

Short abstract
Recent work has revealed a complex network of distinct alternative splicing events in Drosophila as well as drugs that interfere specifically and directly with one class of splicing regulators in human cells.
Abstract
Silencing of splicing regulators by RNA interference, combined with splicing-specific microarrays, has revealed a complex network of distinct alternative splicing events in Drosophila, while a high-throughput screen of more than 6,000 compounds has identified drugs that interfere specifically and directly with one class of splicing regulators in human cells.
The importance of splicing in the control of gene expression is underscored by the realization that the human genome codes for far fewer genes than expected [1]: we do not have many more genes than the nematode Caenorhabditis elegans and have fewer than the plant Arabidopsis thaliana. Alternative splicing, whereby regulated splice-site usage results in the generation of different protein isoforms for the same gene locus, is key to multiplying the diversity of proteins produced from the human transcriptome. Computational alignments of transcript data and high-throughput splicing-specific microarray analyses have estimated that as many as 70% of human genes undergo alternative splicing [2,3].
For most metazoan genes, an orchestra of around 100 proteins and 5 small nuclear ribonucleoproteins performs the daunting task of precisely excising introns and joining exons together to produce the correct mature RNA product. Because of the degeneracy of the branchpoint site and of the classical 5' and 3' splice sites at the exon-intron boundaries, additional cis-regulatory signals are used to aid exon detection. These signals are recognized by splicing regulators, the most common of which are the serine-arginine-rich RNA-binding proteins (SR proteins) and the heterogeneous nuclear ribonucleoproteins (hnRNPs). These splicing regulators modulate splice-site choice by interacting with components of the splicing machinery and binding to the auxiliary exonic and intronic cis-regulatory signals [4,5]. SR proteins are thought to promote splice-site usage by associating with exonic splicing enhancers [6]; hnRNPs, on the other hand, act antagonistically to repress splice-site usage [7-9]. Current models of exon recognition suggest that the regulators create a network of interactions that determines the inclusion and exclusion of particular exons in transcripts. Figure 1 illustrates the cooperative and antagonistic actions of splicing regulators binding to interspersed regulatory elements in pre-mRNA transcripts. Given that the majority of human genes contain introns and undergo alternative splicing, mutations in cis-regulatory splicing elements or splice sites have the potential to produce defective proteins and are the cause of human genetic disorders such as spinal muscular atrophy, ataxia telangiectasia and thalassemia [10-14].
Figure 1.
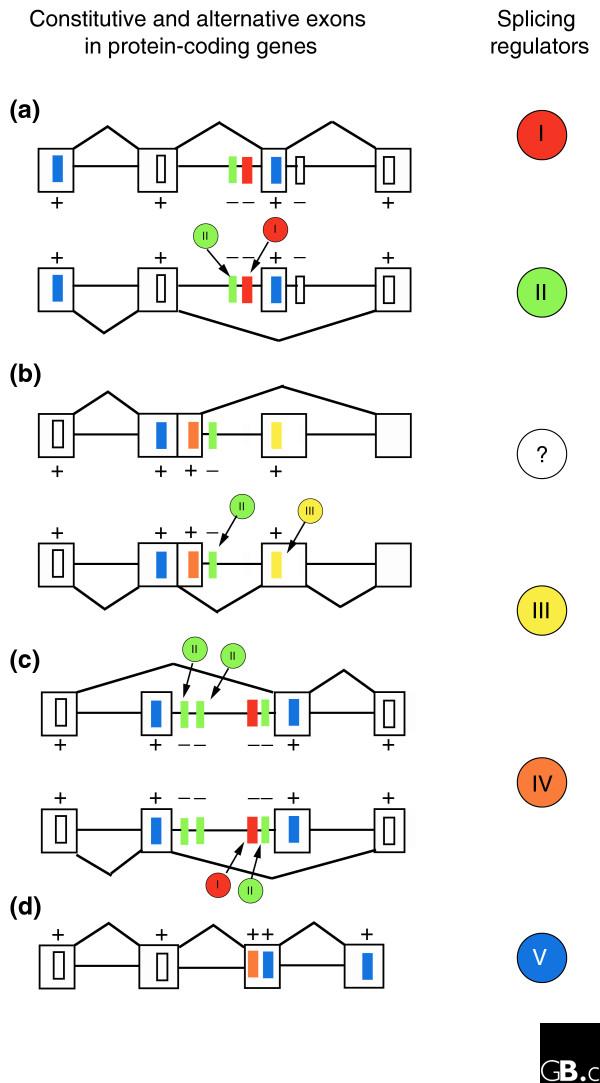
Splicing regulators and their targets. Four different genes are illustrated on the left. Genes (a), (b) and (c) have two different splicing isoforms each, as shown, while (d) is constitutively spliced. Exons are depicted by the outlined boxes and introns by the straight lines connecting them. The splicing regulators are depicted by the colored circles on the right, which bind to the correspondingly colored cis-regulatory elements (upright rectangles) in the exons and introns of the genes, promoting (+) or repressing (-) the use of adjacent splice sites. The white circle and white element represent unknown regulators and unidentified elements. Different combinations of regulators result in differently spliced transcripts, as represented by the zigzag lines joining exons. For example, regulator I regulates the exclusion of alternative exons in (a) and (c). Regulator II is required by regulator I, as indicated by the co-occurrences of their cis-elements in introns near exons in genes (a) and (c), but regulator II does not require regulator I, as in alternative 5' splice site choice in (b), or mutually exclusive skipping of the first regulated exon in (c). Binding of regulator III to its corresponding cis-element in (b) promotes the inclusion of an additional exon.
