

Abstract
Withdrawal from chronic cocaine reduces extracellular glutamate levels in the nucleus accumbens by decreasing cystine/glutamate exchange (xc-). Activating xc- with N-acetylcysteine restores extracellular glutamate and prevents cocaine-induced drug seeking. It was hypothesized that the activation of xc- prevents drug seeking by increasing glutamatergic tone on presynaptic group II metabotropic glutamate receptors (mGluR2/3) and thereby inhibiting excitatory transmission. In the first experiment, the capacity of glutamate derived from xc- to regulate excitatory transmission via mGluR2/3 was determined. Physiological levels of cystine (100-300 nm) were restored to acute tissue slices from the nucleus accumbens or prefrontal cortex. Cystine increased glutamate efflux and decreased miniature EPSC (mEPSC) and spontaneous EPSC (sEPSC) frequency as well as evoked EPSC amplitude. These effects of cystine were presynaptic, because there was no change in mEPSC or sEPSC amplitude, and an increase in the evoked EPSC paired-pulse facilitation ratio. The cystine-induced reduction in EPSCs was reversed by blocking either xc- or mGluR2/3. In the second experiment, blocking mGluR2/3 prevented the ability of N-acetylcystine to inhibit the reinstatement of drug seeking in rats trained to self-administer cocaine. These data demonstrate that nonsynaptic glutamate derived from xc- modulates synaptic glutamate release and thereby regulates cocaine-induced drug seeking.
Keywords: cystine/glutamate exchange, mGluR, cocaine, accumbens, EPSC, glutamate
Introduction
Plasticity in excitatory transmission in the nucleus accumbens (NA) is induced by chronic administration of drugs of abuse (Thomas et al., 2001; Kalivas et al., 2005). One important afferent to the NA in this regard is from the prefrontal cortex (PFC). Changes in PFC-NA glutamate transmission occur after chronic cocaine administration, including reduced basal levels of extracellular glutamate (Pierce et al., 1996; Hotsenpiller et al., 2001; McFarland et al., 2003). In the NA, the basal levels of extracellular glutamate are maintained primarily by the exchange of extracellular cystine for intracellular glutamate via system cystine/glutamate exchange (xc-) (Baker et al., 2002). This heteroexchanger belongs to a superfamily of glycoprotein-associated amino acid transporters, catalyzes the Na+-independent exchange of extracellular cystine for intracellular glutamate in a 1:1 stoichiometric ratio (McBean, 2002), and is functionally expressed in the brain as a heterodimer (Sato et al., 2002).
The reduction in extracellular glutamate by withdrawal from cocaine is a result of compromised xc- (Baker et al., 2003). Restoring extracellular glutamate with systemic administration of cysteine prodrugs prevented the reinstatement of cocaine seeking. Group II metabotropic glutamate receptor (mGluR2/3) agonists also inhibit the reinstatement of cocaine- or heroin-seeking behavior (Baptista et al., 2004; Bossert et al., 2004). Stimulating mGluR2/3 is a well established method for reducing glutamate release, EPSCs, and inducing long-term depression (LTD) (Manzoni et al., 1997; Dietrich et al., 2002; Otani et al., 2002; Robbe et al., 2002), and in vivo administration of mGluR2/3 agonists has potent behavioral effects (Kenny and Markou, 2004). Therefore, a mechanism that may account for the ameliorative effect of cysteine prodrugs on cocaine seeking is that the increase in extracellular glutamate produced by activating xc- restores tone on presynaptic mGluR2/3 and thereby reduces synaptic glutamate release.
Two experiments were conducted to examine the hypothesis that glutamate derived from xc- stimulates inhibitory presynaptic mGluR2/3, thereby reducing synaptic glutamate release and preventing cocaine-primed drug seeking. In the first experiment, acute tissue slices from the NA or PFC were used to determine whether glutamate originating from xc- inhibits miniature EPSCs (mEPSCs), spontaneous EPSCs (sEPSCs), or evoked EPSCs by stimulating mGluR2/3. In the second experiment, cocaine-primed reinstatement of drug seeking was used to determine whether blocking mGluR2/3 prevents the capacity of the cysteine prodrug N-acetylcysteine to inhibit drug seeking.
