

Abstract
Hydrophobic interactions play a major role in binding non-native substrate proteins in the central cavity of the bacterial chaperonin GroEL. The sequence of local conformational changes by which GroEL and its cofactor GroES assist protein folding can be explored using the polarity-sensitive fluorescence probe Nile Red. A specific single-cysteine mutant of GroEL (Cys261), whose cysteine is located inside the central cavity at the apical region of the protein, was covalently labeled with synthetically prepared Nile Red maleimide (NR). Bulk fluorescence spectra of Cys261-NR were measured to examine the effects of binding of the stringent substrate, malate dehydrogenase (MDH), GroES, and nucleotide on the local environment of the probe. After binding denatured substrate, the fluorescence intensity increased by 32±7%, suggesting enhanced hydrophobicity at the position of the label. On the other hand, in the presence of ATP, the fluorescence intensity decreased by 13±3%, implying increased local polarity. In order to explore the sequence of local polarity changes, substrate, GroES, and various nucleotides were added in different orders; the resulting changes in emission intensity provide insight into the sequence of conformational changes occurring during GroEL-mediated protein folding.
Introduction
The ‘chaperonins’1 are oligomeric barrel-shaped-complexes composed of ∼60 kDa protein subunits, which mediate the folding of polypeptide chains in an ATP-dependent fashion.2 The bacterial chaperonin from E. coli, GroEL, in concert with its cap-like cofactor, GroES, has been the subject of many studies (for reviews, see).3-5 The oligomeric complex GroEL is actually a homotetradecamer composed of identical 57 kDa (547 aa) subunits arranged in two stacked heptameric toroidal rings (Figure 1B), as first clearly shown by the crystal structure without nucleotide published in 1994.6 Each subunit of GroEL consists of three domains: an apical domain that binds substrate and GroES, an intermediate or hinge domain, and an equatorial domain, which binds ATP and is closest to the other ring of the structure (red, green, and blue respectively in Figure. 1A). The co-chaperonin, GroES, is a homoheptamer of 10 kDa subunits,7 which binds as a dome-shaped cap on one end of the double-ring GroEL structure. A wide array of biophysical and biochemical studies8-17 have provided useful information on the method by which GroEL/ES assists in folding of substrates, especially the kinetics of the ATPase cycle, which features positive cooperativity among the seven ATP binding sites in one ring, and negative cooperativity for the affinity of the two rings of GroEL for GroES. From all these investigations, a generally accepted view of the overall mode of action of the system has appeared, summarized as follows (after).4 (1) The cycle begins with the empty cis (near-side) ring, which has hydrophobic residues exposed on the inner surface of the apical domains. (2) Non-native substrate protein (likely with exposed hydrophobic residues) binds to the apical domain of the cis ring. (3) ATP binds to the cis ring, causing an increase in affinity for GroES as well as other conformational changes, and a closed folding cavity is fully formed by binding the GroES over the open end of the structure. (4) ATP hydrolysis eventually occurs, and disassembly of the complex is triggered by ATP binding to the trans ring. The released substrate may have achieved the native form; if not, the substrate may bind the GroEL/ES system again.
FIGURE 1.
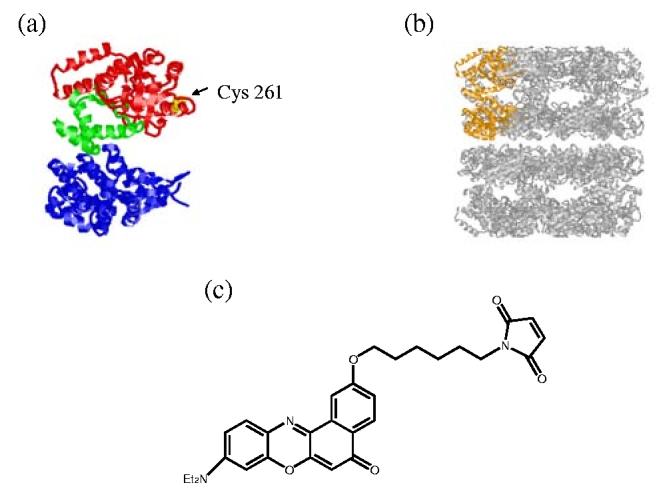
The GroEL tetradecamer, which consists of two rings, is illustrated on the right (Figure 1B), and one of the monomer subunits shown in orange is expanded on the left to indicate the position of the mutation (Figure 1A). This cysteine mutant (Cys261, shown in yellow) was labeled with Nile Red maleimide. Color of the left figure (Figure 1A) corresponds the three different domains in GroEL; apical is red, intermediate is green and equatorial is blue. This crystal structure is from the x-ray structure of apo-GroEL, PDB filename 1GRL.6
Although GroEL/GroES-assisted folding has been studied for more than a decade and many characteristics of the system have been established, the exact role that the chaperonin plays with respect to the substrate is still not fully understood, and new physical measurement techniques are needed. In one model,18-20 GroEL acts as an “unfoldase” upon ATP/GroES binding, actively pulling apart misfolded substrate (but see21). In an alternate model, the step of binding a non-native protein multivalently to an open GroEL ring via hydrophobic interactions is associated with unfolding,22,23 and then ATP/GroES binding produces an environment, encapsulated and hydrophilic, that is conducive to productive folding.24,25 Fully definitive evidence has yet to be provided for these models, and a better mechanistic understanding is central to the elucidation of GroEL/GroES function.
Here we address the question: how can fluorescence spectroscopy of a local probe detect the sequence, size, and type of conformationally-driven local polarity changes that occur in the GroEL folding chamber during the process? Previous studies have shown that the hydrophobic interaction is important in the binding of substrate to GroEL. A set of hydrophobic residues on the inner surface of the apical domain of GroEL is known to be involved in substrate binding.25 Based on the hydrophobic amino acid sequences from strongly binding substrates, workers have even been able to predict potential GroEL substrates from all of the proteins in E. coli.26 More broadly, binding of GroES, substrate and nucleotides may all influence the conformation of GroEL. X-ray and cryo-EM studies have shown that ATP binding promotes small elevation and counterclockwise twisting movements of the apical domain, and GroES binding induces a further upward movement and large clockwise twist (120 degrees) of the apical domain.27-29 Binding of substrate induces conformational changes of GroEL that breaks the symmetry between the two rings of GroEL and reduces the diameter of GroEL at the ends by rearranging apical domains.30 These conformational changes would be also expected to affect the local polarity of the apical domain in GroEL.
