

Short abstract
Recent work has identified the topology of almost all the inner membrane proteins in Escherichia coli, and advances in nuclear magnetic resonance spectroscopy now allow the determination of α-helical membrane protein structures at high resolution.
Abstract
Recent work has identified the topology of almost all the inner membrane proteins in Escherichia coli, and advances in nuclear magnetic resonance spectroscopy now allow the determination of α-helical membrane protein structures at high resolution. Together these developments will help overcome the current limitations of high-throughput determination of membrane protein structures.
The structural genomics initiatives now underway worldwide have the ultimate aim of determining the structures and functions of all proteins. The field has developed rapidly over the past five years and the rate at which structure entries are being deposited in the public databases has increased significantly (Figure 1a). Structural genomics relies primarily on X-ray crystallography, nuclear magnetic resonance (NMR) and computational model building to determine protein structure. High-throughput operations for many of the processes involved have already been developed, and the field is currently funded at a significant level in the United States, Canada, the European Union, Israel, China, and Japan. Genomic sequence analysis predicts that 20-30% of proteins produced by most organisms will be integral membrane proteins, which as a class are critical for many essential cellular functions and constitute 60-70% of current drug targets [1]. Less than 1% of the atomic structures in the Protein Data Bank represent membrane proteins (Figure 1b), however, and this percentage is actually decreasing as more and more structures of soluble proteins are being added every day. Membrane protein structure determination, especially for α-helical membrane proteins in which the transmembrane portion of the protein is in the form of one or more α-helices rather than a β-barrel, may look as though it is falling behind the rest of the field, but several exciting developments over the past year should change this situation.
Figure 1.
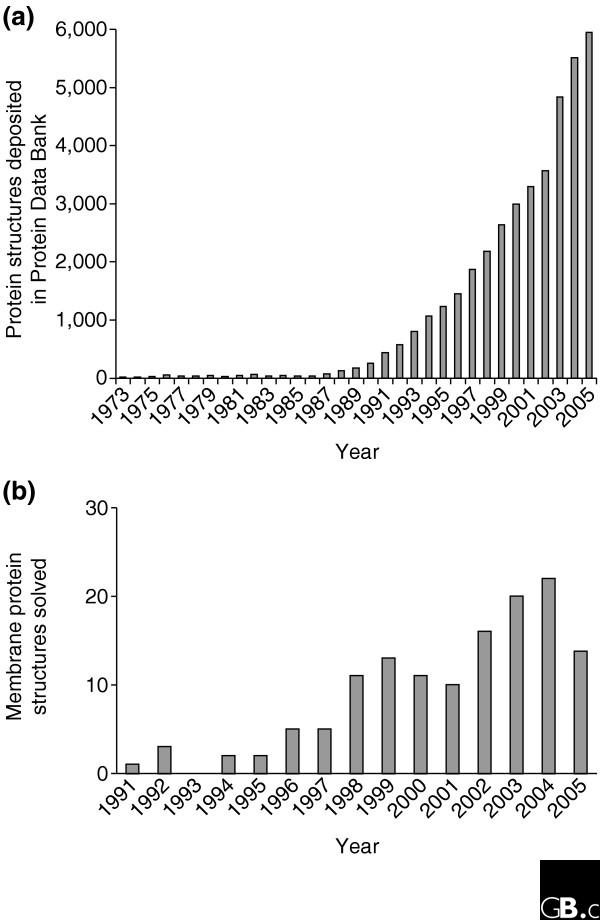
Number of protein structures and membrane protein structures deposited annually in the Protein Data Bank (PDB). (a) The total number of structures deposited in the PDB per year. The data are taken from the PDB website [17], which was last updated on 13 December 2005; the PDB currently holds 31,248 protein structures in total. (b) The number of unique membrane protein structures solved for the years indicated. The data are taken from [18], which was last updated on 11 December 2005.
