding of calstabin2 to PKA-phosphorylated RyR2.
Determinants of Calstabin2 Binding to RyR2.
The findings of this study demonstrate that residue D37 on calstabin2 plays a key role in determining the binding of calstabin2 to PKA-phosphorylated RyR2. Indeed, calstabin2–D37S binds to PKA-phosphorylated RyR2 and restores single-channel properties to those of non-PKA-phosphorylated RyR2 channels (12). From the crystal structure, it is evident that (i) the side chains of residues calstabin2–D41 and calstabin2–D79 have negative charges pointing away from the calstabin2 binding pocket; (ii) calstabin2–D11 and calstabin2–D100 are at the opposite end of the binding pocket; and (iii) calstabin2–D37 has its negatively charged side chain pointing toward the calstabin2 binding pocket (Fig. 1). Calstabin2–D11 is at the bottom of the cone-shaped structure outside the binding pocket, and neutralization of this residue results in minimal binding to PKA-phosphorylated RyR2 compared with the other Asp mutants of calstabin2. Although the negative charge of calstabin2–D41 points away from the binding pocket of calstabin2, it is near calstabin2–D37, and therefore it still may participate in repulsion of the PKA-phosphorylated RyR2–S2808. Residues outside of the calstabin2 binding pocket likely also make contacts with RyR2. Indeed, calstabin2–Q3, calstabin2–R18, and calstabin2–M49 have also been proposed to play a role in bind to RyR2 (22). The existence of multiple interaction sites between calstabin and RyR is likely the reason that binding studies with fragments of RyR have been inconsistent (23, 24).
Catalytic Activity and Binding to RyR2 Are Not Correlated.
Cis-trans peptidyl-prolyl isomerase activity is important for the protein folding and chaperone functions of immunophilins. Previous studies with calstabin1 have shown that some mutants, such as calstabin1–S39A, lose the ability to bind peptidomimetics, such as FK-506, but still maintain isomerase activity (25). Conversely, calstabin1–F36Y binds to FK-506 despite reduced peptide isomerase activity (26). We now show that the isomerase-deficient mutant calstabin2–D37V retains the ability to modulate RyR2 gating, indicating that the functional role of calstabin2 in the RyR2 complex can be dissociated from its enzymatic activity as an isomerase.
The Potential for the Calstabin2–RyR2 Interaction as a Therapeutic Target.
In Tg mice overexpressing WT calstabin2 or calstabin2–D37V, progression of ischemia-induced heart failure was less severe. These data support recently published observations that a new class of drugs known as “calcium channel stabilizers” have protective effects against heart failure (27, 28). The prototype of this new class of drugs, known as JTV-519, is a 1,4-benzothiazapine derivative that enhances calstabin2 binding to RyR2 even when RyR2 is PKA-hyperphosphorylated (7, 27, 28). Treatment of WT but not calstabin2−/− mice after MI with JTV519 prevented the development of severe heart failure (28), confirming that calstabin2 is required for the molecular action of this drug. In addition, JTV-519 was also shown to provide protection from cardiac arrhythmias by the same mechanism (7). Our finding that increased levels of both WT calstabin2 and calstabin2–D37V preserve cardiac function after MI indicates the importance of stoichiometry between calstabin2 and RyR2. In the WT mice the levels of calstabin2 are not sufficient to allow binding to the PKA-hyperphosphorylated RyR2. This deficiency is overcome by expression of either WT calstabin2 or calstabin2–D37V in the heart. We had hypothesized that the calstabin2–D37V Tg mice would exhibit greater protection against cardiac dysfunction after MI. However, it was not possible in the present study to assess differences between overexpression of WT calstabin2 vs. calstabin2–D37V, because the levels of the WT overexpression were high enough to overcome the 2- to 3-fold decrease in binding affinity of calstabin2 to PKA-phosphorylated RyR2. Moreover, the fact that cardiac function was not fully normalized despite relative preservation of function in calstabin2-overexpressing mice after MI is consistent with the presence of large regions of infarcted muscle in these animals. Thus, the improved function in the Tg animals is due to increased performance of the remaining noninfarcted muscle.
In the present study the generation of mutant calstabin2 lacking key negatively charged residues has allowed us to probe potential interactions between negative charges on calstabin2 and negative charges on PKA-phosphorylated RyR2. This approach led to the identification of key negative charges on calstabin2, including Asp-37, that may be involved in reducing the affinity between calstabin2 and PKA-phosphorylated RyR2 during heart failure.