Materials and Methods
Animals. Before initiation of these studies, approval was received by the Institutional Animal Care and Use Committee at the Medical University of South Carolina (MUSC). Our study conforms to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All of the rats (Sprague Dawley-derived albinos; Harlan Laboratories, Indianapolis, IN) used in the electrophysiology experiments were postnatal day 13 (P13) at arrival and were delivered to the Animal Care Facility at MUSC as a litter with the dam. Male rats used in the reinstatement studies arrived weighing 250 g and were housed two per cage. Rats in both sets of experiments were maintained on a 12 h light/dark cycle (lights on at 7:00 A.M.).
Slices and recordings. All of the physiology experiments were conducted using acute, coronal brain slices containing either the PFC or the NA. Rats (P14-P23) were anesthetized with chlorohydrate (400 mg/kg, i.p.) and decapitated. Brains were dissected and immersed in cold (4°C) oxygenated artificial CSF (aCSF) composed of the following (in mm): 200 sucrose, 1.9 KCl, 33 Na2HCO3, 6 MgCl2, 0.5 CaCl2, 10 glucose, and 0.4 ascorbic acid. Tissue slices (300 μm) were obtained with a vibratome and incubated for 1 h at room temperature in aCSF containing the following (in mm): 126 NaCl, 2.5 KCl, 25 NaHCO3, 10 glucose, 4 MgCl2, and 1 CaCl2. Slices were immersed in a recording solution composed of the following (in mm): 126 NaCl, 3 KCl, 26 NaHCO3, 1 MgCl2, 2.3 CaCl2, and 10 glucose, maintained at 28-32°C at a perfusion rate of 1-3 ml/min and viewed by differential interference contrast (DIC) optics. The core compartment of the NA was localized using primarily the anterior commissure as a landmark. The prelimbic cortex of the PFC is flanked by the corpus callosum in coronal sections and the layer V pyramidal neurons identified under DIC optics. Borosilicate pipettes were filled with the following (in mm): 135 CsCl, 2 MgCl2, 10 HEPES, 1 EGTA, 4 NaCl, 2 NaATP, and 0.3 Tris-GTP. N-(2,6-dimethylphenylcarbamoylmethyl) triethylammonium chloride (2 mm) was added to pipettes during the evoked and paired-pulse experiments to block voltage-sensitive Na+ channels generating action potentials. Pipettes were connected to the headstage of a HEKA EPC10 amplifier (ALA Scientific, Westbury, NY), with Ag/AgCl wire. An Ag/AgCl reference wire or pellet was placed in the bath and, by using offset, Vm shifts were corrected. Signals were digitized using Pulse software (ALA Scientific). Voltage-clamp recordings were obtained in continuous single-electrode voltage-clamp mode and filtered at 2.9 kHz. Series resistance and capacitance was compensated automatically by Pulse and optimized manually. Access resistance was monitored continuously, and a 15% change was deemed acceptable.
mEPSCs were collected in the presence of TTX (1 μm; Alomone Labs, Jerusalem, Israel) and bicuculline (10 μm; Sigma, St. Louis, MO), and sEPSCs were collected in the presence of bicuculline (10 μm) alone. Data were collected for 60 s every 2 min during either baseline or test periods and analyzed using Minianalysis software (Synaptosoft, Decatur, GA). In each experiment, l-cystine (Sigma), the xc- blocker (S)-4-carboxyphenylglycine (CPG; Tocris Cookson, Ellsville, MO), the mGluR2/3 antagonist (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid (LY341495; Tocris Cookson), and/or the group I mGluR antagonist RS-1-aminoindan-1,5-dicarboxylic acid (AIDA; Tocris Cookson) were perfused in the bath. A bipolar-stimulating electrode (World Precision Instruments, Sarasota, FL) was placed medial to the lateral ventricle in the prelimbic cortex, and responses were recorded in the NA. Approximately 20 min after break in, evoked EPSC data were collected with 10 sweeps at 20 s intervals during baseline and following 10 min of drug exposure. The pulse was 0.2 ms, and its intensity was set to yield a stable response of 20-150 pA. For paired-pulse analysis, a set of two pulses was delivered with an interpulse interval of 50 ms, and the pairs of pulses were applied every 20 s. The amplitude of each response was compared during baseline and cystine.
Slice dialysis. The active membrane of concentric dialysis probes was between 2 and 3 mm in length and ∼0.22 mm in diameter (McFarland et al., 2003). Probes were inserted into the slice, and dialysis buffer was advanced through the probe at a rate of 2 μl/min via a syringe pump. The concentration of glutamate was determined using HPLC with fluorometric detection (McFarland et al., 2003).
