

Abstract
The controlled formation of ROS (reactive oxygen species) and RNS (reactive nitrogen species) is now known to be critical in cellular redox signalling. As with the more familiar phosphorylation-dependent signal transduction pathways, control of protein function is mediated by the post-translational modification at specific amino acid residues, notably thiols. Two important classes of oxidant-derived signalling molecules are the lipid oxidation products, including those with electrophilic reactive centres, and decomposition products such as lysoPC (lysophosphatidylcholine). The mechanisms can be direct in the case of electrophiles, as they can modify signalling proteins by post-translational modification of thiols. In the case of lysoPC, it appears that secondary generation of ROS/RNS, dependent on intracellular calcium fluxes, can cause the secondary induction of H2O2 in the cell. In either case, the intracellular source of ROS/RNS has not been defined. In this respect, the mitochondrion is particularly interesting since it is now becoming apparent that the formation of superoxide from the respiratory chain can play an important role in cell signalling, and oxidized lipids can stimulate ROS formation from an undefined source. In this short overview, we describe recent experiments that suggest that the cell signalling mediated by lipid oxidation products involves their interaction with mitochondria. The implications of these results for our understanding of adaptation and the response to stress in cardiovascular disease are discussed.
Keywords: 4-hydroxynonenal (4-HNE), lysophosphatidylcholine, mitochondrion, oxidized lipid, reactive oxygen species (ROS), thiol
Introduction
The formation of low molecular mass second messengers as cell-signalling molecules is well established in the redox cell-signalling field. The molecules involved in signalling are structurally diverse and include the ROS (reactive oxygen species) and RNS (reactive nitrogen species), NO, O2•− (superoxide radical), H2O2 and peroxynitrite (ONO2H) [1]. In addition, in the presence of transition metals, a secondary series of reactive lipid products can be formed from the reactions with unsaturated fatty acids (Figure 1A). With identification of three isoforms of the enzyme NOS (nitric oxide synthase) and multiple isoforms of the NADPH oxidases, it is clear that ROS/RNS formation in the cell has the potential to be regulated. In this light, all cellular sources of ROS/RNS are now being scrutinized for their potential to contribute to redox cell signalling. This perspective necessitates that we reexamine some basic concepts in the free radical field. These are as follows.
Figure 1.
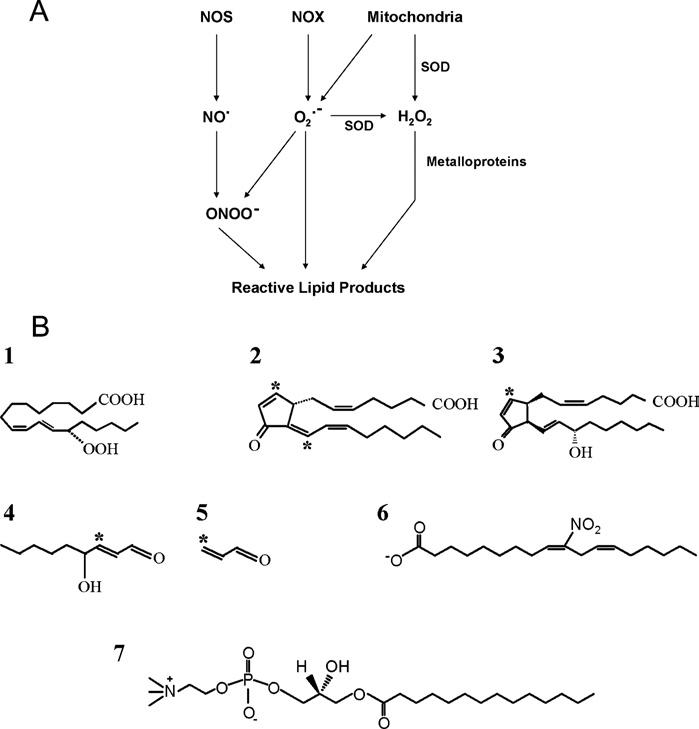
Formation of reactive intermediates and structures of reactive lipid products (A) Primary sources of ROS and RNS are shown. The enzyme NOS generates NO, while the family of NADPH-dependent oxidases (NOX) generate superoxide radicals (O2•−). Mitochondria generate O2•−, which can travel into the cell or can be converted into H2O2 before entering the cytoplasm. These three species – NO, O2•− and H2O2 – are referred to here as the primary reactive species. NO can combine with O2•− to form peroxynitrite (ONO2H), a highly reactive secondary species capable of initiating lipid peroxidation. (B) Reactive lipid products are grouped here into three major classes. First, lipid peroxides are exemplified here by 13S-HpODE (13-hydroxyoctadecadienoic acid) (1), a product derived from the enzymatic action of 15-lipoxygenase on linoleic acid. Secondly, reactive lipids with electrophilic properties are exemplified by 15d-PGJ2 (15-deoxy-△12,14-prostaglandin J2) (2), iso-J2 (isoprostane J2) (3), 4-HNE (4) and acrolein (5). Carbons capable of adducting to nucleophiles present in cellular proteins are indicated by an asterisk. Finally, receptor agonists are the third class. These include LNO2 (nitrolinoleic acid), which can also act as an electrophile and NO donor (6), and lysoPC (7).