Complete genome sequences and microarray technology have made possible the large-scale study of splicing and the detection of alternatively spliced exons [2,15,16], but there is still much to do to uncover the 'splicing code'. This will involve identifying the cis-acting elements in exons and introns, identifying the regulators they bind, and understanding their mode of action in different cell types and at various developmental stages. Associations between regulators and splicing events have traditionally been made by biochemical and genetic methods. Although valuable, these methods are slow and can only study one regulator or one splicing event at a time. At present, a complete 'mapping' between regulators and their target exons via cis-regulatory elements is not yet available for any species. Such a map will be important in revealing the mechanisms involved in context-dependent inclusion and exclusion of alternative exons (as in different cell types or developmental stages), as well as in the design of low-toxicity drugs to alter splicing in the correction of genetic disorders or to disrupt viral gene expression. Two recent publications exemplify high-throughput and systematic strategies: Blanchette and colleagues [17] tackled the identification of targets of four splicing regulators in Drosophila melanogaster with a splicing-sensitive microarray, while Soret et al. [18] screened for chemical compounds that directly bind to SR proteins in human cells and interfere with spliceosomal assembly. The application of these methods to the study of more regulators and more compounds will pave the way for future attempts to develop therapies for diseases arising from aberrant splicing.
Analysis of splicing-regulator targets in Drosophila
In order to identify the alternative splicing events controlled by specific splicing regulators, Blanchette et al. [17] made use of a recent experimental innovation - the combination of the silencing of regulators with splicing-sensitive microarrays to detect the effects. This technology has the potential not only rapidly to identify the targets of these regulators throughout the genome, but also to tease apart their combinatorial control. Blanchette et al. [17] investigated four well-characterized and highly expressed splicing regulators - the SR proteins dASF (Drosophila alternative splicing factor, the homolog of human ASF, also known as splicing factor 2; SF2) and B52 (the Drosophila equivalent of the mammalian SRp55 protein) and the hnRNPs PSI and hrp48. They developed a splicing-sensitive microarray platform to monitor around 3,000 annotated alternatively spliced genes in the Drosophila genome. As in other studies [2,15,19], oligonucleotide probes were designed to span annotated alternatively spliced junctions, with control probes across the relevant constitutive junctions (that is, junctions that are always spliced together) and within exons. In order to silence the regulators by RNA interference (RNAi), Drosophila SL2 cells were treated with double-stranded RNAs specific for each regulator gene. As these regulators are likely to be associated not only with splicing but also with other RNA-processing functions, Blanchette et al. [17] initially feared that a decrease in the levels of the regulators might affect pre-mRNA processing in such a general manner that the array data would be uninterpretable. Fortunately, hierarchical clustering of multiple RNAi experiments showed characteristic and reproducible splicing responses.