Surgery. Before surgery, the animals were given food and water ad libitum. Rats were anesthetized with ketamine HCl (87.5 mg/kg Ketaset; Fort Dodge Animal Health, Fort Dodge, IA) and xylazine (5 mg/kg Rompum; Bayer, Etobicoke, Ontario, Canada) and implanted with indwelling jugular catheters. Threaded guide cannula (C313G; Plastics One, Roanoke, VA) were attached to SILASTIC tubing inserted into the right jugular vein (2.7-3.0 cm) and then run subcutaneously and externalized via a 3 mm biopsy hole. The catheters were flushed daily with 0.2 ml of heparin (100 IU/ml) and cefazolin antibiotic (100 mg/ml) in sterile saline vehicle to help protect against infection and maintain patency. After surgery, the animals were individually housed for a surgical recovery period of 1 week. After the recovery period, food was limited to 20 g/d, and water was provided ad libitum.
Self-administration, extinction, and reinstatement training. Behavioral training began at the end of the 7 d surgical recovery period and was conducted as described previously (McFarland et al., 2003). Briefly, after food training, the subjects began cocaine self-administration training. Correct lever presses resulted in an infusion of cocaine (0.25 mg/kg in 0.05 ml over 3 s) and also illumination of a stimulus light over the lever. After the 20 s timeout, the light was extinguished, and the first press on the active lever again resulted in cocaine delivery. Sessions lasted for 2 h or until 200 reinforcements were acquired. Rats remained in self-administration training until they met a criterion that the average responding over three consecutive sessions varied by <10%.
During extinction sessions, responding on the active lever resulted in illumination of the stimulus light for 20 s but no infusion of cocaine. Sessions lasted for 1 h, followed by a 1 h break (in-house lights were off, and the levers were retracted) and then another 1 h extinction session. This extinction training protocol continued until subjects met the criterion (<15% of self-administration responding) for three consecutive days.
Reinstatement of drug-seeking behavior was assessed following a challenge injection of cocaine (10 mg/kg, i.p.) or saline (0.9%; 1 ml/kg). Four hours before systemic cocaine or saline injection, some subjects received injections of N-acetylcysteine (60 mg/kg, s.c.) or saline vehicle. Rats in select groups received injections of LY341495 (1 mg/kg, i.p.) or saline vehicle. Reinstatement testing lasted for 2 h. During this 2 h period, active lever presses were counted but resulted in saline, not cocaine, delivery.
Statistical analysis. Analysis of mEPSC and sEPSC was performed offline using Minianalysis software (Synaptosoft). The root mean square (RMS) of the noise was computed for each set of data. Data were analyzed manually, and events two SDs above the RMS were used in the analysis. Each detected event was visually inspected to prevent the inclusion of false data. The mean sEPSC and evoked data were compared using an ANOVA with repeated measures and least significant difference post hoc test using Statistica version 5.5 (Statsoft, Tulsa, OK). Kolmogorov-Smirnov tests were used for comparing cumulative probability histograms using Minianalysis software (Synaptosoft). Paired-pulse data were compared using a paired t test (p < 0.05). Microdialysis data were normalized to percentage change from the average of the last three baseline samples and evaluated using a two-way ANOVA with repeated-measures over time.
Results
Cystine increases extracellular glutamate
Restoring physiological levels of extracellular cystine (100-300 nm) (Baker et al., 2003) to PFC or NA tissue slices promoted glutamate efflux as measured by a microdialysis probe inserted into the slice (Fig. 1A,C). It is unlikely that the rise in extracellular glutamate was attributable to cell death, because stable glutamate levels were observed throughout the experiment when the slices were bathed in aCSF (Fig. 1A). Interestingly, when the concentration of extracellular cystine was raised outside of the physiological range to 1 or 10 μm, no increase in extracellular glutamate was measured (Fig. 1A).
Figure 1.
