

Short abstract
A two-step procedure for identifying regulatory motifs in distantly related organisms is described that combines the advantages of sequence alignment and motif detection approaches.
Abstract
Although proven successful in the identification of regulatory motifs, phylogenetic footprinting methods still show some shortcomings. To assess these difficulties, most apparent when applying phylogenetic footprinting to distantly related organisms, we developed a two-step procedure that combines the advantages of sequence alignment and motif detection approaches. The results on well-studied benchmark datasets indicate that the presented method outperforms other methods when the sequences become either too long or too heterogeneous in size.
Background
Phylogenetic footprinting is a comparative method that uses cross-species sequence conservation to identify new regulatory motifs [1]. Based on the observation that functional regulatory motifs evolve more slowly than non-functional sequences, the method identifies potential regulatory motifs by detecting conserved regions in orthologous intergenic sequences [2,3]. The comparison of orthologous sequences from multiple genomes is often based on multiple sequence alignment [4,5] and several alignment algorithms, such as CLUSTALW [6], DIALIGN [7,8], MAVID [9,10] and MLAGAN [11], have proven very useful to identify conserved motifs in closely related higher vertebrate sequences [4,12,13]. Although the comparison of closely related organisms has proven successful, inclusion of more distantly related species can greatly improve the detection of conserved regulatory motifs. By adding more distantly related sequences, the conserved functional motifs can be more easily distinguished from the often highly variable 'background' sequence. Moreover, this leads to the detection of motifs that have a function in a wider variety of organisms, for example, all vertebrates [14-19]. Both Sandelin et al. [20] and Woolfe et al. [21], for instance, performed a whole genome comparison of human and pufferfish, which diverged approximately 450 million years ago (mya) to discover non-coding elements conserved in both organisms. They showed that most of these conserved non-coding elements are located in regions of low gene density (implying long intergenic regions) [21]. Moreover, many of the conserved non-coding elements are located at large distances from the nearest gene [20,21]. These findings led to the conclusion that it is interesting to analyze whole intergenic regions of vertebrate genes, rather than limit the comparative analyses to the promoter region located near the transcription start.
However, vertebrate intergenic regions may differ considerably in size, such as when comparing intergenics of, for example, mammals with those of Fugu [22-24]. Since multiple sequence alignments are often based on global alignment procedures, they will likely fail to correctly align such sequences of heterogeneous length [25].
An alternative for alignment methods is the use of de novo motif detection procedures for phylogenetic footprinting. These are based on either probabilistic or combinatorial algorithms. One such method, FootPrinter [26,27], uses a string based motif representation with dynamic programming to search a phylogenetic tree for motifs that show a minimal number of mismatches. Probabilistic algorithms, such as MEME [28], Consensus [29,30] and Gibbs sampling [31,32], use a matrix representation of the motif (position specific weight matrix). Currently, several implementations of Gibbs sampling are available, such as AlignACE [33,34], ANN-spec [35], BioProspector [36] and MotifSampler [37-40]. However, these algorithms are sensitive to low signal-to-noise ratios, that is, the presence of small motifs (five to eight base pairs (bp)) in long intergenic sequences. This often results in the detection of many false positive motifs. On the other hand, an advantage of these procedures is that, because motif detection comes down to locally aligning the orthologous sequences, non-collinear motifs can still be detected.
Neither motif detection nor multiple alignment methods are optimally suited to correctly align long intergenic sequences of heterogeneous length. Here, we present a simple two-step procedure that identifies conserved regions by combining the advantages of both alignment and motif detection methods. Such highly conserved regions most likely contain transcription factor binding sites or other functional intergenic sequences [41]. To show its efficiency, we applied our two-step approach to well described benchmark datasets. Since regions of strong conservation among divergent vertebrates are often associated with developmental regulators [20,21], we choose mainly these types of genes to test our methodology. The presented approach, however, is applicable to any set of organisms and genes for which one wants to compare the intergenic sequences.