Materials and Methods
Expression of WT and Mutant Calstabins and RyR2 and Calstabin Binding and Isomerase Assays.
WT and mutant calstabin2s and RyR2–S2808D were generated as described in refs. 12 and 29. CSR membranes were prepared as described in refs. 11 and 21. Binding of 35S-labeled calstabins to PKA-phosphorylated RyR2 was performed as described in ref. 12. Calstabin2 cloned into pGEX 4T-2 was expressed in BL21 cells with a GST tag, purified according to the manufacturer’s protocol by using glutathione-Sepharose beads (Amersham Pharmacia Biosciences), and eluted with 10 mM glutathione, and the GST tag was cleaved with thrombin (Calbiochem). Purified calstabin2 was isolated from GST and thrombin by FPLC by using a Sephadex 75 column. The isomerase assay was performed as described in ref. 30.
Immunoprecipitation from Cardiac Lysates and Immunoblotting.
Immunoprecipitations and immunoblotting were performed as described in ref. 31. Membranes were incubated for 1–2 h at room temperature with primary antibodies anti-calstabin (1:1,000) (10), anti-RyR (5029; 1:3,000) (10), or anti-phosphoRyR2-pSer2809 (1:5,000) (31).
Single-Channel Recordings.
Single-channel recordings of recombinant RyR2 incorporated in black-lipid membranes were acquired under voltage-clamp conditions at 0 mV as described in ref. 11. Unless otherwise indicated, single-channel recordings were made in the presence of 150 nM [Ca2+] in the cis (cytoplasmic) chamber. Concentration of free Ca2+ in the cis chamber was adjusted by adding CaCl2 from 20 mM stock and was calculated with the winmaxc program (version 2.40; www.stanford.edu/∼cpatton/maxc.html) (32). Ryanodine (5 μM) was added to the cis chamber to confirm that observed channel currents were modified by ryanodine and hence were due to RyR2. Data were analyzed from digitized current recordings by using fetchan software (Axon Instruments). All data are expressed as mean ± SEM. The unpaired Student t test was used for statistical comparison of mean values between experiments, and analyses of variance were used for multiple comparisons. A P value of <0.05 was considered statistically significant.
Generation of α-MHC–Calstabin2 Tg Mice.
WT and mutant calstabin2 were cloned into the pRSET-A vector (Invitrogen) by PCR amplification of the coding sequence by using PCR primers with a BamH1 site engineered at the 5′ end and XhoI at the 3′ end and the previously constructed pCMV-calstabin2 plasmid (12) as the template. A vector containing the α-MHC promoter (cardiac muscle promoter) pCMCP was used to generate constructs for making Tg mice (33). Digestion of the plasmid constructs with SacII and SalI released a 5.8-kb fragment containing the promoter and cDNA for the transgene. The fragment was purified by CsCl centrifugation and microinjected into fertilized mouse eggs (F1[C57BL/6 × CBA/J] × F1[C57BL/6 × CBA/J] (34).
Mouse Genotyping.
Mice were screened for Tg insertion by PCR by using a 5′ primer with the MHC promoter and a 3′ primer at the end of the calstabin2 coding sequence: MHC exon (forward), 5′-CCA CAT TCT TCA GGA TTC TCT GAA AAG TTA AC-3′; F12.6 (reverse), 5′-CAC TCT AG TTG AGC AGC TCC ACG TCA AAG-3′.
Two positive strains from each calstabin2 transgene were bred by crossing with WT F1 mice to generate heterozygous Tg mice. Transgene expression was confirmed by RT-PCR by using the screening primers. Protein levels in mouse heart lysates were quantified by immunoblotting and densitometry, and the lowest expressers were used for the study. All mice were handled and studied according to protocols approved by the Institutional Animal Care and Use Committee of Columbia University.
Myocardial Infarcts, Echocardiography, and Histology.
Three- to 4-month-old mice were subjected to MI by left anterior descending artery ligation; histology and echocardiography were as reported in ref. 28. Mice with infarct sizes 20% or greater were entered into the study.
Acknowledgments
We thank Arthur Karlin and Steven Marx for critical reading of the manuscript and Jeanine D’Armiento for help with generating Tg mice. This work was supported by grants to A.R.M. from the National Heart, Lung, and Blood Institute.