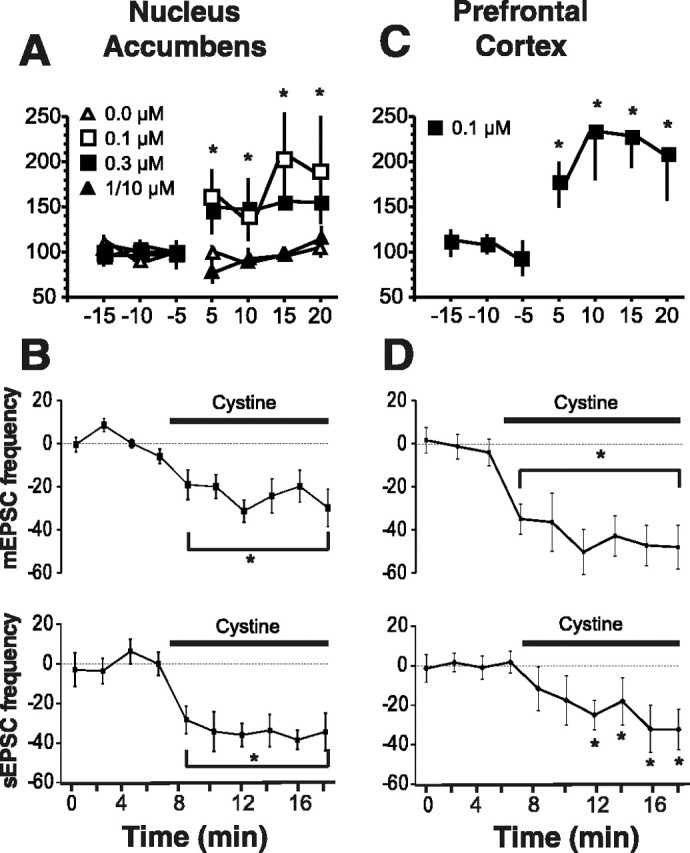
Cystine regulates glutamate levels and excitatory transmission. A, Cystine increased extracellular glutamate in NA slices, as revealed by microdialysis (n = 5 per dose). The 1/10 μm dose is pooled data for perfusion of 1 μm (n = 2) and 10 μm (n = 3). Data were normalized to the average of the three baseline samples and are shown as mean ± SEM percentage change. Basal levels of glutamate (picomoles per sample) are as follows: 0 = 0.176 ± 0.023; 0.1 = 0.134 ± 0.016; 0.3 = 0.125 ± 0.019; 1/10 = 0.136 ± 0.040. B, Cystine (0.3 μm) reduced mEPSC (n = 11; 0.9 ± 0.2 Hz) and sEPSC (n = 9 baseline; 0.8 ± 0.2 Hz) frequency in medium spiny cells (Vm = -80 mV) in the NA. Data are presented as a percentage change from baseline ± SEM. C, Cystine (0.1 μm) increased extracellular glutamate in prefrontal cortex slices (n = 6). Data were normalized to percentage change from baseline; the basal level was 0.084 ± 0.015. D, Cystine (0.1 μm) reduced mEPSC (n = 12, 2.3 ± 0.6 Hz) and sEPSC (n = 8; 4.9 ± 0.8 Hz) frequency in PFC slices. *p < 0.05, comparing cystine treatment to the average baseline value using a one-way ANOVA with repeated measures and least significant difference post hoc comparison.
Cystine decreases EPSCs
To assess the effect of cystine-induced elevations in extracellular glutamate on vesicular excitatory transmission, the frequency of mEPSCs was measured in the NA and PFC. Cystine caused a significant reduction in the frequency of mEPSCs (Fig. 1B,D) without affecting mEPSC amplitude (PFC baseline, 13 ± 1.2 pA; cystine, 12 ± 1.2 pA; NA baseline, 17 ± 1.7 pA; cystine 16 ± 1.7 pA).
The EPSC evoked by stimulating glutamatergic afferents to the NA (Fig. 2A) and the frequency of sEPSC (Fig. 2D) were both reduced by cystine. In parallel with the dialysis data (Fig. 1A), the effect of cystine was biphasic with respect to dose, with 0.1 and 0.3 μm producing significant effects but 0.01 and 1.0 μm without effect (Fig. 2B,E). When cells exposed to 0.3 μm cystine were washed with control buffer, the amplitude of the EPSC and frequency of the sEPSC remained below baseline (Fig. 2A,D). Cumulative probability plots demonstrate the reduction in sEPSC frequency by cystine (0.3 μm) occurred without a change in sEPSC amplitude (Fig. 2F,G). Thus, the effect of cystine on excitatory transmission appears to be via a presynaptic mechanism, because cystine changed mEPSC and sEPSC frequency independent of amplitude. Also, consistent with a presynaptic action, cystine produced a significant increase in the paired-pulse facilitation ratio (Fig. 2C).