In order to sense the local environmental polarity in GroEL, we have chosen the Nile Red fluorophore as a reporter dye. The emission of Nile Red has a pronounced red-shift as the solvent environment becomes more polar, and due to its visible absorption wavelength compared to other polarity reporters like ANS (1-anilinonaphthalene-8-sulfonic acid,),31 Nile Red has been used as a local polarity reporter in host-guest studies of polymers at the single-molecule level.32 In biological systems, Nile Red has been used as a hydrophobic probe of lipids33 or protein hydrophobic surfaces.34,35 In these experiments, Nile Red was added to the solution without any specific covalent bond, and an increase in fluorescence intensity was detected, since the fluorescence of free dye in water is very weak compared to the fluorescence in hydrophobic environments. Recently, covalent attachment of a Nile Red derivative with a reactive functional group to a protein with four cysteine residues was reported,36 and an amine-reactive form was used to observe conformational changes in single calmodulin-peptide complexes.37
Extending an earlier brief report,38 we describe in this paper the sequence of changes in local hydrophobicity of GroEL due to binding of GroES, substrate, and different nucleotides using fluorescence spectroscopy. A maleimide-functionalized Nile Red (Figure 1C) was synthesized for this study. A GroEL mutant with a single cysteine at the inner surface of the apical domain (termed Cys261, Figure 1A) was labeled with Nile Red maleimide. Changes in local polarity were monitored by recording Nile Red fluorescence intensity as a function of time. In order to observe the binding sequence dependence, various reagents such as GroES, substrate, and nucleotide or nucleotide mimics were added in different orders. The stringent substrate malate dehydrogenase (MDH) was selected for these studies, since MDH is known to require a minimum of three consecutive apical domains9 for binding. This requirement increases the probability of interaction with Nile Red, since a low labeling ratio of Nile Red per GroEL (approximately 2 Nile Red molecules per tetradecamer) was employed.
Materials and methods
Proteins
Wild-type GroEL and GroES were expressed and purified as described.39 Briefly, the cells were lysed in a buffer containing 50 mM Na-phosphate (pH 8.0), 300 mM NaCl, 1mM PMSF, 25 μg/mL DNAse. The lysate was loaded onto a Ni-NTA column, and the column was eluted with a buffer containing 50 mM Na-phosphate (pH 8.0), 300 mM NaCl, 200 mM imidazole. The protein was further purified using a MonoQ column with a linear gradient of 0-1 M NaCl in a buffer containing 25 mM HEPES (pH 7.4). Cys261, where all native cysteine residues have been replaced with alanine except one cysteine at position 261 mutated from threonine, was produced by site-directed mutagenesis of GroEL, and purified as described.9 Cys261 was stored in a buffer containing 50 mM Tris-HCl (pH 7.4), 50 mM KCl, 5 mM DTT. Porcine heart malate dehydrogenase (MDH) and Bovine liver rhodanese were purchased from Sigma and used as received.
Synthesis of Nile Red maleimide and Labeling of Cys261
In order to attach the Nile Red fluorophore to a specific residue such as cysteine, a maleimide-functionalized Nile Red was synthesized (Figure 1C). This Nile Red maleimide, with the thiol-reactive maleimide unit spaced from the fluorescent Nile Red chromophore by a hexamethylene chain, was prepared from a Nile Red precursor bearing a phenolic hydroxyl group at the 2-position, 9-diethylamino-2-hydroxy-5H-benzo[a]phenoxazin-5-one.40 The overall synthesis involves preparation of the phenolfunctionalized chromophore and a protected maleimide moiety followed by connection of the two parts via the C6 spacer and finally deprotection to create the free maleimide. Full details of the synthetic method are presented in the Supplementary Material.
The cysteine residue on Cys261 (23 μM) was labeled with Nile Red maleimide (1:1 molar ratio of label to tetradecamer) at room temperature for 2 hr. Before labeling, Cys261 was exchanged into a buffer containing 2 mM TCEP. The labeling reaction was terminated by 5 mM DTT, and the labeled protein was separated from free dye by dilution and concentration in a Microcon YM30 (Amicon, Inc.), followed by gel filtration (Micro bio-spin P30 column; Bio-rad). After labeling, the protein concentration was determined to be ∼10 μM by a BCA assay (Pierce), probably due to the loss of the protein during buffer exchange before and after labeling. Uv-vis absorption spectra were used to determine that the average number of Nile Red fluorophore per Cys261 tetradecamer was ∼ 2, using a value of 20,000 M−1 cm−1 for the measured extinction coefficient of Nile Red maleimide in buffer A (defined below). Therefore, the average value of Nile Red labels per single GroEL ring could be regarded as one. The labeled Cys261 GroEL tetradecamer will be referred to as Cys261-NR. The enzymatic activity of the labeled protein was tested using a standard refolding assay with the substrate rhodanese as described.41 Importantly, this assay showed that the complex of GroES with the labeled GroEL variant is as effective as the wild-type GroEL/ES system in facilitating refolding of the substrate protein. We have explored increases in the number of Nile Red labels per GroEL tetradecamer. With ∼ 4 Nile Red molecules per tetradecamer, GroEL did show normal activity as above. It also showed similar behavior in terms of the approximate fluorescence changes for all the cases described in this study, except that the raw fluorescence values were higher than for the case of 2 Nile Red labels. Using the empty Cys261-NR fluorescence as a reference, the addition of unfolded MDH to the 4-Nile Red-labeled Cys261-NR caused an increase of 50±6% in the emission intensity (data not shown), compared to 32±7% increase for 2 labels. (The fact that these two numbers are not identical can be understood by realizing that when four labels are present (say two per ring), then one MDH molecule can bind to two labels at a time, and this case is not probable when there are only two labels per tetradecamer.) On the other hand, when GroEL has more than 6 Nile Red labels per tetradecamer, the enzymatic activity and binding behavior were altered. In this study, we only present measurements with the case of 2 Nile Red labels per GroEL tetradecamer.
Fluorescence measurements
All of the fluorescence measurements utilized a FluoroMax-2 fluorimeter (Jobin-Yvon ISA Spex, Inc.) with an excitation wavelength at 532 nm. A Cys261-NR solution (0.2 μM, 100 μL) was prepared in buffer A containing 50 mM Tris-HCl (pH 7.4), 50 mM KCl, 5 mM MgCl2, and the molar ratio of the Cys261-NR:MDH:GroES was maintained at 1:1:2.5. To measure fluorescence changes upon addition of reagents, 2 μL of GroES (24.5 μM), 2.5 μL of MDH (8.6 μM), and 1 μL of nucleotide (100 mM) were used to minimize dilution effects on the fluorescence intensity. The transition state analogue (ADP/AlFx) was generated with 1 mM ADP, 0.5 mM Al(NO3)3, and 5 mM NaF. The final Mg2+ concentration was increased to 10 mM when ADP/AlFx was used. MDH was denatured by 2 hr incubation in a buffer containing 3M GdHCl, 50 mM Tris-HCl (pH 7.4), and 5 mM DTT. For the kinetic measurements, the fluorescence intensity was followed for 15-20 minutes in order to reach equilibrium after each reagent was added, and percentage changes were computed relative to the emission from empty Cys261-NR before the addition of any reagent. In case of ADP/AlFx, the fluorescence was measured for 30 minutes, thereby allowing enough time for ADP/AlFx complex formation. All of these measurements were repeated 2-4 times.