Results
A two-step procedure for phylogenetic footprinting
In this study, we aimed to detect regulatory motifs that have been retained over long periods in evolution; in our test case, this applied to mammals to ray-finned fishes such as Fugu. The Fugu genome, however, is very compact and approximately eight or nine times smaller than the human one, although both genomes are assumed to contain a similar repertoire of genes. The compactness of the genome of Fugu is the result of shorter intergenic regions and introns [22,23,42]. On the other hand, the preliminary and still often erroneous annotation of the Fugu genome sometimes results in the selection of very long intergenic regions. Such heterogeneous sizes of the intergenic regions that need to be compared complicate identification of regulatory motifs. Widely used alignment algorithms, such as AVID, LAGAN and others, will usually fail when the sequences that need to be aligned differ too drastically in length. This problem is exacerbated when the sequences have a low overall percent identity. To cope with this, motif detection procedures could offer a solution. However, because regulatory motifs are typically only 6 to 30 bp long, whereas intergenic sequences of vertebrate genes range up to tens of kilobases [43], this results in a low signal-to-noise ratio that complicates the immediate use of de novo motif detection procedures. Therefore, we developed a two-step procedure to combine the advantages of the alignment and motif detection procedures.
We included a first data reduction step based on an alignment method prior to the second motif detection step (see Materials and methods and Figure 1). This data reduction step increases the signal-to-noise ratio in the input set used for motif detection. Data reduction is based on the assumption that longer regions conserved in the orthologs of closely related species are more likely to contain biologically relevant motifs compared to non-conserved regions [21]. Therefore, in our benchmark study, regions conserved among closely related orthologous intergenic sequences of comparable size were preselected as input for motif detection. The mammalian intergenic sequences showed a relatively high overall percent identity and were comparable in length. Subsequently, these selected conserved mammalian subsequences were subjected to motif detection, together with the full-length Fugu intergenic region.
Figure 1.
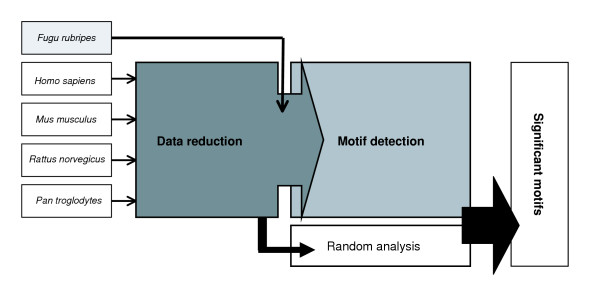
Schematic representation of the two-step procedure for phylogenetic footprinting. In the data reduction step, regions conserved among closely related (mammalian) orthologs are selected. Subsequently, these strongly conserved sequences are combined with a more distant ortholog (for example, Fugu); this set of genes is then subjected to motif detection. Finally, significantly conserved blocks are identified using a threshold defined by a random analysis.
Data reduction
The data reduction procedure preselects subsequences conserved in closely related (mammalian) sequences. It requires a multiple alignment procedure that combines a pairwise alignment (AVID) and a clustering algorithm (Tribe-MCL). Details on this procedure can be found in the Materials and methods section. A resulting cluster consists of unique, non-overlapping subsequences, corresponding to a specific region conserved among the different related orthologs (human, chimp, mouse and rat).
In our benchmark study, we were primarily interested in finding DNA motifs conserved among all input sequences (orthologs). Therefore, only clusters containing conserved subsequences of all mammalian orthologs included in this study (human, chimp, rat and mouse) were retained for further analysis (supplementary website [44]).