Figure 2.

Cystine inhibition of excitatory transmission is concentration dependent. A, Evoked EPSCs were normalized to the average of baseline (2-10 min) and are shown as mean percentage change. Legend and n at each dose are shown in B. Cells in the 0.3 μm group were exposed to a wash period (22-30 min). Representative traces are from a single cell during baseline, cystine, and wash. Calibration: 60 pA, 10 ms. B, Data presented in A are presented as mean ± SEM percentage change from baseline (samples 8-10) by cystine (samples 18-20). Symbols correspond to A, and n at each dose is shown below the symbol. C, Cells exposed to 0.3 μm cystine (Cys) exhibit enhanced paired-pulse facilitation. The data are expressed as a percentage change in the ratio of the second response to the first. D, Normalized sEPSC during baseline and cystine. See E for legend and n at each concentration of cystine. A representative 1 s current trace from a single cell after 10 min of baseline, cystine (0.3 μm), and wash is shown. Calibration: 20 pA, 200 ms. E, Data in D presented as mean ± SEM percentage change from baseline (samples 8-10) by cystine (samples 18-20). Symbols corresponding to D and n at each dose are shown below the symbol. F, The distribution of time intervals between successive sEPSC during baseline (10 min) and cystine (20 min). G, Distribution of sEPSC amplitude during baseline (10 min) and cystine (20 min). The asterisk denotes a significant difference from baseline (p ≤ 0.05).
Cystine/glutamate exchange regulates synaptic transmission by activating mGluR2/3
To determine the underlying mechanism(s) responsible for the cystine-induced reduction in excitatory transmission, an inhibitor of xc- (CPG; 1 μm) was bath applied to the slices. The experiment in Figure 3, A and B, replicated the reduction in EPSC amplitude and sEPSC frequency by cystine (0.3 μm) in NA tissue slices. Although CPG alone did not influence the sEPSC frequency or the magnitude of evoked EPSCs in the NA, it blocked the capacity of cystine to reduce sEPSC frequency and evoked EPSC amplitude (Fig. 3C,D). At higher concentrations, CPG also inhibits group I mGluRs (Schoepp et al., 1999). Figure 3, E and F, shows that although coadministration of the selective group I mGluR antagonist AIDA (1.0 mm) produced a trend toward a reduction in current, it did not inhibit the capacity of cystine (0.3 μm) to reduce sEPSCs or EPSCs. Extracellular glutamate derived from xc- might regulate synaptic glutamate release by stimulating presynaptic mGluR2/3 (Conn and Pin, 1997; Robbe et al., 2002). Although bath application of the mGluR2/3 antagonist LY341495 (300 nm) alone did not alter sEPSCs or EPSCs, it prevented the effects of cystine (0.3 μm) (Fig. 3G,H).
Figure 3.
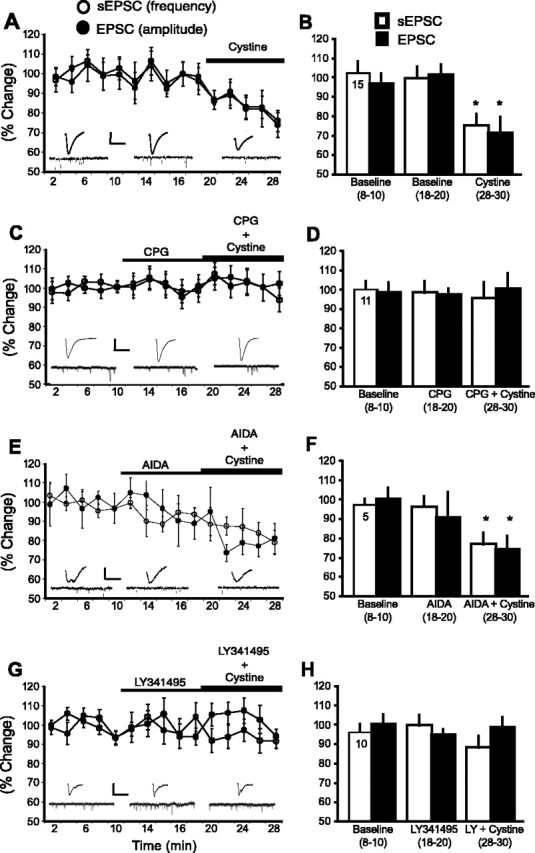