Abbreviations
- Tg
transgenic
- MI
myocardial infarction
- RyR
ryanodine receptor
- SR
sarcoplasmic reticulum
- CSR
cardiac SR
- PKA
protein kinase A.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Hunt S. A., Baker D. W., Chin M. H., Cinquegrani M. P., Feldman A. M., Francis G. S., Ganiats T. G., Goldstein S., Gregoratos G., Jessup M. L., et al. J. Am. Coll. Cardiol. 2001;38:2101–2113. doi: 10.1016/s0735-1097(01)01683-7. [DOI] [PubMed] [Google Scholar]
- 2.Bristow M. R., Hershberger R. E., Port J. D., Gilbert E. M., Sandoval A., Rasmussen R., Cates A. E., Feldman A. M. Circulation. 1990;82:I12–I25. [PubMed] [Google Scholar]
- 3.Wehrens X. H., Lehnart S. E., Marks A. R. Annu. Rev. Physiol. 2005;67:69–98. doi: 10.1146/annurev.physiol.67.040403.114521. [DOI] [PubMed] [Google Scholar]
- 4.Marks A. R., Marx S. O., Reiken S. Trends Cardiovasc. Med. 2002;12:166–170. doi: 10.1016/s1050-1738(02)00156-1. [DOI] [PubMed] [Google Scholar]
- 5.Marks A. R., Tempst P., Hwang K. S., Taubman M. B., Inui M., Chadwick C., Fleischer S., Nadal-Ginard B. Proc. Natl. Acad. Sci. USA. 1989;86:8683–8687. doi: 10.1073/pnas.86.22.8683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brillantes A. B., Ondrias K., Scott A., Kobrinsky E., Ondriasova E., Moschella M. C., Jayaraman T., Landers M., Ehrlich B. E., Marks A. R. Cell. 1994;77:513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- 7.Wehrens X. H., Lehnart S. E., Reiken S. R., Deng S. X., Vest J. A., Cervantes D., Coromilas J., Landry D. W., Marks A. R. Science. 2004;304:292–296. doi: 10.1126/science.1094301. [DOI] [PubMed] [Google Scholar]
- 8.Lam E., Martin M. M., Timerman A. P., Sabers C., Fleischer S., Lukas T., Abraham R. T., O’Keefe S. J., O’Neill E. A., Wiederrecht G. J. J. Biol. Chem. 1995;270:26511–26522. doi: 10.1074/jbc.270.44.26511. [DOI] [PubMed] [Google Scholar]
- 9.Timerman A. P., Wiederrecht G., Marcy A., Fleischer S. J. Biol. Chem. 1995;270:2451–2459. doi: 10.1074/jbc.270.6.2451. [DOI] [PubMed] [Google Scholar]
- 10.Jayaraman T., Brillantes A.-M. B., Timerman A. P., Erdjument-Bromage H., Fleischer S., Tempst P., Marks A. R. J. Biol. Chem. 1992;267:9474–9477. [PubMed] [Google Scholar]
- 11.Marx S. O., Reiken S., Hisamatsu Y., Jayaraman T., Burkhoff D., Rosemblit N., Marks A. R. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 12.Wehrens X. H., Lehnart S. E., Huang F., Vest J. A., Reiken S. R., Mohler P. J., Sun J., Guatimosim S., Song L. S., Rosemblit N., et al. Cell. 2003;113:829–840. doi: 10.1016/s0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 13.Jiang D., Xiao B., Yang D., Wang R., Choi P., Zhang L., Cheng H., Chen S. R. Proc. Natl. Acad. Sci. USA. 2004;101:13062–13067. doi: 10.1073/pnas.0402388101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao B., Sutherland C., Walsh M. P., Chen S. R. Circ. Res. 2004;94:487–495. doi: 10.1161/01.RES.0000115945.89741.22. [DOI] [PubMed] [Google Scholar]
- 15.Stange M., Xu L., Balshaw D., Yamaguchi N., Meissner G. J. Biol. Chem. 2003;278:51693–51702. doi: 10.1074/jbc.M310406200. [DOI] [PubMed] [Google Scholar]