Results and Discussion
Fluorescence measurements of Nile Red maleimide in various solvents
First, the polarity-sensitive fluorescence of the maleimide-functionalized Nile Red was measured in various solvents. The emission maximum shifted from 564 nm (in a nonpolar solvent, toluene) to 656 nm (in a polar solvent, water) as the solvent hydrophobicity decreases, and the emission intensity in water decreased to 300 times less than that in toluene, the similar behavior for the parent molecule.34 The wavelength shifts of both absorption and emission in different solvents can be examined with a parameter describing the solvent polarity, the orientation polarizability Δf, which is defined as Δf = (ε − 1)/(2ε + 1) − (n2 − 1)/(2n2 + 1), where ε is the dielectric constant and n is the refractive index of the solvent. Lippert42 showed that the spectral shifts are proportional to Δf (the so-called Lippert equation43 ), because solvent reorientation affects the energy difference between ground and excited state of the fluorophore. Using this equation, the solvent polarity of an unknown solution can be estimated in principle by comparing the wavelength shifts with those of known solvents. However, our measurements of Nile Red labeled Cys261 (Cys261-NR) showed little spectral shift after the addition of substrate, nucleotides and GroES (Figure 4) due to the limited polarity range of the local environment in the protein, while the emission intensity did show changes.
FIGURE 4.

Fluorescence spectra of the Nile Red-labeled mutant GroEL (Cys261-NR, 0.2 μM) before and after the single addition of substrate, GroES, and different nucleotides (532 nm excitation). (A) Cys261-NR (rectangles) was incubated with the same amount of either unfolded (open triangles) or native MDH (open circles) at 25°C, for 20 minutes. No change in fluorescence was observed upon addition of native MDH, while unfolded MDH produced a significant increase. (B) Cys261-NR was incubated with either nucleotides (ATP (triangles), ADP (open circles), or ADP/AlFx (rectangles)) or GroES (open diamonds, 25°C, incubation times as indicated). All nucleotides and GroES induced a decrease in fluorescence compared to nucleotide-free Cys261-NR.
In order to quantify the relationship between solvent polarity and emission intensity, we used a binary solvent mixture to vary the polarity in a limited range, and measured Nile Red maleimide fluorescence. A methanol/water mixture was chosen for the following reasons. First, the emission maximum of Nile Red maleimide in methanol (635 nm) is close to Cys261-NR (625 nm). Second, the emission intensity of Nile Red in methanol/water mixtures was observed to be linearly dependent on solvent Δf in an earlier study.44 Figure 2A shows the emission spectra of Nile Red maleimide in various molar ratios of methanol dissolved in water. The emission maximum red-shifts and the intensity decreases as the molar ratio of methanol is decreased (decreasing hydrophobicity). This trend is consistent with the behavior in different pure solvents of varying polarity. A specific polarity range of the methanol/water mixture, in which the Nile Red maleimide emission intensity was comparable to Cys261-NR, was examined more closely, as can be seen in Figure 2B. In this range, the maximum emission wavelength of Nile Red maleimide in methanol/water mixture was not altered due to the limited polarity changes. However, the emission intensity decreased as solvent polarity increased. The Δf values of the solvent mixture were calculated by using literature values of refractive index45 and dielectric constant.46 As shown in the inset of Figure 2B, the decrease of emission intensity was proportional to Δf in this range. Therefore, the emission intensity of Cys261-NR, lying in this region, can be expected to respond linearly to the solvent polarity.
FIGURE 2.

Fluorescence spectra of Nile Red maleimide in methanol/water mixtures. (A) Fluorescence spectra of Nile Red maleimide were measured in water with various molar ratios of methanol. The emission maximum was red-shifted and the emission intensity decreased as solvent hydrophobicity decreased. The molar ratios of methanol in water decreasing from the top are 1 (pure methanol), 0.85, 0.65, 0.5, 0.35, 0.25, 0.2, 0.18, 0.15, 0.13, 0.1, 0.06, 0.03, 0 (pure water). (B) Expanded plot of (A), where the emission intensities of Nile Red maleimide are comparable to Cys261-NR. Inset shows that the emission intensity linearly decreases as a function of Δf in this range. Spectra of Cys261-NR and Cys261-NR with unfolded MDH are shown for comparison.
We now compare these results with the emission spectra of the protein Cys261-NR in the absence or presence of unfolded MDH (Figure 2B). These spectra were recorded with equal absorbance solutions at the excitation wavelength (532 nm). In general, both the emission wavelength and intensity of a fluorescent solute can reflect the polarity of a solution. In particular, when emission wavelength is also utilized to extract the polarity information, it is important to recognize that other effects such as hydrogen bonding and spectral relaxation can also cause a shift of emission wavelength. Moreover, the behavior of a fluorophore in a protein can be quite different from that in a solvent due to protein relaxation. In other words, the environment of Nile Red in Cys261 might not be same as Nile Red in a solvent, since dielectric relaxation of the protein can occur in ∼ns depending on the structure,47 which is comparable to the excited state lifetime of Nile Red. In this case, spectral relaxation toward longer wavelength due to solvation might not be complete, and spectral relaxation effects should be considered before making a direct polarity comparison using maximum emission wavelength. In our case, the maximum wavelength of Cys261-NR was shorter than the Nile Red maleimide in methanol/water mixture (Figure 2B), while the emission intensity was comparable. This difference can be explained by the relatively slow relaxation in the protein, and experimentally ‘red edge effects’ can be used to measure the dynamics of the spectral relaxation.48 In fact, studies of polarity-sensitive probes such as 2,6-TNS and PRODAN (6-propionyl-2-dimethylaminonaphthalene) in proteins showed that the emission maximum shifted to longer wavelengths with longer wavelength excitation, due to slow relaxation of the environment.49,50 The dependence of maximum emission wavelength on excitation wavelength was measured for Nile Red maleimide and Cys261-NR (Figure 3). The emission maximum of Cys261-NR did shift from 621 nm to 646 nm as the excitation wavelength changed from 475 nm to 625 nm, while the emission maximum of Nile Red maleimide in 0.10 molar methanol in water did not shift at all. This result suggests that the protein relaxation affects the emission wavelength shift, and thus for fixed excitation at 532 nm, we are justified in interpreting changes in the brightness of Cys261-NR emission as primarily reflecting changes in local polarity near the Δf value of 0.317.