The pathology associated with ROS/RNS formation may be due to loss of control of cell signalling rather than oxidative stress [2].
Post-translational modification of proteins by ROS/RNS may signify that the protein in question is a ‘receptor’ for the redox second messenger: for example, the thiol redox switches that are present on a number of important cell-signalling proteins [3].
Intracellular antioxidants such as glutathione may establish a ‘redox tone’ by controlling the availability of redox active proteins to participate in cell signalling [4].
Redox signalling may involve irreversible covalent modification of proteins [5].
In this short overview, we will examine the concept that the mitochondrion plays a role in redox cell signalling using the interaction with oxidized lipids as an example [6].
Lipid oxidation product-dependent protein modification
ROS and RNS can react with cellular lipids directly or indirectly, generating a spectrum of products, many of which contain functional groups capable of modifying proteins (Figure 1B). Oxidized lipids can activate cell-signalling pathways in three different ways: (i) by non-covalent mechanisms involving binding to a protein receptor; (ii) covalent pathways leading to direct protein modification by the oxidized lipids or (iii) the activation of pathways leading to calcium influx and intracellular ROS/RNS formation.
Currently, the most detailed mechanisms relate to the covalent and non-covalent interactions of lipids with proteins. For example, protein adducts are formed through the reaction of lipid aldehydes with nucleophilic protein centres, such as cysteine, lysine and histidine, resulting in Schiff base products [7,8]. Another mechanism of protein adduction is Michael addition, where thiolate groups of cysteine residues react with electrophilic carbon centres present in α-β unsaturated carbonyls. This functional group is present in the cyclopentenone prostaglandins, enzymatic products formed from the cyclo-oxygenase pathway and the nonenzymatic products known as the isoprostanes (Figure 1B). Simpler oxidation products containing this same reactive group, resulting from β-scission, include 4-HNE (4-hydroxynonenal) and acrolein.
Direct evidence for the modification of cell-signalling molecules with lipid electrophiles has been obtained in a number of systems; we have termed this group of proteins the ‘electrophile-responsive proteome’. For example, adduct formation with Keap-1 [kelch-like ECH (erythroid cell-derived protein with CNC homology)-associated protein-1] has been observed with the cyclopentenone 15-deoxy-△12,14-prostaglandin J2, resulting in up-regulation of cellular antioxidant proteins under the control of the electrophile response element [5,9,10]. Thus reactive lipid products may transduce ROS/RNS into cellular signals through post-translational modification of critical cellular proteins. Clearly, in this model, the origin of intracellular ROS/RNS and their location relative to a source of oxidizable lipid is critical. Interestingly, mitochondria are a major source of ROS in the cell and also contain a complex lipid structure with numerous intercalated redox active proteins.
Involvement of mitochondrial ROS in cell signalling
Recent studies are increasingly supporting the concept that mitochondrially produced ROS may act as signalling molecules in various physiological settings, including tumour necrosis factor α [11], leptin [12] and endothelin [13] signalling. Interestingly, many of these studies suggested that the MAPK (mitogen-activated protein kinase) pathway is one of the pathways regulated by mitochondrial ROS [13-18]. The precise mechanisms underlying the mitochondrial ROS-dependent initiation of the signal transduction pathways are largely unknown.