- 16.Deivanayagam C. C., Carson M., Thotakura A., Narayana S. V., Chodavarapu R. S. Acta Crystallogr. D Biol. Crystallogr. 2000;56:266–271. doi: 10.1107/s0907444999016571. [DOI] [PubMed] [Google Scholar]
- 17.Huse M., Chen Y. G., Massague J., Kuriyan J. Cell. 1999;96:425–436. doi: 10.1016/s0092-8674(00)80555-3. [DOI] [PubMed] [Google Scholar]
- 18.Futer O., DeCenzo M. T., Aldape R. A., Livingston D. J. J. Biol. Chem. 1995;270:18935–18940. doi: 10.1074/jbc.270.32.18935. [DOI] [PubMed] [Google Scholar]
- 19.Xin H. B., Rogers K., Qi Y., Kanematsu T., Fleischer S. J. Biol. Chem. 1999;274:15315–15319. doi: 10.1074/jbc.274.22.15315. [DOI] [PubMed] [Google Scholar]
- 20.Tradler T., Stoller G., Rucknagel K. P., Schierhorn A., Rahfeld J. U., Fischer G. FEBS Lett. 1997;407:184–190. doi: 10.1016/s0014-5793(97)00345-1. [DOI] [PubMed] [Google Scholar]
- 21.Kaftan E., Marks A. R., Ehrlich B. E. Circ. Res. 1996;78:990–997. doi: 10.1161/01.res.78.6.990. [DOI] [PubMed] [Google Scholar]
- 22.Lee E. H., Rho S. H., Kwon S. J., Eom S. H., Allen P. D., Kim do H. J. Biol. Chem. 2004;279:26481–26488. doi: 10.1074/jbc.M309574200. [DOI] [PubMed] [Google Scholar]
- 23.Masumiya H., Wang R., Zhang J., Xiao B., Chen S. R. J. Biol. Chem. 2003;278:3786–3792. doi: 10.1074/jbc.M210962200. [DOI] [PubMed] [Google Scholar]
- 24.Zissimopoulos S., Lai F. A. J. Biol. Chem. 2004;280:5475–5485. doi: 10.1074/jbc.M412954200. [DOI] [PubMed] [Google Scholar]
- 25.Park S. T., Aldape R. A., Futer O., DeCenzo M. T., Livingston D. J. J. Biol. Chem. 1992;267:3316–3324. [PubMed] [Google Scholar]
- 26.Wiederrecht G., Hung S., Chan H., Marcy A., Martin M., Calaycay J., Boulton D., Sigal N., Kincaid R., Siekierka J. J. Biol. Chem. 1992;267:21753–21760. [PubMed] [Google Scholar]
- 27.Kohno M., Yano M., Kobayashi S., Doi M., Oda T., Tokuhisa T., Okuda S., Ohkusa T., Matsuzaki M. Am. J. Physiol. 2003;284:H1035–H1042. doi: 10.1152/ajpheart.00722.2002. [DOI] [PubMed] [Google Scholar]
- 28.Wehrens X. H., Lehnart S. E., Reiken S., van der Nagel R., Morales R., Sun J., Cheng Z., Deng S. X., de Windt L. J., Landry D. W., Marks A. R. Proc. Natl. Acad. Sci. USA. 2005;102:9607–9612. doi: 10.1073/pnas.0500353102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gaburjakova M., Gaburjakova J., Reiken S., Huang F., Marx S. O., Rosemblit N., Marks A. R. J. Biol. Chem. 2001;276:16931–16935. doi: 10.1074/jbc.M100856200. [DOI] [PubMed] [Google Scholar]
- 30.Siekierka J. J., Hung S. H. Y., Poe M., Lin C. S., Sigal N. H. Nature. 1989;341:755–757. doi: 10.1038/341755a0. [DOI] [PubMed] [Google Scholar]
- 31.Reiken S., Lacampagne A., Zhou H., Kherani A., Lehnart S. E., Ward C., Huang F., Gaburjakova M., Gaburjakova J., Rosemblit N., et al. J. Cell Biol. 2003;160:919–928. doi: 10.1083/jcb.200211012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bers D. M., Patton C. W., Nuccitelli R. Methods Cell Biol. 1994;40:3–29. doi: 10.1016/s0091-679x(08)61108-5. [DOI] [PubMed] [Google Scholar]
- 33.Kim H. E., Dalal S. S., Young E., Legato M. J., Weisfeldt M. L., D’Armiento J. J. Clin. Invest. 2000;106:857–866. doi: 10.1172/JCI8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hogan B., Costantini F., Lacy E. Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Lab. Press; 1986. [Google Scholar]