Motif detection
The motif detection step aims at identifying motifs that are statistically over-represented in the reduced set of orthologous intergenic sequences. To this end, we extended a previously developed Gibbs sampling based motif detection approach, MotifSampler [37-39] (see Materials and methods). The adapted implementation allows the user to choose a core sequence. A potential motif is only retained when it occurs in this core sequence. Indeed, the input data for motif detection consists of a set of (mammalian) subsequences and a complete Fugu intergenic sequence. This Fugu sequence shows a relatively low overall percent of identity with the other sequences. Due to the high sequence conservation (strong data dependence) between the mammalian subsequences, the original implementation of MotifSampler is not appropriate for detecting motifs in the most divergent sequence: the cost function (log likelihood score) that is optimized in the original MotifSampler offers a trade-off between the degree of conservation of the motif and the number of occurrences of the motif [45]. This results in the detection of motifs that are highly conserved between the highly similar (mammalian) sequences but that show little or no conservation with the Fugu intergenic sequence. Therefore, to ensure the detection of motifs conserved among all sequences, we introduced the concept of a core sequence. By selecting the most divergent ortholog (the Fugu sequence) as the core sequence, the algorithm is forced to only detect motifs that are also present in the most distantly related organism.
The adapted implementation was also redesigned to search for long conserved blocks instead of searching for short conserved motifs only. In datasets consisting of orthologs, not only the motif itself is conserved but also the local context of the motif [21,45]. For this reason, we designed BlockSampler to extend motifs and search for the longest conserved blocks. A motif is thus used as a seed to generate ungapped multiple local alignments. Looking for longer motifs/blocks also increases the specificity of motif detection (less false positives). Finally, since it was previously shown that choosing a background model increases the performance of motif detection [37], we adapted the algorithm such that it uses for each ortholog in the dataset an organism-specific background model.
Results of developed methodology on benchmark datasets
To evaluate its performance, we applied our two-step motif detection procedure to several benchmark datasets. Since we were primarily interested in detecting regulatory motifs over large evolutionary distances, that is, conserved between Fugu and mammalian genomes, we compiled sets of evolutionarily divergent vertebrate orthologs that had been described to contain conserved motifs.
In vertebrate organisms, large conserved regions tend to be associated with genes encoding regulators of development [20,21]. Since our strategy aims at detecting such conserved blocks, we tested the methodology on three sets of orthologous genes that function in the regulation of development, containing motifs described in the literature: hoxb2 [46], pax6 [47] and scl [48]. We also included in the analysis one gene, cfos, not related to developmental processes [26].
All the benchmark sets consisted of orthologous genes that contain evolutionarily retained motifs described in the literature that have, to a large extent, been experimentally verified. These known motifs were used to evaluate the performance of our approach and to compare it to other algorithms. Additionally, we monitored whether our procedure was capable of detecting as yet unknown motifs.
Using the two-step procedure we detected 8 significant blocks for hoxb2, 13 for pax6, 1 for scl and none for the cfos dataset (Table 1). The consensus scores of each of these 22 blocks are given in Tables 2, 3, 4 for each benchmark dataset, respectively. The location of these blocks on the complete intergenic region of the respective Fugu orthologs is shown in Figure 2; alignments can be found in [44].
Table 1.
Conserved blocks detected in benchmark datasets
| Gene | Number of blocks | ||
| Two-step | UCSC | UCR | |
| cfos | 0 | 0 | 0 |
| hoxb2 | 8 | 5 | 0 |
| pax6 | 13 | 11 | 0 |
| scl | 1 | 0 | 0 |
| Total | 22 | 16 | 0 |
Number of blocks two-step: number of conserved blocks identified using the two-step procedure. For more details on the blocks see Tables 2 (hoxb2), 3 (pax6) and 4 (scl). Number of blocks UCSC: the number of blocks detected by the two-step procedure that were recovered in the USCS genome browser (aligned between mammals and Fugu) [51]. Number of blocks UCR: the number of blocks detected by the two-step procedure that correspond to an ultra-conserved region [20].
Table 2.