Genome-wide membrane topology determination
As noted in a previous review [2], the major bottlenecks in membrane protein structural genomics are the identification of potential membrane proteins in selected genomes and the production of the milligram quantities of protein necessary for most structure determination techniques. In most cases, accurate homology-based prediction of protein type and function is not possible for membrane proteins, as currently available bioinformatic tools detect membrane proteins in genomes solely on the basis of predicting transmembrane segments [3], and predictions from different programs sometimes do not agree with one another. To provide more information for identifying and characterizing predicted membrane proteins, Daley and colleagues [4] recently used a combination of bioinformatic and experimental approaches to develop a successful method for the topology analysis of almost all the inner membrane proteins in the Escherichia coli genome. Topological models of membrane proteins describe the numbers of transmembrane segments and the orientation of the protein with respect to the lipid bilayer. An accurate topology model of a membrane protein not only provides reliable information to aid the identification of membrane proteins but is also important for functional protein analysis.
Experimental approaches to determining topology usually deal with proteins individually and are very time-consuming. In contrast, Daley et al. [4] first used a simple and reliable experimental approach to determine the location of the carboxyl termini of nearly all the inner membrane proteins in E. coli. They genetically fused the reporter tags alkaline phosphatase (PhoA) or green fluorescent protein (GFP) to the carboxyl terminus of each prospective membrane protein sequence to exploit the fact that PhoA activity can only be detected in the periplasm (the space between the inner and outer membranes of E. coli), whereas GFP only fluoresces in the cytoplasm. The location of the carboxyl terminus of a membrane protein with respect to the cytoplasmic membrane can thus be accurately determined. The authors then used the experimentally determined carboxyl terminus location as a constraint for the widely used hidden Markov model (HMM) program TMHMM for transmembrane topology prediction [5] to generate a topology model for each protein.
Out of approximately 1,000 genes predicted by TMHMM to be inner membrane proteins in the E. coli genome, Daley and coworkers [4] focused on 737 proteins. Other proteins predicted to have a single transmembrane segment (monotopic proteins) were left out of the study, as it remains a major challenge to distinguish secreted proteins from monotopic integral membrane proteins; even so, Daley et al. [4] were able to determine the locations of the carboxyl termini of 502 proteins out of 665 proteins whose genes could be cloned into the vectors used. In addition, the carboxy-terminal location of another 99 proteins out of the 737 proteins was determined by finding their homologs among the 502 experimentally determined proteins. When the resulting set of 601 proteins was compared with 71 proteins for which the location of the carboxyl terminus was known previously, 69 agreed with previous assignments. Further studies are needed to resolve the discrepancies associated with the remaining two proteins. This brings the success rate of the carboxyl terminus assignment in the study by Daley et al. [4] to the order of 99% or higher. The accuracy of carboxyl terminus prediction using TMHMM alone was tested for all the 601 proteins, and was only 78%. Significant improvements in the quality of the topology models for these inner membrane proteins have therefore been achieved by using the experimentally determined constraints. This combination of bioinformatic and experimental approaches has laid a foundation for the functional analysis of these inner membrane proteins, and the method can be readily applied to integral membrane proteins of other genomes. An interesting finding by Daley et al. [4] is that 57% of the 601 proteins studied have both their amino and carboxyl termini on the cytoplasmic side of the membrane. This indicates that two closely spaced transmembrane helices separated by a short hydrophilic loop ('helical hairpin') might be a basic building block of membrane proteins.
Overexpression of membrane proteins in bacteria
One of the major concerns for membrane protein production in bacteria is the potential toxicity of these proteins to the host, limiting the ability to express proteins at high level [2]. Another very important finding of Daley et al. [4] is therefore that the overexpression of a vast majority of the membrane proteins fusion constructs had only a limited effect on cell growth. Not only are these proteins typically not toxic, but it was also estimated that about 50% of the GFP fusion proteins could be overexpressed with little harmful effects - a rate similar to the overexpression usually achieved for soluble proteins. There are many possible reasons why the other 50% of these proteins were not overexpressed; their low stability in the host cells might be one of them. In a study of the attempted expression of 99 putative membrane proteins from Mycobacterium tuberculosis in E. coli, not a single case of cell lysis was observed [6]. In the case of the mycobacterial proteins, the use of E. coli codons and strains, the T7 promoter, and short His-tags as reporters, together with the choice of strain for the expression host, was shown to allow the expression of 'foreign' proteins with a broad range of molecular weights and number of transmembrane helices. Some 50% of the 99 putative mycobacterial protein sequences were expressed and 25% were overexpressed, in good agreement with the results of Daley et al. [4].