xc- and mGluR2/3 regulate excitatory transmission in the NA. A, C, E, G, Data were normalized to the average of baseline and are shown as mean ± SEM percentage change. Data in B, D, F, and H were collapsed during 8-10, 18-20, and 28-30 min of each experiment and compared with baseline (n is shown in the first bar). A, B, Cystine (0.3 μm) reduced sEPSC frequency and evoked EPSC amplitude after 20 min of baseline. Evoked EPSC traces from a single cell (calibration: 300 pA, 10 ms) and 1 s current traces from the same cell during aforementioned treatments are shown. C, D, The xc- antagonist CPG (1 μm) prevented cystine (0.3 μm) from reducing sEPSC frequency and EPSC amplitude. Evoked EPSC traces from a single cell (calibration: 200 pA, 10 ms) and 1 s current traces from the same cell during aforementioned treatments are shown. E, F, Cystine (0.3 μm) reduces sEPSC frequency and EPSC amplitude in the presence of the group I metabotropic glutamate receptor antagonist AIDA (1 mm). Evoked EPSC traces from a single cell (calibration: 100 pA, 10 ms) and 1 s current traces from the same cell during aforementioned treatments are shown. G, H, The mGluR2/3 antagonist LY341495 (LY) (0.3 μm) prevented cystine (0.3 μm) from reducing sEPSC frequency and EPSC amplitude. Evoked EPSC traces from a single cell (calibration: 100 pA, 10 ms) and 1 s current traces from the same cell during aforementioned treatments are shown. *p < 0.05, comparing cystine treatment with the average baseline value using a one-way ANOVA with repeated measures and least significant difference post hoc comparison.
mGluR2/3 is essential for activation of cystine/glutamate exchange to prevent cocaine-primed reinstatement
To determine whether the stimulation of mGluR2/3 by glutamate derived from xc- contributed to the capacity of the procysteine drug N-acetylcystine to inhibit cocaine-primed reinstatement (Baker et al., 2003), animals were trained to self-administer cocaine, and their behavior was extinguished to criterion. As expected, a cocaine injection (10 mg/kg, i.p.) reinstated active lever pressing, and this effect was blocked by pretreatment with N-acetylcysteine (60 mg/kg, s.c.) (Fig. 4). Coadministration of the mGluR2/3 antagonist LY341495 (1 mg/kg, i.p.) together with N-acetylcysteine prevented the blockade of cocaine-induced reinstatement by N-acetylcysteine (Fig. 4). Importantly, LY341495 alone did not alter the reinstatement of lever pressing by a cocaine-priming injection.
Figure 4.

Blocking mGluR2/3 prevents the capacity of N-acetylcysteine to inhibit cocaine-induced reinstatement. Each rat was given injections of either N-acetylcysteine (60 mg/kg, s.c.) or saline 4 h before a cocaine injection (10 mg/kg, i.p.). In addition, rats were given injections once every hour after the injection of N-acetylcysteine or saline with either saline or the mGluR2/3 antagonist LY341495 (1 mg/kg, i.p.) before the cocaine priming injection. The data are shown as the mean ± SEM total number of active lever presses during the 120 min after a cocaine-priming injection (n > 5 for each treatment). *p < 0.05, compared with control (first bar on the left) using a one-way ANOVA followed by a Dunnett's test.
Discussion
The elevation of extracellular glutamate produced in NA tissue slices by restoring physiological levels of extracellular cystine was shown to inhibit excitatory synaptic activity by stimulating mGluR2/3. Moreover, xc- regulation of synaptically released glutamate was shown to be functionally relevant in vivo using the reinstatement model of cocaine seeking. Thus, activation of xc- with N-acetylcysteine inhibits cocaine-induced reinstatement (Baker et al., 2003), and this effect was prevented by blocking mGluR2/3.