List of the significant blocks detected in the hoxb2 dataset
| Block | Consensus sequence and possible binding sites |
| Hoxb2 1.1 (-) | AATTCTTTGATGCAATCGGAGGGAGCTGTCAGGGGGCTAAGATTGATCGCCTCATsTCCT |
| *Meis (CTGTCA), CTGTCA: 26-31 + | |
| *Hox/Pbx, AGATTGATCG: 40-49 + | |
| Cap, M00253, NCANHNNN: 39-46 - (0.937); 22-29 - (0.918) | |
| CDP CR1, M00104, NATCGATCGS: 41-50 + (0.964) | |
| CDP CR3+HD, M00106, NATYGATSSS: 41-50 + (0.992) | |
| CdxA, M00101, AWTWMTR: 1-7 + (0.919); 6-12 + (0.903) | |
| HSF2, M00147, NGAANNWTCK: 40-49 + (0.925) | |
| MEIS1, M00419, NNNTGACAGNNN: 23-34 - (0.951) | |
| TGIF, M00418, AGCTGTCANNA: 24-34 + (0.966) | |
| Pbx1, M00096, ANCAATCAW: 39-47 - (0.909) | |
| Hoxb2 2.1 (-) | TTGCACTTrGAGTTTACATTTTAATG |
| *Octamer-motif (ATTTgCAT), GTTTACAT: 12-19 + | |
| *Adhf-2a (TGCACTgAGA), TGCACTTrGA: 2-11 + | |
| CdxA, M00101, AWTWMTR: 20-26 + (0.978); 19-25 - (0.905); 17-23 - (0.927) | |
| SRY, M00148, AAACWAM: 14-20 - (0.905) | |
| Hoxb2 2.2 (UCSC) | AAAAnTGTACTTTTTTAGTATTTACyT |
| *HoxA5 (TTTAaTAaTTA), TTTAGTATTTA: 14-24 + | |
| CdxA, M00101, AWTWMTR: 16-22 - (0.979) | |
| SRY, M00148, AAACWAM: 7-13 - (0.928) | |
| Hoxb2 2.3 (UCSC) | GTGTGTTCTAGTGAACATTTTCATATATATTTATTGGTTAT |
| *Glucocorticoid receptor, AGTGAACA: 10-17 + | |
| *CCAAT BOX, ATTGGTT: 27-33 + | |
| Cap, M00253, NCANHNNN: 15-22 + (0.919); 21-28 + (0.906); 7-14 - (0.919) | |
| CdxA, M00101, AWTWMTR: 23-29 + (0.958); 29-35 + (0.940); 28-34 - (0.956); 26-32 - (0.951); 24-30 - (0.958); 22-28 - (0.960) | |
| FOXJ2, M00422, NNNWAAAYAAAYANNNNN: 23-40 - (0.932) | |
| HFH-3, M00289, KNNTRTTTRTTTA: 25-37 + (0.908) | |
| NF-Y, M00185, TRRCCAATSRN: 30-40 - (0.914) | |
| Oct-1, M00162, CWNAWTKWSATRYN: 14-27 + (0.913) | |
| Pbx-1, M00096, ANCAATCAW: 30-38 - (0.948) | |
| Hoxb2 2.4 (UCSC) | GTGAACATTTTCATATATATTTATTGGTTATAGCCTGTTAAAATATTTTCTTTT |
| *GATA 1, TTATAGCC: 28-35 + | |
| *CCAAT BOX, ATTGGTT: 23-29 + | |
| Cap, M00253, NCANHNNN: 5-12 + (0.919); 11-18 + (0.906) | |
| CCAAT box, M00254, NNNRRCCAATSA: 21-32 - (0.940) | |
| CdxA, M00101, AWTWMTR: 13-19 + (0.958); 19-25 + (0.940); 39-45 + (0.925); 46-52 + (0.901); 36-42 - (0.930); 18-24 - (0.957); 16-22 - (0.951); 14-20 - (0.958); 12-18 - (0.960) | |
| FOXD3, M00130, NAWTGTTTRTTT: 41-52 + (0.924) | |
| FOXJ2, M00422, NNNWAAAYAAAYANNNNN: 13-30 - (0.932) | |
| HFH-3, M00289, KNNTRTTTRTTTA: 15-27 + (0.908) | |
| HNF-3beta, M00131, KGNANTRTTTRYTTW: 39-53 + (0.920) | |
| NF-Y, M00185, TRRCCAATSRN: 20-30 - (0.914) | |