FIGURE 3.

The Dependence of fluorescence maximum emission wavelength on the excitation wavelength. While Nile Red maleimide in a 0.1 molar ratio of methanol in water (triangles) did not change its emission wavelength, both Cys261-NR (circles) and unfolded MDH loaded Cys261-NR (open diamonds) showed red shifts in maximum emission wavelength as excitation wavelength increased.
Single-reagent experiments: Changes of fluorescence spectrum with MDH or nucleotides
Using the stringent substrate MDH and various nucleotides, bulk fluorescence spectra of Cys261-NR were measured to examine the effects of binding of the reagent on the local environment of the probe. In all cases, the fluorescence from (empty) Cys261-NR was monitored first in buffer A, and then either native or unfolded MDH was added (Figure 4A). After the addition of each reagent, all spectra reached steady-state within 30 minutes, and had nearly the same maximum wavelength near 625 nm within 1-2 nm. Although the spectrum did not shift appreciably due to the limited polarity change, changes in polarity could still be easily followed by the peak or integrated fluorescence intensity. The addition of the unfolded substrate to the Cys261-NR caused an increase of about 32±7% in the emission intensity, suggesting enhanced hydrophobicity at the labeling position. By contrast, when native substrate was added, the fluorescence intensity did not change significantly, understandable since properly-folded MDH does not bind to GroEL. These results suggest two possibilities: (1) exposed hydrophobic residues from unfolded MDH can interact with the Nile Red fluorophore in the apical domain of GroEL, or (2) unfolded MDH binding to GroEL induces conformational changes at the labeling position such that the dye samples a more hydrophobic environment.
The effects of singly-added nucleotides (ATP, ADP, and the transition state mimic ADP/AlFx) and GroES on the fluorescence intensity were also investigated. In contrast to unfolded MDH, the addition of nucleotides or GroES leads to a decrease in emission intensity compared to the level of fluorescence from the empty Cys261-NR (Figure 4B). GroES induced a small decrease of the fluorescence (5±3%) similar to the ADP case, in spite of the lack of affinity of GroES for GroEL in the absence of nucleotide.51 ATP reduced the fluorescence intensity by the largest amount, 13±3%, implying increased local polarity at the position of the probe. A rasmol comparison between the x-ray apo structure6 and the ATP-bound cryo-EM structure28 shows that apical domain is twisted counterclockwise after ATP binding, causing more hydrophilic amino acids near the apical domain to move closer to the probe position at residue 261. The other nucleotides used in this study, ADP and ADP/AlFx, showed a similar, but smaller decrease in emission intensity (6±2% for ADP, 11±3% for ADP/AlFx, see Figure 4B) compared to ATP, although ADP/AlFx had a comparable value to ATP. This finding will be discussed in more detail below.
Kinetic measurements of the Cys261-NR fluorescence intensity
In order to comprehensively explore the sequence of local conformational changes in the full catalytic cycle of GroEL/ES, the three reagents, unfolded MDH, GroES, and nucleotide nt (chosen from ATP, ADP, ADP/AlFx), were added to empty Csy261-NR in different orders – and all six possible sequences were investigated for each of the three nucleotides (nt): MDH-ES-nt, MDH-nt-ES, ES-MDH-nt, ES-nt-MDH, nt-MDH-ES, and nt-ES-MDH. For the cases with ATP addition, since ATP is continuously hydrolyzed on the time scale of 15-20 seconds10,52 and thereby drives the folding cycle continuously, the measurements with ATP average over all possible states, and eventually lead to actual refolding of MDH. Therefore, it may be difficult to obtain sequence-dependent information from measurements with ATP. Nevertheless, addition of non-hydrolyzable nucleotides such as ADP and ADP/AlFx should be able to reveal a certain degree of sequence information on the GroEL/GroES-mediated folding mechanism, since they prevent the continuous cycling of the enzyme. Since all of the spectra showed the similar peak wavelength and shape, only the peak emission intensity was followed as a function of time, always relative to the starting value, i.e., the emission from empty Cys261-NR before the addition of any reagent. Using a concentration of ∼0.2 μM for both MDH and GroEL (Cys261-NR), one may estimate from previous work that about one hour is needed for MDH to regain full activity in the presence of GroEL/GroES/ATP.53 In our experiments, the components are added serially, one by one, and it would only be in the last ∼15-20 minute interval that assisted folding could begin. As a consequence, most of the observed fluorescence changes reflect protein-protein binding/interaction effects or conformational changes of the labeled GroEL.
Considering the data from all sequences, in general, there was little dependence on the order of reagent addition except for the very last reagent. Thus, for reagents added first or second, a 32±7% increase in fluorescence intensity was observed when MDH was added and a 13±3% decrease was observed by adding ATP, with a smaller decrease of 6±2% for ADP and 11±3% for ADP/AlFx (Figure 5 and data not shown). However, the fluorescence intensity changes produced by the last reagent showed different behavior compared to the single-reagent experiments. To describe this, the six sequences can be considered in three groups, according to the last reagent added: nucleotide (MDH-ES-nt, ES-MDH-nt), GroES (MDH-nt-ES, nt-MDH-ES), and MDH (ES-nt-MDH, nt-ES-MDH). The first two groups will be discussed together, since they have a common feature in the sense that MDH binds to the apical domain of GroEL before the nucleotide-dependent GroEL-GroES association.
FIGURE 5.

(A) Kinetic measurements of the relative peak fluorescence intensity of Cys261-NR induced by the addition of denatured MDH, GroES and different nucleotides. The fluorescence intensity at maximum was monitored as a function of time, 10 s per point. MDH was added first, then GroES, and finally various nucleotides (ATP (open rectangles), ADP (open circles), or ADP/AlFx (open triangles)). Five additional analogous experiments with different sequences of adding MDH, GroES and nucleotide were also performed to observe the sequence dependence (see text). (B) Comparison of fluorescence reductions by (GroES + nt) for the two sequences of MDH-ES-nt (black bars), and ES-nt (gray bars) in the same units of part (A).