Extracellular, nonsynaptic glutamate in the NA arises primarily from xc- (Baker et al., 2002). Two previous studies indicate that glutamate derived from nonvesicular stores modulates glutamate transmission. Jabaudon et al. (1999) demonstrated an increase in NMDA currents following blockade of glutamate transport that was independent of synaptically released glutamate. Warr et al. (1999) showed that extracellular glutamate derived from stimulating xc- with high concentrations of cystine (1 mm) elicits postsynaptic NMDA currents in cerebellar slices. Although these studies point to the possible involvement of xc- derived glutamate in stimulating postsynaptic glutamate receptors under pharmacological conditions, the present study used physiological concentrations of extracellular cystine (100-300 nm) (Baker et al., 2003) to promote xc- activity. Under these conditions, xc- mediated elevation in extracellular glutamate inhibited synaptically released glutamate by stimulated mGluR2/3 presynaptic autoreceptors. Although in the physiological range and consistent with concentrations of cystine used in vivo to elevate extracellular glutamate in cocaine-withdrawn rats (Baker et al., 2003), the concentration of cystine used in the present study was lower than the reported Km values (∼2-100 μm) for [35]S-cystine or Na+-independent [3]H-glutamate uptake via xc- in experiments using cultured cells (Patel et al., 2004). The reason for this difference between in vivo and in vitro physiological studies and uptake measurements made in vitro is unclear but may reflect differences in the phosphorylation or trafficking of the exchanger between preparations (Gochenauer and Robinson, 2001; Baker et al., 2003).
Glutamate transmission in the NA is critical for the expression of behaviors associated with addiction, such as drug seeking and sensitization (Kalivas et al., 2005). Specifically, release of glutamate in the projection from the PFC to the NA provokes rein-statement of drug seeking in animals trained to self-administer cocaine (McFarland et al., 2003). Interestingly, the dependence of drug seeking on the enhanced release of synaptic glutamate in this projection is accompanied by reduced basal levels of extracellular, nonsynaptic glutamate in the NA (Pierce et al., 1996; Hotsenpiller et al., 2001; McFarland et al., 2003). The reduction in extracellular, nonsynaptic glutamate results from decreased activity of xc-, and restoration of extracellular glutamate by stimulating xc- blocks cocaine-primed reinstatement of drug seeking (Baker et al., 2003). The present study demonstrates that activating xc- prevents drug seeking via increasing extracellular, nonsynaptic glutamate tone on mGluR2/3, thereby inhibiting synaptic glutamate release. Accordingly, recent studies found that mGluR2/3 agonists inhibit cue- and heroin-primed reinstatement of drug seeking (Baptista et al., 2004; Bossert et al., 2004).
Anatomical studies reveal a presynaptic location of mGluR2/3 outside of the synaptic cleft in corticostriatal synapses (Robbe et al., 2002), and physiological studies show that mGluR2/3 are particularly susceptible to changes in tonic levels of nonsynaptic, extracellular glutamate (Petralia et al., 1996; Alagarsamy et al., 2001; Dietrich et al., 2002; Bandrowski et al., 2003). Presynaptic mGluR2/3 regulate classic forms of synaptic plasticity, such as LTD (Robbe et al., 2002), and may contribute to cocaine-induced synaptic plasticity in the NA (Thomas et al., 2001; Kalivas et al., 2005). Indeed, the inability to wash out the effect of cystine on EPSCs (Fig. 2A,D) is similar to the inability to wash the effect of mGluR2/3 agonists and may indicate the induction of LTD by cystine (Robbe et al., 2002).
Activation of xc- reduces excitatory transmission in NA and PFC tissue slices by increasing the concentration of nonsynaptic, extracellular glutamate and thereby stimulating inhibitory presynaptic mGluR2/3. This mechanism for regulating excitatory transmission is compromised after withdrawal from repeated cocaine administration and contributes to the reinstatement of drug seeking in rats trained to self-administer cocaine.
Footnotes
This work was supported in part by United States Public Health Service Grants DA03906 (P.W.K.), DA05369 (P.W.K.), and MH65924 (J.K.S.) and National Research Service Award Predoctoral Fellowship DA018459 (M.M.M.). We thank Laurence Neely for his technical assistance.