| Oct-1, M00162, CWNAWTKWSATRYN: 4-17 + (0.913) | |
| Pbx-1, M00096, ANCAATCAW: 20-28 - (0.948) | |
| SRY, M00148, AAACWAM: 47-53 - (0.961) | |
| Hoxb2 2.5 (UCSC) | AATTCyCTCTTGGAACTTTCTTTGTTCTTCmGTAG |
| HSF1, M00146, AGAANRTTCN: 12-21 + (0.915); 12-21 - (0.930) | |
| HSF2, M00147, NGAANNWTCK: 12-21 + (0.948); 12-21 - (0.930) | |
| SRY, M00148, AAACWAM: 17-23 - (0.961) | |
| Hoxb2 3.1 (UCSC) | GGCCnAGACnAGCGATTGGCGGAGrCCGGTCCCGTGACCAnGAATTCCCTGyAATTT |
| NF-Y, M00185, TRRCCAATSRN: 12-22 - (0.915) | |
| USF, M00187, CYCACGTGNC: 29-38 - (0.957) | |
| USF, M00217, NCACGTGN: 30-37 + (0.902) | |
| Hoxb2 3.2 (-) | TCCCGTGACCAnGAATTCCCTGyAATTTCGnyGGAGTCC |
| USF, M00217, NCACGTGN: 1-8 + (0.902) |
For each block, the consensus sequence is given followed by the possible binding sites situated in this block: motifs previously described in the literature [46] are marked with an asterisk. The motifs are summarized by their motif name (in bold), by their consensus sequence, if known, as described in the original article, by the sequence of the motif instance in our search, by the positions of the motif instance relative to the consensus sequence of the entire block and by the strand (indicated by a '+' or a '-') on which the motif occurred. Motif hits derived by Transfac are indicated by their matrix accession number, the consensus of this binding site and the instances of this motif in our search. These are further characterized by their positions relative to the consensus sequence of the entire block, by the strand on which the motif occurred and by the corresponding MotifLocator score (in parentheses). The blocks identified by the UCSC genome browser as conserved between mammals and Fugu are marked with 'UCSC', while the blocks detected by our two-step methodology but not present in the UCSC genome browser are indicated with a '-'.
Table 3.
List of the significant blocks detected in the pax6 dataset
| Block | Consensus sequence and possible binding sites |
| pax6 1.1 (UCSC) | CTTAATGATGAGAGATCTTTCCGCTCATTGCCCATTCAAATACAATTGTAGATCGAAGCCGGCCTT GTCAsGTTGAGAAAAAGTGAATTTCTAACATCCAGGACGTGCCTGTCTACT |
| *Minimal fragment for expression in lens and cornea as described in [46]: 11-117 + | |
| Cap, M00253, NCANHNNN: 25-32 + (0.940); 79-86 - (0.964); 4-11 - (0.946); 1-8 - (0.903) | |
| CCAAT box, M00254, NNNRRCCAATSA: 27-38 + (0.901) | |
| *CdxA, M00100, 'MTTTATR': 1-7 + (0.921)*; 87-93 + (0.913) | |
| *CdxA, M00101, AWTWMTR: 1-7 + (0.934); 4-10 + (0.921); 38-44 + (0.905), 87-93 + (0.988) | |