(a) The cases where the last reagent is either nucleotide (MDH-ES-nt, ES-MDH-nt) or GroES (MDH-nt-ES, nt-MDH-ES)
A typical set of kinetic scans showing the relative peak intensity as a function of time is shown in Figure 5A for one specific sequence of adding reagents, MDH-ES-nt. As described above, we assumed that the fluorescence intensity of Cys261-NR linearly responds to polarity changes, therefore all plots were normalized to the same standard condition (empty Cys261-NR). The fluorescence changes produced by adding MDH and GroES before nucleotides were similar to the single-reagent experiments. However, the fluorescence reductions by nucleotide addition, especially with ATP and ADP/AlFx, increased (Figure 5A, ∼20% and ∼17%, rather than 13% and 11%), while ADP showed a similar decrease to single-reagent experiments (∼8%). To be consistent, all fluorescence changes in Figure 5B are reported after 15 min incubation, even though steady state is not reached with the ATP case. Interestingly, the fluorescence decay transients upon either nt or GroES addition at the end showed both a fast change, and then a slow drop (Figure 5A, data not shown), while the single addition of each component did not show slow kinetics. This behavior will be discussed more in detail below. Similar results were obtained upon nucleotide addition for the ES-MDH-nt case (∼19% decrease for both ATP and ADP/AlFx, and ∼9% decrease for ADP, specific traces not shown). Also, for the cases where the GroES was the last reagent (MDH-nt-ES, nt-MDH-ES), the fluorescence decreases by adding GroES were larger (22±5%, data not shown), while the observed fluorescence decrease by GroES binding to the empty GroEL was not significant (Figure 4B).
One can consider the effect of MDH release into the cavity triggered by nucleotides with GroES, to understand these fluorescence differences caused by nucleotides compared to single-reagent experiments. In order to investigate the effect of MDH on the fluorescence, the fluorescence changes induced by (GroES + nt) in the absence of MDH were calculated and represented as gray bars in Figure 5B. The values of summed fluorescence intensity reduction by GroES and nucleotides were roughly similar to the sum of the fluorescence drop for each reagent added singly. If the fluorescence decreases by (GroES+nt) in Figure 5B originates from the formation of GroES-GroEL complex, the additional decreases in Figure 5A, which are presented as black bars in Figure 5B can be understood by the presence of MDH in addition to the GroEL-GroES complex formation. Actually, in order to fully associate GroEL with GroES, (1) nucleotides should be present, considering that the affinity of GroES for GroEL is large only when nucleotide has been added,14,51,52 and (2) MDH has to dissociate from the apical domains to which it is bound (ideally to three consecutive GroEL subunits), since it is known that the binding sites for both GroES and substrate overlap.27 Therefore, in the case of Figure 5A, nucleotide addition at the end induces the formation of GroEL-GroES complex, which leads MDH to release from the GroEL apical domain into the cavity, a process that will serve to negate the fluorescence increase induced by MDH binding. In other words, the net fluorescence decreases by addition of nucleotides should have two effects, (1) nucleotide binding, a fast process, and (2) GroEL-GroES formation and MDH release, which should take a longer time. Moreover, the large change toward increased polarity at the probe location can be understood in light of the fact that a relatively nonpolar binding surface (from MDH) is replaced by a more polar binding surface (from GroES). From this point of view, all four sequences (MDH-ES-nt, ES-MDH-nt, MDH-nt-ES, nt-MDH-ES) should have similar results due to the nature of GroEL-GroES association; the displacement of MDH by GroES can only begin at the end of these sequences in the presence of nucleotides.
In fact, MDH release into the cavity also depends on the type of nucleotides added. The difference between black (with MDH addition first) and gray bar (without MDH addition) in Figure 5B can be regarded as the degree of MDH release into the cavity by the GroEL-GroEL association. ADP/AlFx showed more or less similar fluorescence changes (the difference between black and gray bar, 10±5%) as ATP (12±3%), while ADP (6±3%) showed a small change. Therefore, ADP/AlFx seems to have a similar ability to release MDH as ATP, and this correlates well with a recent study, observing both productive folding and substrate release in GroEL with ADP/AlFx.54 ADP/AlFx stabilizes the transition state, by forming a trigonal bipyramidal conformation in the nucleotide pocket, which mimics ATP undergoing hydrolysis. 55 From anisotropy measurements and gel filtration with labeled fluorescence detection, the additional γ-phosphate provided by adding AlFx to ADP is known to contribute to a stabilized GroEL-GroES complex.54 Additional support for our interpretation is provided by work showing that the addition of AlFx in the presence of ADP induces further apical movement comparable to the ATP case.56
Based on these results, we can understand the behavior as follows. First, a substrate like MDH can bind to the GroEL apical domain in GroEL, causing increased hydrophobicity. Although GroES exists in the solution, it cannot bind tightly to the GroEL apical domain due to the absence of nucleotides. After addition of nucleotides, GroES can finally bind to GroEL, and the nucleotide type determines the extent of the conformational changes of the apical domain by GroEL-GroES complex, inducing MDH release into the cavity. Furthermore, as noted alone, the fluorescence decay after the final addition in Figure 5A shows a fast change followed by a slow drop. The common feature of all these sequences is that the compact GroEL-GroES complex formation can only start at the end of the sequence and therefore, the displacement of MDH by GroES should always occur at the end. From this point of view, the slow kinetics can be understood to relate to the GroEL-GroES association rate. Having both fast and slow kinetics is consistent with a recent study in which time dependent changes of GroEL apical movement by GroES binding were slowed in the presence of the substrate.56,57
(b) The cases where the last reagent was MDH (nt-ES-MDH, ES-nt-MDH)
Intriguing results were obtained for the two sequences in which MDH was the last reagent added, and the nucleotide used was ADP/AlFx (Figure 6). Specifically, if ADP/AlFx was added after GroES (ES-ADP/AlFx-MDH), the fluorescence increase induced by MDH (“the MDH effect”) was large (20±1%, Figure 6A), while if ADP/AlFx was added before GroES (ADP/AlFx-ES-MDH), the MDH effect was much smaller than in all other cases (7±2%, Figure 6B). On the other hand, when the nucleotide added is either ATP or ADP, the MDH effect was independent of the order of adding nt and ES (i.e., for the sequences nt-ES-MDH and ES-nt-MDH). In these two situations, the fluorescence increase induced by MDH was ∼31% and ∼28% for ATP and ADP, respectively.
FIGURE 6.

Sequence dependence of the time-dependent fluorescence intensity changes of Cys261-NR with ADP/AlFx. The fluorescence intensity at maximum was monitored while adding GroES and ADP/AlFx, with unfolded MDH addition at the end. (A). The sequence ES-ADP/AlFx-MDH. (B). The sequence ADP/AlFx-ES-MDH. (C) Same as (A), except long incubation (45 minutes) with ADP/AlFx, (D) Same as (B), except 1:1 molar ratio of GroEL:GroES.