Mechanism of ROS production by mitochondria
ROS production in mitochondria can occur at several sites, namely complex I and ubiquinone of the electron-transport chain and the tricarboxylic acid cycle dehydrogenases [19,20]. Importantly, it is now becoming clear that each of the sites has the potential to be regulated. Identification of physiological regulators of mitochondrial ROS production is thus important in the light of redox cell signalling. Several molecules/factors have been suggested as regulators of mitochondrial ROS production, which include an elevated inner membrane potential (△ψ) [21], Ca2+ [22] and NO [6,23]. In addition, ROS production through the ubiquinone/ubiquinol redox pool can be regulated by NO. Complex I can also produce ROS itself, and in combination with complex II through a process known as ‘reversed electron transport’ [20]. Coupling of mitochondrial ROS formation through the process of redox tone can occur through S-glutathiolation [24]. Beyond the electron-transport chain, the tricarboxylic acid cycle dehydrogenases, specifically the 2-oxoglutarate dehydrogenase and possibly pyruvate dehydrogenase, have been shown to generate ROS when the NADH/NAD+ ratio is high [25,26]. The exogenous stimulators of mitochondrial ROS are not clear but, from our recent studies, it is evident that lipid oxidation products play a major role.
Mitochondrial ROS-mediated signal transduction by oxidized lipids
Oxidized lipids can be derived from both exogenous and endogenous sources. For example, oxLDL (oxidized low-density lipoprotein), which is involved in the pathogenesis of cardiovascular disease, contains a complex mixture of reactive aldehydes, electrophilic lipids and other oxidized phospholipids. Although it has been demonstrated in a number of laboratories that exposure of endothelial cells to oxLDL induces ROS/RNS formation [27], we have recently demonstrated that the mitochondrion is a major contributor to this oxidative signal, as shown in Figure 2 [28].
Figure 2.
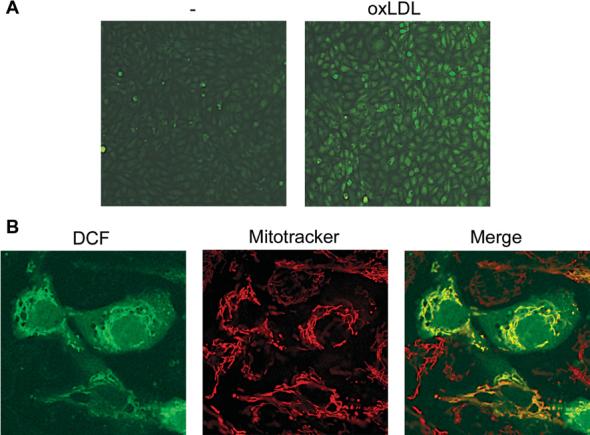
OxLDL-induced production of ROS in mitochondria Bovine aortic endothelial cells were loaded with 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) followed by 30 min exposure to oxLDL (75μg/ml) and 15 min exposure to Mitotracker Red. Cells were then washed and either epifluorescence (A) or confocal (B) images were acquired. Exposure of cells to oxLDL revealed a substantial increase in DCF fluorescence (A) compared with untreated cells. (B) The Figure demonstrates that DCF fluorescence co-localized with a mitochondrial marker (Mitotracker).
Furthermore, the individual electrophilic lipids in oxLDL such as 4-HNE cause ROS production and activate MAPK pathways in endothelial cells [29]. One of the major targets of 4-HNE is the mitochondrion, where it selectively inactivates proteins containing reactive thiols, such as 2-oxoglutarate dehydrogenase and pyruvate dehydrogenase [30], and can thereby inhibit complex I-dependent (NADH-linked) respiration [31].
In addition to 4-HNE, we have demonstrated that a chemically inert oxidized lipid, lysoPC (lysophosphatidylcholine), can also induce mitochondrial ROS production through a Ca-dependent process that leads to the selective activation the ERK (extracellular-signal-regulated kinase)/MAPK pathway. The mechanism remains to be defined in detail but appears to involve an interplay between the Ca2+-dependent mitochondrial dehydrogenases and complex I.
Summary
In this short overview, we have summarized the evidence that mitochondria can integrate redox cell signalling initiated by oxidized lipids. The scheme in Figure 3 summarizes the potential mechanisms through which both direct modification of proteins by electrophilic lipids and indirect stimulation of mitochondrial ROS can result in the initiation of cell signalling. This model offers the potential for mitochondrial therapeutics through the targeting of the regulatory mechanisms that underlie mitochondrial ROS formation.
Figure 3.