The RNA isolated from the siRNA-treated cells was then hybridized on the microarrays. Each regulator affected distinct sets of splice junctions, but the results revealed significant overlap in the targets of dASF/SF2 and B52/SRp55, consistent with the observation that SR proteins can complement one another on particular targets (see references cited in [17]). The authors also observed that almost all the events affected by PSI are also controlled by hrp48, suggesting that hrp48 is a required partner for PSI. Interestingly, this relationship does not seem to be mutual, but further experiments are necessary to confirm this finding. Figure 1 illustrates this scenario: regulator II is an obligatory partner to I, but I is not required for II.
Consistent with the notion that dASF/SF2 is a general regulator of alternative splicing, its knockdown affected the largest number of events (319 events). Conversely, PSI, a more specific regulator of alternative splicing, affected the fewest events (43 events). In order to obtain evidence for direct binding of B52/SRp55, a positional weight matrix of an identified binding site for B52/SRp55 was used to scan the exon-intron boundaries of splicing events affected by knockdown of the factor. The motif was indeed specifically over-represented at the 5' splice sites of exons at which splicing is reduced when B52/SRp55 is knocked down. Together, the findings revealed a network of tens to hundreds of alternative splicing events that are regulated by individual or combinations of splicing regulators.
As acknowledged by Blanchette et al. [17], questions still remain about why some targets are affected similarly by different regulators. There might be co-occurring binding sites in the same set of exons, or the regulators might have overlapping binding specificities, or the regulators might be interacting with another common regulator already present. Knockdowns and overexpression of more splicing factors, and parallel analyses of the sequence similarity of regulated exons (and flanking introns), as well as the RNA binding domain characteristics of the splicing regulators, should clarify these questions.
Regulating the regulators with small-molecule inhibitors
Various ways of correcting splicing defects have been sought. Disease-associated exons can be induced to skip by antisense oligonucleotides [20,21], or exon inclusion can be restored by synthetic exon-specific effectors [22] or mediated by RNA trans-splicing [23]. Although they are effective in correcting the splicing defect, these methods are not readily adaptable to high-throughput analysis with a view to finding drugs that rescue aberrant splicing events.
In another attempt to correct splicing defects, Sorel et al. [18] investigated the inhibition of the recombinant SR protein ASF/SF2 by small-molecule compounds. This approach lends itself well to high-throughput methods. In earlier work the authors had found that drugs interfering with the kinase activity of topoisomerase I (topo I) affect the phosphorylation status of SR proteins and prevent spliceosomal assembly (see reference cited in [18]). Building on this, they screened 2,500 chemical compounds for the inhibition of topo I phosphorylation of ASF/SF2, and obtained 28 potent inhibitors (Figure 2a). To determine whether these compounds selectively inhibit in vitro splicing of pre-mRNAs, the authors tested these 28 compounds on two pre-mRNA substrates: a synthetic single-intron pre-mRNA derived from the adenovirus major late-transcription unit (Minx), and a derivative of the human β-globin gene that contains three exonic splicing enhancers resembling a high-affinity ASF/SF2-binding site (βglo-3S). Three compounds had strong inhibitory effects on βglo-3S, whose splicing depends on ESE sequences, but only one of the three affected Minx pre-mRNA splicing, which is independent of ESE sequences (Figure 2a).
Figure 2.
