

Short abstract
Recent work shows that the inhibition of the SOS stress response in Escherichia coli reduces the development of resistance to the antibiotics ciprofloxacin and rifampicin.
Abstract
Recent work shows that the inhibition of the SOS stress response in Escherichia coli reduces the development of resistance to the antibiotics ciprofloxacin and rifampicin. This finding may help in the battle against the rise of resistance to antimicrobial drugs.
Bacterial resistance to antimicrobial drugs is currently receiving much publicity and political attention. The cost to health services around the world is counted in billions of dollars; an increase in morbidity and mortality are the costs to those infected. The problem is getting worse and treatment options for combating bacteria resistant to multiple drugs are narrowing. Unless something is done, we may well return to the horrors of the pre-antibiotic era. In a recent article, Cirz and colleagues [1] report the interesting finding that the rate of resistance to some drugs in Escherichia coli can be greatly reduced by interfering with a bacterial stress response. This article sets the work by Cirz et al. [1] in the general context of antimicrobial drug resistance and discusses whether this new finding could be helpful in the battle against the rise of drug-resistant bacteria.
Are 'mutation-busting' drugs the answer to the problem of drug resistance?
Resistance to antimicrobials occurs in four main ways (Figure 1). The first possible mechanism is the mutation of the drug's target; a classic example of this is the mutation of gyrA, encoding the essential DNA gyrase A subunit, the major target of quinolones such as ciprofloxacin in E. coli [2]. A second mechanism is a bypass of the drug's target by the acquisition of a similar but insensitive target protein. A good example here would be the acquisition of a plasmid-borne dihydrofolate reductase (DHFR) insensitive to trimethoprim; the acquired DHFR compensates for the inhibition of the host's DHFR in the presence of trimethoprim and is the predominant cause of resistance to this antimicrobial drug in E. coli [3]. A third mechanism is the enzymatic degradation or modification of the drug; a well-known example is the destruction of β-lactam antibiotics by plasmid-mediated TEM β-lactamase, which accounts for around 90% of all ampicillin resistance in E. coli [4]. And fourth, resistance can be caused by a nonspecific reduced permeability to antimicrobial drugs. This is typically caused by reduced production of porins, the protein channels that allow antimicrobials through the outer bacterial membrane, and/or an increased production of drug-efflux pumps, which remove drugs from both cytoplasm and periplasm [5]. These two events are often coordinated, for example through the Mar regulon in E. coli, which - when constitutively activated by mutation - leads to resistance to multiple antimicrobial drugs [6].
Figure 1.
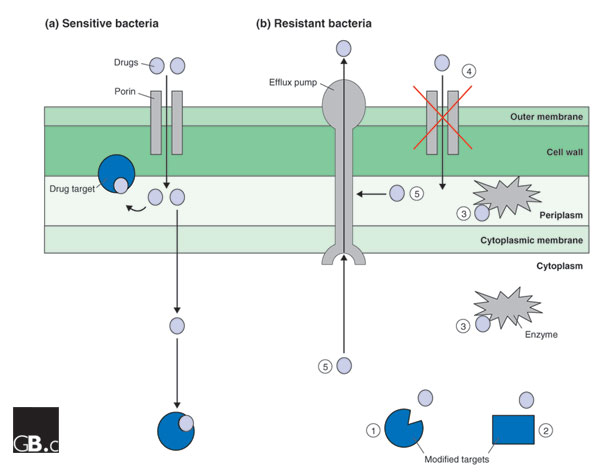
Antimicrobial drug-resistance mechanisms. A typical Gram-negative bacterial cell envelope is shown, consisting of the outer membrane, the peptidoglycan cell wall, the periplasm, which contains enzymes required to synthesize the cell wall, and the cytoplasmic membrane. (a) The entry point and targets of antimicrobial drugs in a non-resistant bacterium. Drugs enter the periplasm through porins in the outer membrane, and some drugs exert their effect in the periplasm; for example, ampicillin and the other β-lactams interfere with the synthesis of the cell wall. Other drugs cross the cytoplasmic membrane and inhibit cytoplasmic targets. (b) Possible resistance mechanisms: (1) Mutation of a target so that it is no longer inhibited by the drug; (2) acquisition, for example on a plasmid, of a novel target that is not sensitive to the actions of the drug; (3) enzymatic destruction or modification of the drug either in the cytoplasm, for example, the inactivation of gentamicin by aminoglycoside-modifying enzymes, or in the periplasm, for example, the destruction of β-lactams by β-lactamase; (4) reduction of the cytoplasmic, and usually periplasmic, concentration of the drug through reduction in the expression of porin genes or loss-of-function mutations in porin genes; (5) removal of drugs from the periplasm or cytoplasm by efflux pumps. In some cases, drug resistance is due to a combination of these mechanisms.