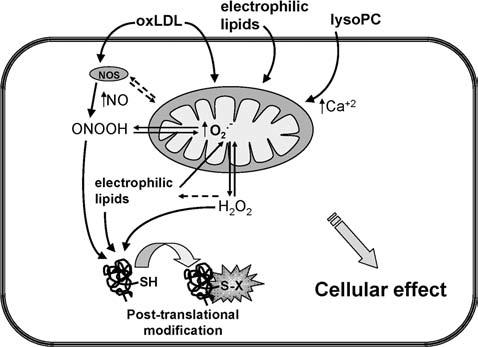
Mechanisms of mitochondrial-dependent redox signalling OxLDL, electrophilic lipids or lysoPC induce O2•− generation in mitochondria that may then be converted into H2O2. OxLDL also activates NOS and increases production of NO; in the presence of available O2•−, oxLDL may contribute to peroxynitrite formation. In addition, lysoPC can elevate intracellular calcium levels. Furthermore, ROS, RNS and electrophilic lipids contribute to the post-translational modification of protein thiols (protein-SH) to form protein-S-X. Moreover, it is postulated that oxidative stress increases the intracellular pool of electrophilic lipids.
Acknowledgments
The studies described in this review were supported by NIH (National Institutes of Health) grants ES10167 and HL58031 to V.M.D.-U. and by an External Research Program grant from Philip Morris to D.A.D.
Abbreviations used:
- 4-HNE
4-hydroxynonenal
- lysoPC
lysophosphatidylcholine
- MAPK
mitogen-activated protein kinase
- NOS
nitric oxide synthase
- oxLDL
oxidized low-density lipoprotein
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
References
- 1.Patel RP, Moellering D, Murphy-Ullrich J, Jo H, Beckman JS, Darley-Usmar VM. Free Radical Biol. Med. 2000;28:1780–1794. doi: 10.1016/s0891-5849(00)00235-5. [DOI] [PubMed] [Google Scholar]
- 2.Shiva S, Moellering D, Ramachandran A, Levonen AL, Landar A, Venkatraman A, Ceaser E, Ulasova E, Crawford JH, Brookes PS, et al. Biochem. Soc. Symp. 2004;71:107–120. doi: 10.1042/bss0710107. [DOI] [PubMed] [Google Scholar]
- 3.Cooper CE, Patel RP, Brookes PS, Darley-Usmar VM. Trends Biochem. Sci. 2002;27:489–492. doi: 10.1016/s0968-0004(02)02191-6. [DOI] [PubMed] [Google Scholar]
- 4.Landar A, Darley-Usmar VM. Amino Acids. 2003;25:313–321. doi: 10.1007/s00726-003-0019-7. [DOI] [PubMed] [Google Scholar]
- 5.Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, Zanoni G, Morrow JD, Darley-Usmar VM. Biochem. J. 2004;378:373–382. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brookes PS, Levonen AL, Shiva S, Sarti P, Darley-Usmar VM. Free Radical Biol. Med. 2002;33:755–764. doi: 10.1016/s0891-5849(02)00901-2. [DOI] [PubMed] [Google Scholar]
- 7.Isom AL, Barnes S, Wilson L, Kirk M, Coward L, Darley-Usmar V. J. Am. Soc. Mass Spectrom. 2004;15:1136–1147. doi: 10.1016/j.jasms.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 8.Uchida K. Prog. Lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 9.Sanchez-Gomez FJ, Cernuda-Morollon E, Stamatakis K, Perez-Sala D. Mol. Pharmacol. 2004;66:1349–1358. doi: 10.1124/mol.104.002824. [DOI] [PubMed] [Google Scholar]
- 10.Ceaser EK, Moellering DR, Shiva S, Ramachandran A, Landar A, Venkartraman A, Crawford J, Patel R, Dickinson DA, Ulasova E, et al. Biochem. Soc. Trans. 2004;32:151–155. doi: 10.1042/bst0320151. [DOI] [PubMed] [Google Scholar]
- 11.Rogers RJ, Monnier JM, Nick HS. J. Biol. Chem. 2001;276:20419–20427. doi: 10.1074/jbc.M008915200. [DOI] [PubMed] [Google Scholar]