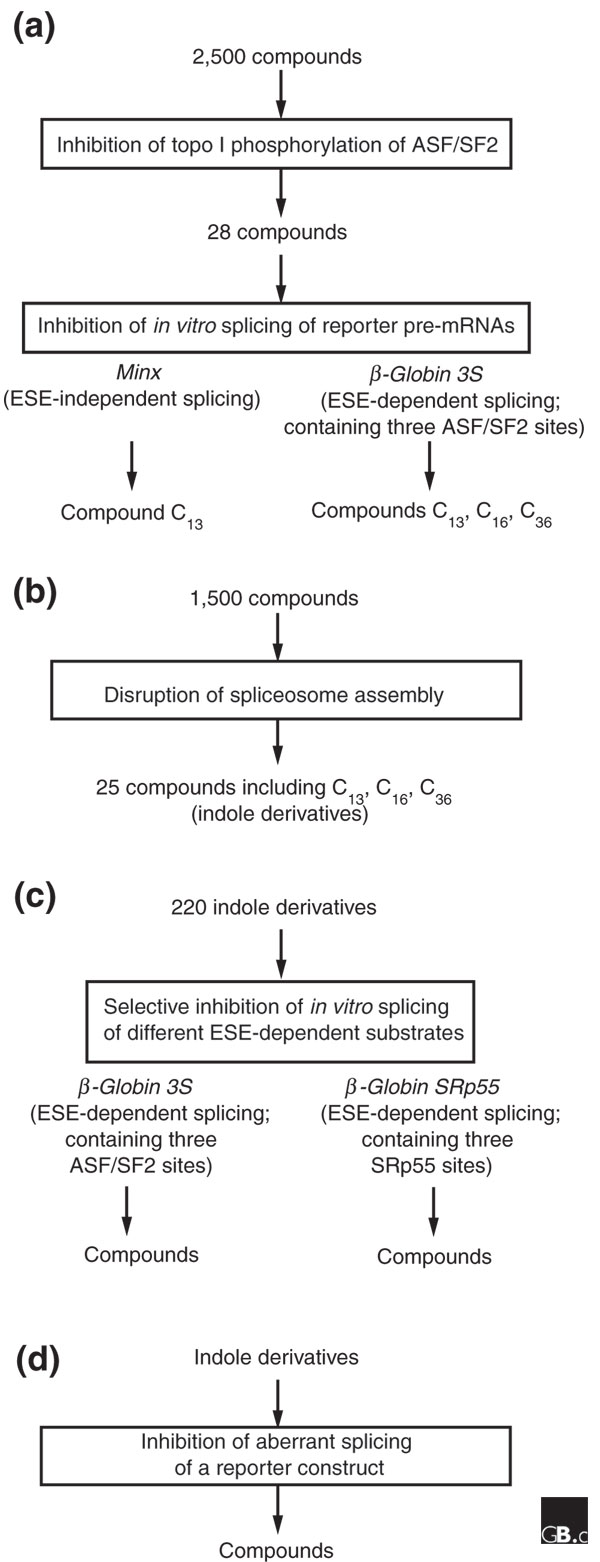
Screening strategies for small molecules that inhibit SR protein-mediated splicing [18]. (a) Compounds were screened for their ability to inhibit topoisomerase I phosphorylation of ASF/SF2, and then for repression of splicing of reporter constructs. (b) Compounds were screened for disruption of spliceosomal assembly, resulting in compounds that were indole derivatives. (c) Indole derivatives were screened for selective disruption of reporter pre-mRNAs where splicing was ASF/SF2 or SRp55 dependent. (d) Indole derivatives were screened for the ability to inhibit aberrant splicing. See text for further details.
To determine which stage of spliceosome assembly was affected, Soret et al. [18] then added 32P-labeled βglo-3S pre-mRNA to HeLa nuclear extracts and incubated the mixture with each of the three compounds. An early step of assembly must have been disrupted, as no splicing complexes were formed. Realizing that monitoring spliceosome assembly would be a more straightforward way of identifying compounds that affect splicing, the authors screened 1,500 small molecules and identified 25 candidates (Figure 2b). Surprisingly, these all have similar structures, being indole derivatives of the pyridocarbazole, benzopyridoindole or pyrido-pyrrolo-isoquinoline classes.