Since the fluorescence increase is caused by MDH binding to the apical domain of the Cys261-NR, the observed smaller increase implies that MDH binding is hindered. There are several possible explanations for this effect. First, the incubation time after adding ADP/AlFx was chosen to be 30 minutes to guarantee binding of the Al salt, 58 therefore this incubation time was longer than for ATP (15 minutes) or ADP (20 minutes). However, this might allow different degrees of ADP/AlFx complex formation, i.e., if the time for ADP/AlFx complex formation is longer than 30 minutes, the fluorescence increase upon addition of MDH in Figure 6AB might depend on the sequence of ADP/AlFx addition. In order to test this, the experiment shown in Figure 6A was repeated, but now ADP/AlFx was added and incubated 45 minutes (Figure 6C). The large fluorescence increase (17±3% increase) as in Figure 6A was maintained. Hence, one can exclude that the incubation time for complex formation affects MDH binding.
Another possibility for the sequence dependence shown in Figure 6AB might be the formation of a symmetric complex (GroES-GroEL-GroES), known as the “American football” structure.59 A symmetric complex was observed in both electron microscopy and cross-linking experiments15,59-61 in the presence of ATP or AMP-PNP, and this was ascribed to the formation of the RR allosteric state (illustrated in Figure 7B). This symmetric structure is stable until ATP hydrolyzes to ADP. Therefore, in the presence of ADP alone, no symmetric complex can be formed. However, since ADP/AlFx is a transition state analogue, imitating a state between ATP and ADP (such as ADP-Pi), this nucleotide might therefore allow the formation of the symmetric complex if GroEL reaches the RR state before GroES binding. As illustrated in Figure 7B, our observations might be understood if both of the apical domains are blocked by GroES and thus MDH binding to the inner apical surface would be significantly hindered. To further test this hypothesis, GroES was added in an equimolar amount to GroEL (1:1 ratio GroEL:GroES, rather than 1:2.5 as in Figure 6ABC) to prevent formation of the symmetric complex with both ends capped. As shown in Figure 6D, the large fluorescence increase upon MDH addition (14±2%) is recovered as in Figure 6A. This result supports the interpretation that the small MDH effect in Figure 6B is due to the formation of the doubly-capped symmetric complex. A recent study from Taguchi et al. also reported a symmetric complex, but with ATP/BeFx.62
FIGURE 7.

Proposed scheme for the formation of symmetric/asymmetric complex of GroEL/GroES with ADP/AlFx. (A). When GroES is added first, GroES can only bind after adding ADP/AlFx, and the binding affinity of ADP/AlFx to the trans ring may be reduced because of GroES binding to the cis ring. This prevents the formation of the symmetric complex. (B). When ADP/AlFx is added first (before GroES), hindered MDH binding to the GroEL can be explained by the formation of the symmetric football complex.
Within the picture of cooperativity based on TT/RR states of the enzyme, it is also possible to address the behavior when GroES is added before ADP/AlFx (Figure 6AC). In this situation, GroEL would begin in the TT state, since nucleotides have not been added and GroES does not strongly bind to GroEL without them (Figure 7A).51 After adding ADP/AlFx, however, GroES can bind to GroEL, and form either a symmetric complex (GroES-GroEL-GroES) or an asymmetric complex in terms of GroES binding, (single GroES binding to GroEL,) known as the “bullet complex”.59 The large fluorescence increase induced by MDH addition Figure 6A suggests that the asymmetric bullet complex seems to prevail, leaving open a binding site for unfolded MDH in the trans ring. This might be explained with a reduced binding affinity of ADP/AlFx to the trans ring when GroES is already bound to the cis ring. By analogy, for the eukaryotic chaperonin TriC it is known that ADP/AlFx induces lid closure in only one ring of the TriC complex.58 Recent work has shown that for the ATP bullet complex, no stable binding of either GroES or substrate to the trans ring is observed,10 but in that work an ATP hydrolysis-deficient mutant GroEL was used. It might be possible that like the ATP bullet complex, the ADP/AlFx bullet has less affinity for the substrate binding to the trans ring, since the MDH effects for both ES-ADP/AlFx-MDH and ADP/AlFx-ES-MDH sequences are smaller (20±1%, 7± 2%, respectively) than the average MDH effect (32±7%), but one cannot easily explain the much smaller MDH effect for the case of ADP/AlFx-ES-MDH without symmetric complex formation. It is worth noting that both symmetric and asymmetric complexes seem to be present together in the case of ADP/AlFx-ES-MDH, since a fluorescence increase after MDH addition (Figure 6B) was still observable even though it was smaller than other cases, and the sum of the fluorescence decrease by (ADP/AlFx + ES) in Figure 6B was not twice as much as compared in Figure 6A.
Even though the symmetric complex can be detected under various conditions, as shown in this study and by other methods such as electron microscopy and cross linking, it is still controversial whether this complex is an intermediate of the chaperonin cycle, or just an off-pathway product.63-65 Due to allosteric interactions, the symmetric complex may not be favorable under certain conditions. However, it is known that the binding affinity of GroES to the cis ring is increased in the presence of ATP in the same ring. Oppositely, GroES is released from the cis ring in the presence of ATP in the trans ring. Therefore, the symmetric complex can be observed during the natural GroEL/GroES cycle if the second GroES binding rate to the trans ring is faster than the GroES dissociation rate from the cis ring, both of which can only occur only after the binding ATP to the trans ring.10
Conclusion
Using a mutant of GroEL to which a polarity-sensitive fluorophore, Nile Red, has been covalently attached at a specific location, we have observed a variety of local polarity changes upon the addition of substrate MDH, nucleotides, and GroES. For most cases, a large shift to less polarity is observed when unfolded MDH is added, while addition of nucleotide and GroES produce shifts toward increasing polarity. For sequences in which nucleotide or GroES is added last, we observed competition between GroES and substrate for the binding sites of the apical domain, and the degree of this competition, which ultimately induces the substrate release into the cavity, is determined by the nucleotides. For the special case in which the substrate was added last and the nucleotide was the transition state mimic ADP/AlFx, the fluorescence changes depended upon the order of addition of the first two components, the nucleotide and GroES. This behavior is consistent with the formation of a doubly-capped symmetric complex when ADP/AlFx is added first.