Mutations are an unavoidable fact of life, but it has long been known that gyrA mutations leading to ciprofloxacin resistance in E. coli occur at a higher frequency than one might expect given E. coli's general rate of mutation [7]. One process that leads to increased mutation in E. coli is the SOS response, which is triggered in response to a wide range of stress conditions. It is known to cause increased mutation rates, particularly following DNA damage. The unblocking of stalled replication forks, which are a common and potentially lethal result of DNA damage, requires the SOS response, for example. The trigger for the SOS response is the autolytic degradation of the transcriptional repressor/protease hybrid, LexA, thus derepressing the expression of a group of genes whose products are responsible for DNA repair and unblocking of stalled replication forks. These proteins include error-prone DNA polymerases, whose activities lead to the increased frequency of mutation seen in cells during the SOS response [8].
In their recent article, Cirz et al. [1] postulate that ciprofloxacin induces DNA damage and so instigates the SOS response, thereby increasing the frequency at which ciprofloxacin-resistant mutants arise in E. coli. Through a series of in vitro experiments, they confirmed that this is the case, and that mutations in lexA that block the SOS response result in a reduction in the apparent frequency of mutation to ciprofloxacin resistance in vitro. Another lexA mutant strain had the ciprofloxacin mutation frequency apparently reduced to zero compared to the wild-type parent, when tested in a murine model of infection [1].
The gyrA mutations caused by ciprofloxacin-mediated induction of the SOS response were all confirmed as being base substitutions, typical of those seen in the clinic. Cirz et al. [1] postulate, however, that ciprofloxacin also induces some other repair system, which causes small deletions, as some of the ciprofloxacin-resistant mutants had deletions of entire triplets in gyrA. Such mutants are not seen clinically, and it is highly likely that they come with an extreme fitness cost.
The authors found the same pattern of results when looking at the frequency of occurrence of rifampicin-resistant mutants caused by point mutations in the rpoB gene encoding the beta subunit of RNA polymerase. This is perhaps surprising, because rifampicin does not cause DNA damage, being an inhibitor of RNA polymerase. Several antibiotics are, however, known to cause metabolic stress and induce the SOS response without overtly causing DNA damage; these include for example, the β-lactams [9].
The work of Cirz et al. [1] at least opens up the possibility that inhibitors of the SOS response might represent drugs that reduce point mutation rates in bacteria, and so reduce the frequency with which drug-resistant mutations occur. But would this really be of benefit? Taking the example of quinolone resistance in E. coli, the answer is 'yes and no'. As well as target-site mutations, efflux pump and/or porin regulatory mutations can cause antimicrobial drug resistance [5], so mutation-busting drugs could be doubly helpful. On the other hand, mutations are not the only cause of resistance. For example, mobile genetic elements can cause quinolone resistance in two main ways. First, the insertion of mobile IS elements can derepress efflux-pump gene expression and disrupt porin genes [5]. Second, plasmid-mediated quinolone-resistance determinants are becoming increasingly common; these encode proteins that bind to the active sites of quinolone targets and occlude the drugs [10]. Indeed, there are no classes of antimicrobial drug for which point mutations are the sole reason for the development of resistance. So the best one can say is that mutation-busting drugs would reduce the development of resistance due to mutation. They would, however, do nothing to prevent the development of resistance due to mobile genetic elements. Furthermore, it is likely that by the time they have been developed, many of the mutations they are designed to stop will already have occurred.
Other approaches to solving the problem of antimicrobial drug resistance
Other strategies for combating antimicrobial drug resistance fall into three main types. First, simply develop new drugs. The post-genomic era has led to the discovery of a whole host of essential genes in bacteria whose products might represent targets for novel antimicrobial drugs. But the exploitation of these targets is proving very difficult. More useful has been the adaptation of known drug scaffolds so that they overcome existing resistance mechanisms [11,12]. It is, however, unlikely that permeability-mediated resistance mechanisms of Gram-negative bacteria will be overcome by these new drug variants, as these resistance mechanisms affect a broad spectrum of antibiotics [5]. The second approach is to stop using a particular drug and reintroduce it when resistance levels have fallen. This idea derives from the assumption that resistance mechanisms come with a fitness cost and that in the absence of selection, resistant strains will be out-competed by sensitive strains. Recent work has revealed, however, that most resistance mechanisms impose no significant fitness cost; indeed some may provide a fitness advantage [13,14], and this may explain why sulfonamide-resistance levels in E. coli did not fall in the UK even 10 years after the use of sulfonamides had been discontinued [15]. The third strategy is to learn more about the resistance mechanisms themselves. This area of research is focused on degradative enzymes and efflux pumps. The β-lactamase inhibitors already used clinically have been most successful but do not inhibit a large swathe of these enzymes, so more are required [16]; efflux-pump inhibitors exist but are not currently in a clinically useful form [17].
In conclusion, the problem of antimicrobial drug resistance is very real, and is set to get worse before it gets better. The more we learn about the responses of bacteria to antimicrobial challenge, and about the fundamental mechanisms of drug resistance in bacteria, the more likely we are to be able to develop strategies for reducing the burden of resistance. The availability of large amounts of complete bacterial genome sequence data, coupled with the development of post-genomic technologies aimed at comparing gene complements and gene-expression patterns in resistant and non-resistant bacteria (for example [18,19]), gives us an excellent platform to study resistance mechanisms. So, the dawn of the post-genomic era may help to delay a return to the pre-antibiotic era. Only time will tell.