The next question was whether these compounds were targeting ASF/SF2 directly, or the kinase activity of topo I kinase, or both. Taking advantage of the strong intrinsic fluorescence of one of the compounds, the authors found that it interacted with ASF/SF2 directly rather than with topo I (80% of drug fluorescence was quenched upon binding to ASF/SF2). They found that while the overall structure of ASF/SF2 is important, the RS domain of the protein is the major drug-binding element. The RS domain contains the repeated arginine (R)-serine (S) dipeptides that characterize SR proteins. These domains are present not only in the SR proteins but also in canonical splicing factors such as the U1-snRNP-specific proteins U1-70K and U2AF. Their inhibition could have effects on general RNA splicing and this emphasizes the importance of identifying all possible targets of potential drugs to avoid problems of toxicity. Figure 3 illustrates three possible classes of drug, from the most potentially toxic to the most specific and safest.
Figure 3.
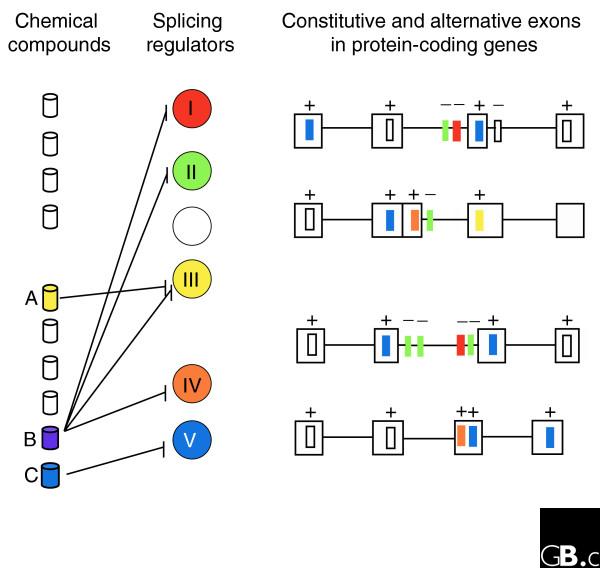
Predicting the toxicity of drugs that affect splicing. Four genes with cis-regulatory elements depicted as in Figure 1 are on the right, with the corresponding splicing regulators symbolized as circles. Chemical compounds are illustrated as cylinders on the left and are connected to the regulator(s) with which they interfere. The most toxic drugs would be those that affect a common regulator that binds to elements common to many genes, for example, drug C affecting regulator V. Drugs that affect several regulators with uncommon target elements would be less toxic: for example, drug B interfering with regulators I, II, III, and IV. The least toxic class would be drugs that specifically affect an uncommon regulator, such as drug A affecting regulator III.
In a subsequent experiment, Soret et al. [18] screened more than 200 indole derivatives for their ability to inhibit the splicing of βglo-3S by ASF/SF2 or the splicing of βglo-SRp55 by SRp55 (Figure 2c). In the latter construct, the sequences with high affinity for ASF/SF2 have been replaced by the optimal binding site for SRp55. Satisfyingly, they discovered that some drugs are specific for one of the SR proteins while some affected both. In a further experiment, they created a gain-of-function mutation in HeLa cells, in which a G-to-A change in an intron of the E1α pyruvate dehydrogenase (PDH) gene generates an ESE that binds the SR protein SC35, and activates a cryptic 5' splice site downstream of the mutation in the same intron. Consistent with indole derivatives selectively inhibiting splicing depending on the SR proteins involved, two drugs were shown to inhibit the use of the cryptic splice site, presumably by affecting the binding of SC35 (Figure 2d). The type of mutation created in PDH is likely to be similar to splicing mutations that generate defective proteins in humans, and provides an avenue for therapeutic intervention (Figure 4).
Figure 4.