Another significant challenge for structural genomics is the production of purified membrane proteins in large quantities from cloned genes. As just discussed, Daley et al. [4] and others [6] have shown that a significant percentage of prokaryotic integral membrane proteins can be readily produced. The GFP fusion construct used by Daley et al. [4] has a cleavable His8-tag, which allows the proteins to be purified by Ni-affinity chromatography by a standard protocol. It thus seems that the production of membrane proteins in large enough quantities for structure determination can be achieved in bacteria, and this may no longer be the rate-limiting step for membrane protein structural genomics.
Advances in NMR technology
It was noted by Daley et al. [4] that most of the E. coli membrane proteins whose function is still unknown have fewer than six transmembrane helices. This indicates a systematic lack of studies with the smaller integral membrane proteins and reflects the fact that most of the membrane protein structures obtained by X-ray diffraction represent large membrane proteins or membrane protein complexes. This bias is likely to be because the larger proteins form crystals more easily than smaller proteins. The larger the protein, the larger the ratio of protein volume to the protein surface area in contact with lipid, which is more favorable to the development of electrostatic contacts between unit cells in a crystal. The smaller the ratio, the more difficult it is to develop these electrostatic contacts. On the other hand, solution and solid-state NMR spectroscopy may be better suited for determining the structures of smaller proteins, and are therefore largely complementary to X-ray crystallography [2]. Each of these NMR methodologies has its advantages, and very significant breakthroughs have been made in the past year in both technologies. For example, detailed comparisons of a wide range of detergents have guided improved sample preparation protocols for solution NMR [7]. Further sample optimization for expression testing, purification and NMR sample preparation was reported by Tian and colleagues [8]. Today, excellent tools are in place for obtaining excellent samples of membrane proteins of modest molecular weight. Slightly anisotropic (directionally dependent) samples of detergent-solubilized membrane proteins represent specific structural challenges, but methods for preparing such samples have recently become better [9,10], and the characterization of helical tilt and orientation has also been improved [11].
After several decades of hard work, high-resolution structures of α-helical membrane proteins have finally been determined by solution NMR. Most recently, several new structures obtained by solution NMR have appeared that foreshadow a new wave of membrane-protein structures. Oxenoid and Chou [12] have determined the structure at atomic resolution of an α-helical membrane protein, human phospholamban pentamer, embedded in oriented aggregates (micelles) of the detergent dodecylphosphocholine, which substitutes for the lipid membrane. α-Helical membrane proteins are those in which the transmembrane portion of the protein is in the form of one or more α helices rather than a β barrel. The structure revealed that the phospholamban pentamer forms a channel that allows many physiologically relevant small ions, such as Na+, K+ and Cl-, to pass through the membrane. Howell et al. [13] have solved the backbone structure of the two α-helix membrane protein MerF, a component of the bacterial mercury detoxification system. These studies show that solution NMR spectroscopy can be used for structural determination of small and medium-sized α-helical membrane proteins.
It has long been thought that bicelles (bilayered mixed micelles) would be an ideal system in which to study membrane proteins, but in practice they have been used primarily to study synthetic peptides. An exciting development in this context is the optimization by De Angelis and colleagues [14] of the use of magnetically aligned bicelles for high-resolution structural determination of membrane proteins by solid-state NMR spectroscopy. The key to these workers' success is the use of nonhydrolyzable ether-linked lipids to prepare stable bicelles. They showed that purified small molecular membrane proteins in bicelles undergo rapid rotational diffusion around an axis perpendicular to the bilayer; high-resolution structure determination then becomes possible because of the averaging of the nuclear spin interactions, which would otherwise give a very broad NMR signal. Careful studies indicated that the membrane proteins were embedded in bicelles with little or no structural distortion, which often occurs in micelle preparation. Structural characterization is aided by the observation of a helical wheel-like pattern of the resonances in the spectra, called the PISA wheel [15,16]. The structure of MerF in bicelles is close to being finished (S. Opella, personal communication). It will provide an ideal system for studying the structure and mechanism of action of this and other membrane proteins in a lipid bilayer environment under fully hydrated physiological conditions.
The current rate at which unique structures are being solved for membrane proteins resembles the situation for soluble proteins 20 years ago (see Figure 1). As the international efforts of structural genomics start to focus on membrane proteins it is reasonable to expect that more and more high-resolution structures will become available. The time may finally have come for membrane protein structural genomics to move forward at the same pace as the rest of the field, and both solution and solid-state NMR spectroscopy will be technologies central in achieving this goal.