Supplementary Material
Acknowledgement
We thank David Fromm and Stephan Hess for assistance at the early stages of this project. This work was supported in part by the National Science Foundation Grant No. MCB-0212503, by the BioX-Interdisciplinary Initiatives Program at Stanford University, and by the National Institutes of Health, Grant No. 1-R21-GM075166-01.
Footnotes
Supporting Information Available: Full details of the Nile Red maleimide synthesis are presented. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Frydman J. Annu. Rev. Biochem. 2001;70:603–647. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- 2.Martin J, Langer T, Boteva R, Schramel A, Horwich AL, Hartl FU. Nature. 1991;352:36–42. doi: 10.1038/352036a0. [DOI] [PubMed] [Google Scholar]
- 3.Hartl FU. Nature. 1996;381:571–580. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 4.Bukau B, Horwich AL. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- 5.Sigler PB, Xu Z, Rye HS, Burston SG, Fenton WA, Horwich AL. Annu. Rev. Biochem. 1998;67:581–608. doi: 10.1146/annurev.biochem.67.1.581. [DOI] [PubMed] [Google Scholar]
- 6.Braig K, Otwinowski Z, Hegde R, Boisvert DC, Joachimiak A, Horwich AL, Sigler PB. Nature. 1994;371:578–586. doi: 10.1038/371578a0. [DOI] [PubMed] [Google Scholar]
- 7.Hunt JF, Weaver AJ, Landry SJ, Gierasch L, Diesenhofer J. Nature. 1996;379:37–45. doi: 10.1038/379037a0. [DOI] [PubMed] [Google Scholar]
- 8.Rye HS, Burston SG, Fenton WA, Beechem JM, Xu Z, Sigler PB, Horwich AL. Nature. 1997;388:792–798. doi: 10.1038/42047. [DOI] [PubMed] [Google Scholar]
- 9.Farr GW, Furtak K, Rowland MB, Ranson NA, Saibil HR, Kirchhausen T, Horwich AL. Cell. 2000;100:561–573. doi: 10.1016/s0092-8674(00)80692-3. [DOI] [PubMed] [Google Scholar]
- 10.Rye HS, Roseman AM, Chen S, Furtak K, Fenton WA, Saibil HR, Horwich AL. Cell. 1999;97:325–338. doi: 10.1016/s0092-8674(00)80742-4. [DOI] [PubMed] [Google Scholar]
- 11.Weissman JS, Kashi Y, Fenton WA, Horwich AL. Cell. 1994;78:693–702. doi: 10.1016/0092-8674(94)90533-9. [DOI] [PubMed] [Google Scholar]
- 12.Weissman JS, Hohl CM, Kovalenko O, Kashi Y, S. C, Braig K, Saibil HR, Fenton WA, Horwich AL. Cell. 1995;83:577–587. doi: 10.1016/0092-8674(95)90098-5. [DOI] [PubMed] [Google Scholar]
- 13.Weissman JS, Rye HS, Fenton WA, Beechem JM, Horwich AL. Cell. 1996;84:481–490. doi: 10.1016/s0092-8674(00)81293-3. [DOI] [PubMed] [Google Scholar]
- 14.Jackson GS, Staniforth RA, Halsall DJ, Atkinson T, Holbrook JJ, Clarke AR, Burston SG. Biochemistry. 1993;32:2554–2563. doi: 10.1021/bi00061a013. [DOI] [PubMed] [Google Scholar]
- 15.Todd MJ, Viitanen PV, Lorimer GH. Science. 1994;265:659–666. doi: 10.1126/science.7913555. [DOI] [PubMed] [Google Scholar]
- 16.Ranson NA, Dunster NJ, Burston SG, Clarke AR. J. Mol. Biol. 1995;250:581–586. doi: 10.1006/jmbi.1995.0399. [DOI] [PubMed] [Google Scholar]
- 17.Yifrach O, Horovitz A. Biochemistry. 1995;34:5303–5308. doi: 10.1021/bi00016a001. [DOI] [PubMed] [Google Scholar]
- 18.Shtilerman M, Lorimer GH, Englander SW. Science. 1999;284:822–825. doi: 10.1126/science.284.5415.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thirumalai D, Lorimer GH. Annu. Rev. Biophys. Biomol. Struct. 2001;30:245–269. doi: 10.1146/annurev.biophys.30.1.245. [DOI] [PubMed] [Google Scholar]
- 20.Wang J, Boisvert DC. J. Mol. Biol. 2003;327:843–855. doi: 10.1016/s0022-2836(03)00184-0. [DOI] [PubMed] [Google Scholar]
- 21.Park ES, Fenton WA, Horwich AL. FEBS Lett. 2005;579:1183–1186. doi: 10.1016/j.febslet.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 22.Zahn R, Plueckthun A. J. Mol. Biol. 1994;242:165–174. doi: 10.1006/jmbi.1994.1567. [DOI] [PubMed] [Google Scholar]
- 23.Zahn R, Perrett S, Fersht AR. J. Mol. Biol. 1996;261:43–61. doi: 10.1006/jmbi.1996.0440. [DOI] [PubMed] [Google Scholar]
- 24.Brinker A, Pfeifer G, Kerner MJ, Naylor DJ, Hartl FU, HayerHartl M. Cell. 2001;107:223–233. doi: 10.1016/s0092-8674(01)00517-7. [DOI] [PubMed] [Google Scholar]
- 25.Fenton WA, Kashi Y, Furtak K, Horwich AL. Nature. 1994;371:614–619. doi: 10.1038/371614a0. [DOI] [PubMed] [Google Scholar]
- 26.Stan G, Brooks BR, Lorimer GH, Thirumalai D. Protein Science. 2005;14:193–201. doi: 10.1110/ps.04933205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu Z, Horwich AL, Sigler PB. Nature. 1997;388:741–750. doi: 10.1038/41944. [DOI] [PubMed] [Google Scholar]
- 28.Ranson NA, Farr GW, Roseman AM, Gowen B, Fenton WA, Horwich AL, Saibil HR. Cell. 2001;107:869–879. doi: 10.1016/s0092-8674(01)00617-1. [DOI] [PubMed] [Google Scholar]
- 29.Chaudhry C, Horwich AL, Brunger AT, Adams PD. J. Mol. Biol. 2004;342:229–245. doi: 10.1016/j.jmb.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 30.Falke S, Fisher MT, Gogol EP. J. Mol. Biol. 2001;308:569–577. doi: 10.1006/jmbi.2001.4613. [DOI] [PubMed] [Google Scholar]