- 12.Yamagishi SI, Edelstein D, Du XL, Kaneda Y, Guzman M, Brownlee M. J. Biol. Chem. 2001;276:25096–25100. doi: 10.1074/jbc.M007383200. [DOI] [PubMed] [Google Scholar]
- 13.Touyz RM, Yao G, Viel E, Amiri F, Schiffrin EL. J. Hypertens. 2004;22:1141–1149. doi: 10.1097/00004872-200406000-00015. [DOI] [PubMed] [Google Scholar]
- 14.Dougherty CJ, Kubasiak LA, Frazier DP, Li H, Xiong WC, Bishopric NH, Webster KA. FASEB J. 2004;18:1060–1070. doi: 10.1096/fj.04-1505com. [DOI] [PubMed] [Google Scholar]
- 15.Chen K, Thomas SR, Albano A, Murphy MP, Keaney JF., Jr J. Biol. Chem. 2004;279:35079–35086. doi: 10.1074/jbc.M404859200. [DOI] [PubMed] [Google Scholar]
- 16.Nemoto S, Takeda K, Yu ZX, Ferrans VJ, Finkel T. Mol. Cell. Biol. 2000;20:7311–7318. doi: 10.1128/mcb.20.19.7311-7318.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bogoyevitch MA, Ng DC, Court NW, Draper KA, Dhillon A, Abas L. J. Mol. Cell. Cardiol. 2000;32:1469–1480. doi: 10.1006/jmcc.2000.1187. [DOI] [PubMed] [Google Scholar]
- 18.Fang Y, Han SI, Mitchell C, Gupta S, Studer E, Grant S, Hylemon PB, Dent P. Hepatology. 2004;40:961–971. doi: 10.1002/hep.20385. [DOI] [PubMed] [Google Scholar]
- 19.Young TA, Cunningham CC, Bailey SM. Arch. Biochem. Biophys. 2002;405:65–72. doi: 10.1016/s0003-9861(02)00338-7. [DOI] [PubMed] [Google Scholar]
- 20.Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker N. Free Radical Biol. Med. 2004;37:755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 21.Kadenbach B. Biochim. Biophys. Acta. 2003;1604:77–94. doi: 10.1016/s0005-2728(03)00027-6. [DOI] [PubMed] [Google Scholar]
- 22.Kanno T, Sato EE, Muranaka S, Fujita H, Fujiwara T, Utsumi T, Inoue M, Utsumi K. Free Radical Res. 2004;38:27–35. doi: 10.1080/10715760310001626266. [DOI] [PubMed] [Google Scholar]
- 23.Brookes P, Darley-Usmar VM. Free Radical Biol. Med. 2002;32:370–374. doi: 10.1016/s0891-5849(01)00805-x. [DOI] [PubMed] [Google Scholar]
- 24.Taylor ER, Hurrell F, Shannon RJ, Lin TK, Hirst J, Murphy MP. J. Biol. Chem. 2003;278:19603–19610. doi: 10.1074/jbc.M209359200. [DOI] [PubMed] [Google Scholar]
- 25.Tretter L, Adam-Vizi V. J. Neurosci. 2004;24:7771–7778. doi: 10.1523/JNEUROSCI.1842-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. J. Neurosci. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Go YM, Levonen AL, Moellering D, Ramachandran A, Patel RP, Jo H, Darley-Usmar VM. Am. J. Physiol. Heart Circ. Physiol. 2001;281:H2705–H2713. doi: 10.1152/ajpheart.2001.281.6.H2705. [DOI] [PubMed] [Google Scholar]
- 28.Zmijewski JW, Moellering DR, Le Goffe C, Landar A, Ramachandran A, Darley-Usmar VM. Am. J. Physiol. Heart Circ. Physiol. 2005;289:H852–H861. doi: 10.1152/ajpheart.00015.2005. [DOI] [PubMed] [Google Scholar]
- 29.Uchida K, Shiraishi M, Naito Y, Torii Y, Nakamura Y, Osawa T. J. Biol. Chem. 1999;274:2234–2242. doi: 10.1074/jbc.274.4.2234. [DOI] [PubMed] [Google Scholar]
- 30.Humphries KM, Szweda LI. Biochemistry. 1998;37:15835–15841. doi: 10.1021/bi981512h. [DOI] [PubMed] [Google Scholar]
- 31.Humphries KM, Yoo Y, Szweda LI. Biochemistry. 1998;37:552–557. doi: 10.1021/bi971958i. [DOI] [PubMed] [Google Scholar]