Correspondence should be addressed to Dr. Peter W. Kalivas, Department of Neurosciences, Medical University of South Carolina, 173 Ashley Avenue, BSB 403C, Charleston, SC 29425. E-mail: kalivasp@musc.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256389-05$15.00/0
References
- Alagarsamy S, Sorensen SD, Conn PJ (2001) Coordinate regulation of metabotropic glutamate receptors. Curr Opin Neurobiol 11: 357-362. [DOI] [PubMed] [Google Scholar]
- Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW (2002) The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci 22: 9134-9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, Kalivas PW (2003) Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci 6: 743-749. [DOI] [PubMed] [Google Scholar]
- Bandrowski AE, Huguenard JR, Prince DA (2003) Baseline glutamate levels affect group I and II mGluRs in layer V pyramidal neurons of rat sensorimotor cortex. J Neurophysiol 89: 1308-1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista MA, Martin-Fardon R, Weiss F (2004) Preferential effects of the metabotropic glutamate 2/3 receptor agonist LY379268 on conditioned reinstatement versus primary reinforcement: comparison between cocaine and a potent conventional reinforcer. J Neurosci 24: 4723-4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossert JM, Liu SY, Lu L, Shaham Y (2004) A role of ventral tegmental area glutamate in contextual cue-induced relapse to heroin seeking. J Neurosci 24: 10726-10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Pin J-P (1997) Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37: 205-237. [DOI] [PubMed] [Google Scholar]
- Dietrich D, Kral T, Clusmann H, Friedl M, Schramm J (2002) Presynaptic group II metabotropic glutamate receptors reduce stimulated and spontaneous transmitter release in human dentate gyrus. Neuropharmacology 42: 297-305. [DOI] [PubMed] [Google Scholar]
- Gochenauer GE, Robinson MB (2001) Dibutyryl-cAMP (dbcAMP) upregulates astrocytic chlorinde-dependent L-[3H]glutamate transport and expression in both system xc- subunits. J Neurochem 78: 276-286. [DOI] [PubMed] [Google Scholar]
- Hotsenpiller G, Giorgetti M, Wolf ME (2001) Alterations in behaviour and glutamate transmission following presentation of stimuli previously associated with cocaine exposure. Eur J Neurosci 14: 1843-1855. [DOI] [PubMed] [Google Scholar]
- Jabaudon D, Shimamoto K, Yasuda-Kamatani Y, Scanziani M, Gahwiler B, Gerber U (1999) Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc Natl Acad Sci USA 96: 8733-8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Volkow N, Seamans J (2005) Unmanageable motivation in addiction: a pathology in prefrontal-accumbens glutamate transmission. Neuron 45: 647-650. [DOI] [PubMed] [Google Scholar]
- Kenny PJ, Markou A (2004) The ups and downs of addiction: role of metabotropic glutamate receptors. Trends in Pharmacol Sci 25: 265-272. [DOI] [PubMed] [Google Scholar]
- Manzoni O, Michel J-M, Bockaert J (1997) Metabotropic glutamate receptors in the rat nucleus accumbens. Eur J Neurosci 9: 1514-1523. [DOI] [PubMed] [Google Scholar]
- McBean GJ (2002) Cerebral cystine uptake: a tale of two transporters. Trends Pharmacol Sci 23: 299-302. [DOI] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW (2003) Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced rein-statement of drug-seeking behavior. J Neurosci 23: 3531-3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani S, Daniel H, Takita M, Crepel F (2002) Long-term depression induced by postsynaptic group II metabotropic glutamate receptors linked to phospholipase C and intracellular calcium rises in rat prefrontal cortex. J Neurosci 22: 3434-3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SA, Warren BA, Rhoderick JF, Bridges RJ (2004) Differentiation of substrate and non-substrate inhibitors of transport system xc-: an obligate exchanger of l-glutamate and l-cystine. Neuropharmacology 46: 273-284. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Wang Y-X, Niedzielski AS, Wenthold RJ (1996) The metabotropic glutamate receptors, mGLUR2 and mGLUR3, show unique postsynaptic, presynaptic and glial localizations. Neuroscience 71: 949-976. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Bell K, Duffy P, Kalivas PW (1996) Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci 16: 1550-1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Chaumont S, Bockaert J, Manzoni O (2002) Role of P/Q-Ca2+ channels in metabotropic glutamate receptor 2/3 dependent presynaptic long-term depression at nucleus accumbens synapses. J Neurosci 2: 4346-4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Tamba M, Okuno S, Sato K, Keino-Masu K, Masu M, Bannai S (2002) Distribution of cystine/glutamate exchange transporter, system x(c)-, in the mouse brain. J Neurosci 22: 8028-8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepp DD, Jane DE, Monn JA (1999) Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology 38: 1431-1476. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC (2001) Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci 4: 1217-1223. [DOI] [PubMed] [Google Scholar]
- Warr O, Takahashi M, Attwell D (1999) Modulation of extracellular glutamate concentration in rat brain slices by cystine-glutamate exchange. J Physiol (Lond) 514: 783-793. [DOI] [PMC free article] [PubMed] [Google Scholar]