- 31.Slavik J. Biochim. Biophys. Acta. 1982;694:1–25. doi: 10.1016/0304-4157(82)90012-0. [DOI] [PubMed] [Google Scholar]
- 32.Hou Y, Bardo AM, Martinez C, Higgins DA. J. Phys. Chem. B. 2000;104:212–219. [Google Scholar]
- 33.Klinkner AM, Bugelski PJ, Waites CR, Louden C, Hart TK, Kerns WD. J. Histochem. Cytochem. 1997;45:743–753. doi: 10.1177/002215549704500513. [DOI] [PubMed] [Google Scholar]
- 34.Sackett DL, Wolff J. Anal. Biochem. 1987;167:228–234. doi: 10.1016/0003-2697(87)90157-6. [DOI] [PubMed] [Google Scholar]
- 35.Sackett DL, Knutson JR, Wolff J. J. Biol. Chem. 1990;265:14899–14906. [PubMed] [Google Scholar]
- 36.Nakanishi J, Nakajima T, Sato M, Ozawa T, Tohda K, Umezawa Y. Anal. Chem. 2001;73:2920–2928. doi: 10.1021/ac001528p. [DOI] [PubMed] [Google Scholar]
- 37.Tang J, Mei E, Green C, Kaplan J, DeGrado WF, Smith AB, Hochstrasser RM. J. Phys. Chem. B. 2004;108:15910–15918. [Google Scholar]
- 38.Kim S, Hess S, Fromm D, Twieg RJ, Farr GW, Horwich AL, Frydman J, Moerner WE. Biophys. J. 2003;84:27a. [Google Scholar]
- 39.Hayer-Hartl M, Weber F, Hartl FU. EMBO Journal. 1996;15:6111. [PMC free article] [PubMed] [Google Scholar]
- 40.Briggs MSJ, Bruce I, Miller JN, Moody CJ, Simmonds AC, Swann E. J. Chem. Soc. Perkin Trans. 1997;1:1051–1058. [Google Scholar]
- 41.Weber F, Hayer-Hartl M. Refolding of bovine mitochondrial rhodanese by chaperonins GroEL and GroES. In: Schneider C, editor. Methods in Molecular Biology. Vol. 140. Humana Press; Totowa, NJ: 2000. pp. 117–132. [DOI] [PubMed] [Google Scholar]
- 42.Von Lippert E. Z. Electrochem. 1957;61:962–975. [Google Scholar]
- 43.Lakowicz JR. Principles of fluorescence spectroscopy. Kluwer Academic; New York: 1999. [Google Scholar]
- 44.Dutta AK, Kamada K, Ohta K. J. Photochem. Photobio. A. 1996;93:57–64. [Google Scholar]
- 45.Lide DR, editor. Handbook of chemistry and physics. Vol. 82. CRC press; Baton Rouge: 20012002. [Google Scholar]
- 46.Marcus Y. Solvent mixtures: Properties and selective solvation. Marcel Dekker Inc; New York: 2002. [Google Scholar]
- 47.Lakowicz JR. Photochem. and Photobio. 2000;72:421–437. doi: 10.1562/0031-8655(2000)072<0421:OSRIP>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Demchenko AP. Luminescence. 2002;17:19–72. doi: 10.1002/bio.671. [DOI] [PubMed] [Google Scholar]
- 49.Demchenko AP. Biophys. Chem. 1982;15:101–109. doi: 10.1016/0301-4622(82)80022-7. [DOI] [PubMed] [Google Scholar]
- 50.Massey JB, She HS, Pownall HJ. Biochemistry. 1985;24:6973–6978. doi: 10.1021/bi00345a033. [DOI] [PubMed] [Google Scholar]
- 51.Engel A, Hayer-Hartl M, Goldie KN, Pfeifer G, Hegerl R, Muller S, Da Silva ACR, Baumeister W, Hartl FU. Science. 1995;269:832–836. doi: 10.1126/science.7638600. [DOI] [PubMed] [Google Scholar]
- 52.Burston SG, Ranson NA, Clarke AR. J. Mol. Biol. 1995;249:138–152. doi: 10.1006/jmbi.1995.0285. [DOI] [PubMed] [Google Scholar]
- 53.Miller AD, Maghlaoui K, Albanese G, Kleinjan DA, Smith C. Biochem. J. 1993;291:139–144. doi: 10.1042/bj2910139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chaudhry C, Farr GW, Todd MJ, Rye HS, Brunger AT, Adams PD, Horwich AL, Sigler PB. EMBO Journal. 2003;22:4877–4887. doi: 10.1093/emboj/cdg477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fisher AJ, Smith CA, Thoden JB, Smith R, Sutoh K, Holden HM, Rayment I. Biochemistry. 1995;34:8960–8972. doi: 10.1021/bi00028a004. [DOI] [PubMed] [Google Scholar]
- 56.Motojima F, Chaudhry C, Fenton WA, Farr GW, Horwich AL. PNAS. 2004;101:15005–15012. doi: 10.1073/pnas.0406132101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Motojima F, Yoshida M. J. Biol. Chem. 2003;278:26648–26654. doi: 10.1074/jbc.M300806200. [DOI] [PubMed] [Google Scholar]
- 58.Meyer AS, Gillespie JR, Walther D, Millet IS, Doniach S, Frydman J. Cell. 2003;2:369–381. doi: 10.1016/s0092-8674(03)00307-6. [DOI] [PubMed] [Google Scholar]
- 59.Schmidt M, K. R, Rachel R, Pfeifer G, Jaenicke R, Viitanen P, Lorimer G, Buchner J. Science. 1994;265:656–659. doi: 10.1126/science.7913554. [DOI] [PubMed] [Google Scholar]
- 60.Llorca O, Marco S, Carrascosa JL, Valpuesta JM. J. Structural Biol. 1997;118:31–42. doi: 10.1006/jsbi.1996.3832. [DOI] [PubMed] [Google Scholar]
- 61.Azem A, Kessel M, Goloubinoff P. Science. 1994;265:653–656. doi: 10.1126/science.7913553. [DOI] [PubMed] [Google Scholar]
- 62.Taguchi H, Tsukuda K, Motojima F, Koike-Takeshita A, Yoshida M. J. Biol. Chem. 2004;279:45737–45743. doi: 10.1074/jbc.M406795200. [DOI] [PubMed] [Google Scholar]
- 63.Walter S. Cell. Mol. Life. Sci. 2002;59:1589–1597. doi: 10.1007/PL00012485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grallert H, Buchner J. J. Struct. Biol. 2001;13:95–103. doi: 10.1006/jsbi.2001.4387. [DOI] [PubMed] [Google Scholar]
- 65.Torok Z, Vigh L, Goloubinoff P. J. Biol. Chem. 1996;271:16180–16186. doi: 10.1074/jbc.271.27.16180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.