Acknowledgments
Acknowledgements
I thank the Biotechnology and Biological Research Council, the Medical Research Council, the Wellcome Trust, the British Society for Antimicrobial Chemotherapy and the Royal Society for funding.
References
- Cirz RT, Chin JK, Andes DR, de Crecy-Lagard V, Craig WA, Romesberg FE. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol. 2005;3:e176. doi: 10.1371/journal.pbio.0030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkey PM. Mechanisms of quinolone action and microbial response. J Antimicrob Chemother. 2003;51(Suppl 1):29–35. doi: 10.1093/jac/dkg207. [DOI] [PubMed] [Google Scholar]
- Huovinen P, Sundström L, Swedburg G, Sköld O. Trimethoprim and sulfonamide resistance. Antimicrob Agents Chemother. 1995;39:279–289. doi: 10.1128/aac.39.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford PA. Extended-spectrum beta-lactamases in the 21st century: characterization, epidemiology, and detection of this important resistance threat. Clin Microbiol Rev. 2001;14:933–951. doi: 10.1128/CMR.14.4.933-951.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole K. Efflux-mediated antimicrobial resistance. J Antimicrob Chemother. 2005;56:20–51. doi: 10.1093/jac/dki171. [DOI] [PubMed] [Google Scholar]
- Alekshun MN, Levy SB. The mar regulon: multiple resistance to antibiotics and other toxic chemicals. Trends Microbiol. 1999;7:410–413. doi: 10.1016/s0966-842x(99)01589-9. [DOI] [PubMed] [Google Scholar]
- Riesenfeld C, Everett M, Piddock LJV, Hall BG. Adaptive mutations produce resistance to ciprofloxacin. Antimicrob Agents Chemother. 1997;41:2059–2060. doi: 10.1128/aac.41.9.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. Stress responses and genetic variation in bacteria. Mutat Res. 2005;569:3–11. doi: 10.1016/j.mrfmmm.2004.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C, Thompson LE, Gaggero C, Mosseri R, Ingmer H, Cohen SN. SOS response induction by β-lactams and bacterial defence against antibiotic lethality. Science. 2004;305:1629–1631. doi: 10.1126/science.1101630. [DOI] [PubMed] [Google Scholar]
- Nordmann P, Poirel L. Emergence of plasmid-mediated resistance to quinolones in Enterobacteriaceae. J Antimicrob Chemother. 2005;56:463–469. doi: 10.1093/jac/dki245. [DOI] [PubMed] [Google Scholar]
- Schmid MB. Seeing is believing: the impact of structural genomics on antimicrobial drug discovery. Nat Rev Microbiol. 2004;2:739–748. doi: 10.1038/nrmicro978. [DOI] [PubMed] [Google Scholar]
- Livermore DM. Minimising antibiotic resistance. Lancet Infect Dis. 2005;5:450–459. doi: 10.1016/S1473-3099(05)70166-3. [DOI] [PubMed] [Google Scholar]
- Enne VI, Delsol AA, Davis GR, Hayward SL, Roe JM, Bennett PM. Assessment of the fitness impacts on Escherichia coli of acquisition of antibiotic resistance genes encoded by different types of genetic element. J Antimicrob Chemother. 2005;56:544–551. doi: 10.1093/jac/dki255. [DOI] [PubMed] [Google Scholar]
- Enne VI, Bennett PM, Livermore DM, Hall LM. Enhancement of host fitness by the sul2-coding plasmid p9123 in the absence of selective pressure. J Antimicrob Chemother. 2004;53:958–963. doi: 10.1093/jac/dkh217. [DOI] [PubMed] [Google Scholar]
- Enne VI, Livermore DM, Stephens P, Hall LM. Persistence of sulphonamide resistance in Escherichia coli in the UK despite national prescribing restriction. Lancet. 2001;357:1325–1328. doi: 10.1016/S0140-6736(00)04519-0. [DOI] [PubMed] [Google Scholar]
- Bush K. The impact of β-lactamases on the development of novel antimicrobial agents. Curr Opin Investig Drugs. 2002;3:1284–1290. [PubMed] [Google Scholar]
- Kaatz GW. Bacterial efflux pump inhibition. Curr Opin Investig Drugs. 2005;6:191–198. [PubMed] [Google Scholar]
- Avison MB, Bennett PM, Howe RA, Walsh TR. Preliminary analysis of the genetic basis for vancomycin resistance in Staphylococcus aureus strain Mu50. J Antimicrob Chemother. 2002;49:255–260. doi: 10.1093/jac/49.2.255. [DOI] [PubMed] [Google Scholar]
- Cui L, Lian JQ, Neoh HM, Reyes E, Hiramatsu K. DNA microarray-based identification of genes associated with glycopeptide resistance in Staphylococcus aureus. Antimicrob Agents Chemother. 2005;49:3504–3513. doi: 10.1128/AAC.49.8.3404-3413.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]