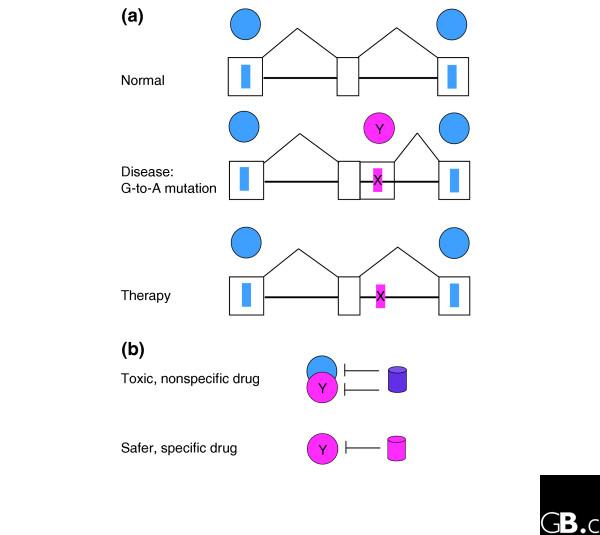
Rescuing aberrant splicing with small-molecule drugs. (a) A gene that is normally spliced with three exons is depicted in the top row. The filled circles represent splicing regulators that act at the sites depicted by the filled rectangles to promote splicing. Exons are depicted as outlined boxes. The second row shows the effects of a G-to-A mutation in the downstream intron that creates a binding site (filled rectangle named X) for a regulator (circle Y) which activates a cryptic 5' splice site, leading to the splicing of an additional sequence into the final mRNA and the production of a defective protein. The third row shows the effects of therapy with a drug that abolishes binding of the Y regulator and restores normal splicing. (b) A drug (cylinder) that nonspecifically inhibits both regulator Y and other common regulators will correct the effects of mutation X but will be more toxic than a drug that inhibits only regulator Y.
As icing on the cake, Soret et al. [18] looked at the effects of their indole derivatives on the splicing of the pre-mRNA of the human immunodeficiency virus HIV-1. This pre-mRNA is regulated by alternative splicing involving SR proteins such as ASF/SF2 and SC35. Chronically infected human promonocytic U1 cell lines, which can be stimulated to produce large quantities of HIV-1 pre-mRNA, were treated with a number of indole derivatives [18]. Several of these were shown specifically to affect HIV-1 alternative splicing, and abolished HIV-1 virion production. Neither cell viability nor the splicing profiles of endogenous genes were affected, indicating the potentially low toxicity of this treatment. This remarkable discovery opens new approaches to treatment of HIV-1 infection by indole derivatives via the interruption of ESE-mediated alternative splicing.
The studies of Blanchette et al. [17] and Soret et al. [18] are the first large-scale screens for the genome-wide targets of a small set of splicing regulators and for compounds that disrupt a specific class of splicing regulators. One can envisage combining the power of the two methods. For example, splicing-specific microarrays can query alternative splicing events affected by particular compounds. Comparing these events to alternative events affected by knockdowns of particular regulators may identify the candidate regulators affected by the compounds. More comprehensive siRNA screens of other RNA-binding proteins [24] using splicing-specific microarrays could point to new roles for splicing modulation. Looking forward, a large-scale mapping of regulators to targets in humans, and of compounds to regulators, combined with computational extraction of potential cis-regulatory binding sites, will be essential in the screening and correction of splicing defects in human disease.
Acknowledgments
Acknowledgements
G.Y. is supported by the Crick-Jacobs fellowship at the Salk Institute. G.Y. acknowledges Xiang-dong Xu and Nicole Coufal for helpful comments on the manuscript.