Acknowledgments
Acknowledgements
The authors thank S.J. Opella for helpful discussions. The work is supported by funding from the National Institutes of Health (P01-GM64676).
References
- Lundstrom K. Structural genomics on membrane proteins: the MePNet approach. Curr Opin Drug Discov Devel. 2004;7:342–346. [PubMed] [Google Scholar]
- Walian P, Cross TA, Jap BK. Structural genomics of membrane proteins. Genome Biol. 2004;5:215. doi: 10.1186/gb-2004-5-4-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Expert Protein Analysis System ExPASy Molecular Biology Server http://www.expasy.ch
- Daley DO, Rapp M, Granseth E, Melen K, Drew D, von Hejne G. Global topology analysis of the Escherichia coli inner membrane proteome. Science. 2005;308:1321–1323. doi: 10.1126/science.1109730. [DOI] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heilne G, Sonnhammer E. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–680. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Korepanova A, Gao FP, Hua Y, Qin H, Nakamoto RK, Cross TA. Cloning and expression of multiple integral membrane proteins from Mycobacterium tuberculosis in Escherichia coli. Protein Sci. 2005;14:148–158. doi: 10.1110/ps.041022305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger-Koplin RD, Sorgen PL, Krueger-Koplin ST, Rivera-Torres IO, Cahill SM, Hicks DB, Grinius L, Krulwich , Girvin ME. An evaluation of detergents for NMR studies of membrane proteins. J Biomol NMR. 2004;28:43–57. doi: 10.1023/B:JNMR.0000012875.80898.8f. [DOI] [PubMed] [Google Scholar]
- Tian C, Karra MD, Ellis CD, Jacob J, Oxenoid K, Sonnichsen F, Sanders CR. Membrane protein preparation for TROSY NMR screening. Methods Enzymol. 2005;394:321–324. doi: 10.1016/S0076-6879(05)94012-3. [DOI] [PubMed] [Google Scholar]
- Jones DH, Opella SJ. Weak alignment of membrane proteins in stressed polyacrylamide gels. J Magn Reson. 2004;171:258–269. doi: 10.1016/j.jmr.2004.08.022. [DOI] [PubMed] [Google Scholar]
- Cierpicki T, Bushweller JH. Charged gels as oriented media for measurement of residue dipolar couplings in soluble and membrane proteins. J Am Chem Soc. 2004;126:16259–16266. doi: 10.1021/ja046054g. [DOI] [PubMed] [Google Scholar]
- Nevzorov AA, Mesleh MF, Opella SJ. Structure determination of aligned samples of membrane proteins by NMR spectroscopy. Magn Reson Chem. 2004;42:162–171. doi: 10.1002/mrc.1320. [DOI] [PubMed] [Google Scholar]
- Oxenoid K, Chou JJ. The structure of phospholamban pentamer reveals a channel-like architecture in membranes. Proc Natl Acad Sci USA. 2005;102:10870–10875. doi: 10.1073/pnas.0504920102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell SC, Mesleh MF, Opella SJ. NMR structure determination of a membrane protein with two transmembrane helices in micelles: MerF of the bacterial mercury detoxification system. Biochemistry. 2005;44:5196–5206. doi: 10.1021/bi048095v. [DOI] [PubMed] [Google Scholar]
- De Angelis AA, Nevzorov AA, Park SH, Howell SC, Mrse AA, Opella SJ. High-resolution NMR spectroscopy of membrane proteins in aligned bicelles. J Am Chem Soc. 2004;126:15340–15341. doi: 10.1021/ja045631y. [DOI] [PubMed] [Google Scholar]
- Wang J, Denny J, Tian C, Kim S, Mo Y, Kovacs F, Song Z, Nishimura K, Gan Z, Fu R, et al. Imaging membrane protein helical wheels. J Magn Reson. 2000;144:162–167. doi: 10.1006/jmre.2000.2037. [DOI] [PubMed] [Google Scholar]
- Marassi FM, Opella SJ. A solid-state NMR index of helical membrane protein structure and topology. J Magn Reson. 2000;144:150–155. doi: 10.1006/jmre.2000.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The RCSB Protein Data Bank http://www.rcsb.org/pdb
- Membrane proteins of known structure http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html