References
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003;302:2141–2144. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- Modrek B, Resch A, Grasso C, Lee C. Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res. 2001;29:2850–2859. doi: 10.1093/nar/29.13.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladd AN, Cooper TA. Finding signals that regulate alternative splicing in the post-genomic era. Genome Biol. 2002;3:reviews0008.1–0008.16. doi: 10.1186/gb-2002-3-11-reviews0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng ZM. Regulation of alternative RNA splicing by exon definition and exon sequences in viral and mammalian gene expression. J Biomed Sci. 2004;11:278–294. doi: 10.1159/000077096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR. Sorting out the complexity of SR protein functions. RNA. 2000;6:1197–1211. doi: 10.1017/S1355838200000960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Gatto-Konczak F, Olive M, Gesnel MC, Breathnach R. hnRNP A1 recruited to an exon in vivo can function as an exon splicing silencer. Mol Cell Biol. 1999;19:251–260. doi: 10.1128/mcb.19.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Mayeda A, Krainer AR. Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol Cell. 2001;8:1351–1361. doi: 10.1016/S1097-2765(01)00409-9. [DOI] [PubMed] [Google Scholar]
- Caceres JF, Stamm S, Helfman DM, Krainer AR. Regulation of alternative splicing in vivo by overexpression of antagonistic splicing factors. Science. 1994;265:1706–1709. doi: 10.1126/science.8085156. [DOI] [PubMed] [Google Scholar]
- Musunuru K. Cell-specific RNA-binding proteins in human disease. Trends Cardiovasc Med. 2003;13:188–195. doi: 10.1016/S1050-1738(03)00075-6. [DOI] [PubMed] [Google Scholar]
- Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev. 2003;17:419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- Caceres JF, Kornblihtt AR. Alternative splicing: multiple control mechanisms and involvement in human disease. Trends Genet. 2002;18:186–193. doi: 10.1016/S0168-9525(01)02626-9. [DOI] [PubMed] [Google Scholar]
- Garcia-Blanco MA, Baraniak AP, Lasda EL. Alternative splicing in disease and therapy. Nat Biotechnol. 2004;22:535–546. doi: 10.1038/nbt964. [DOI] [PubMed] [Google Scholar]
- Pan Q, Shai O, Misquitta C, Zhang W, Saltzman AL, Mohammad N, Babak T, Siu H, Hughes TR, Morris QD, et al. Revealing global regulatory features of mammalian alternative splicing using a quantitative microarray platform. Mol Cell. 2004;16:929–941. doi: 10.1016/j.molcel.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Clark TA, Sugnet CW, Ares M., Jr Genomewide analysis of mRNA processing in yeast using splicing-specific microarrays. Science. 2002;296:907–910. doi: 10.1126/science.1069415. [DOI] [PubMed] [Google Scholar]
- Blanchette M, Green RE, Brenner SE, Rio DC. Global analysis of positive and negative pre-mRNA splicing regulators in Drosophila. Genes Dev. 2005;19:1306–1314. doi: 10.1101/gad.1314205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soret J, Bakkour N, Maire S, Durand S, Zekri L, Gabut M, Fic W, Divita G, Rivalle C, Dauzonne D, et al. Selective modification of alternative splicing by indole derivatives that target serine-arginine-rich protein splicing factors. Proc Natl Acad Sci USA. 2005;102:8764–8769. doi: 10.1073/pnas.0409829102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castle J, Garrett-Engele P, Armour CD, Duenwald SJ, Loerch PM, Meyer MR, Schadt EE, Stoughton R, Parrish ML, Shoemaker DD, Johnson JM. Optimization of oligonucleotide arrays and RNA amplification protocols for analysis of transcript structure and alternative splicing. Genome Biol. 2003;4:R66. doi: 10.1186/gb-2003-4-10-r66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skordis LA, Dunckley MG, Yue B, Eperon IC, Muntoni F. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc Natl Acad Sci USA. 2003;100:4114–4119. doi: 10.1073/pnas.0633863100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunckley MG, Manoharan M, Villiet P, Eperon IC, Dickson G. Modification of splicing in the dystrophin gene in cultured Mdx muscle cells by antisense oligoribonucleotides. Hum Mol Genet. 1998;7:1083–1090. doi: 10.1093/hmg/7.7.1083. [DOI] [PubMed] [Google Scholar]
- Cartegni L, Krainer AR. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat Struct Biol. 2003;10:120–125. doi: 10.1038/nsb887. [DOI] [PubMed] [Google Scholar]
- Garcia-Blanco MA. Messenger RNA reprogramming by spliceosome-mediated RNA trans-splicing. J Clin Invest. 2003;112:474–480. doi: 10.1172/JCI200319462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Parisky K, Celotto AM, Reenan RA, Graveley BR. Identification of alternative splicing regulators by RNA interference in Drosophila. Proc Natl Acad Sci USA. 2004;101:15974–15979. doi: 10.1073/pnas.0407004101. [DOI] [PMC free article] [PubMed] [Google Scholar]