| c-Ets-1(p54), M00032, NCMGGAWGYN: 98-107 + (0.906) | |
| c-Ets-1(p54), M00074, NNACMGGAWRTNN: 92-104 - (0.901) | |
| En-1, M00396, GTANTNN: 37-43 - (0.967) | |
| GATA-3, M00351, ANAGATMWWA: 11-20 + (0.920) | |
| HSF2, M00147, NGAANNWTCK: 13-22 - (0.933) | |
| p53, M00272, NGRCWTGYCY: 101-110 + (0.949) | |
| pax6 1.2 (UCSC) | CATTATTGTTGCCAGCACGAAGCATCACAATCAATCATAAGGAAGTCCAGTTGGCAGGTGTCAATCTTG |
| CdxA, M00101, AWTWMTR: 1-7 - (0.995) | |
| Cap, M00253, NCANHNNN: 25-32 + (0.934); 31-38 + (0.903); 35-42 + (0.903); 47-54 + (0.908); 61-68 + (0.937) | |
| CDP CR3+HD, M00106, NATYGATSSS: 27-36 - (0.907) | |
| c-Ets-1(p54), M00074, NNACMGGAWRTNN: 36-48 + (0.902) | |
| *HOXA3, M00395, CNTANNNKN: 1-9 + (0.905) | |
| MyoD, M00184, NNCACCTGNY: 53-62 - (0.956) | |
| *Pbx-1, M00096, ANCAATCAW: 30-38 + (0.986); 2-10 - (0.923) | |
| Sox-5, M00042, NNAACAATNN: 3-12 - (0.932) | |
| SRY, M00148, AAACWAM: 33-39 + (0.910) | |
| USF, M00122, NNRNCACGTGNYNN: 51-64 + (0.913); 51-64 - (0.908) | |
| pax6 1.3 (UCSC) | GAAAAAGTGAATTTCTAACATCCAGGACGTGCCTGTCTACTTTCAGwGAATTGCATCCAATCACCCC |
| Cap, M00253, NCANHNNN: 3-10 - 0.964 | |
| CCAAT box, M00254, NNNRRCCAATSA: 52-63 + (0.949) | |
| CdxA, M00100, 'MTTTATR': 11-17 + (0.913) | |
| CdxA, M00101, AWTWMTR: 11-17 + (0.988) | |
| c-Ets-1(p54), M00032, NCMGGAWGYN: 22-31 + (0.906) | |
| c-Ets-1(p54), M00074, NNACMGGAWRTNN:16-28 - (0.901) | |
| En-1, M00396, GTANTNN: 58-64 - (0.948) | |
| GATA-1, M00075, SNNGATNNNN: 56-65 - (0.930) | |
| GATA-3, M00077, NNGATARNG: 56-64 - (0.917) | |
| NF-Y, M00185, TRRCCAATSRN: 54-64 + (0.910) | |
| p53, M00272, NGRCWTGYCY: 25-34 + (0.949) | |
| SRY, M00148, AAACWAM: 59-65 + (0.917) | |
| pax6 1.4 (UCSC) | GTCTATATTTAATCCAATTATAAGGGTCACGGAGTAAGTGC |
| *Motif containing homeoboxes described in [46], TTTAATCCAATTATAA: 8-23 + | |
| Cap, M00253, NCANHNNN: 34-41 - (0.904) | |
| CdxA, M00100, 'MTTTATR': 16-22 + (0.907) | |
| CdxA, M00101, AWTWMTR: 16-22 + (0.995); 16-22 - (0.906); 6-12 - (0.931); 4-10 - (0.951) | |
| En-1, M00396, GTANTNN: 15-21 - (0.948) | |
| Nkx2-5, M00240, TYAAGTG: 34-40 + (0.927) | |
| RORalpha1, M00156, NWAWNNAGGTCAN: 18-30 + (0.919) | |
| TCF11, M00285, GTCATNNWNNNNN: 26-38 + (0.906) | |
| pax6 1.5 (UCSC) | GCATCCAATCACCCCCAGGG |
| Cap, M00253, NCANHNNN: 9-16 + (0.965) | |
| En-1, M00396, GTANTNN: 6-12 - (0.948) | |
| GATA-3, M00077, NNGATARNG: 4-12 - (0.917) | |
| SRY, M00148, AAACWAM: 7-13 + (0.917) | |
| pax6 1.6 (UCSC) | CAsGTTGAGAAAAAGTGAATTTCTAACATCCAGGACGTGCCTGTCTACTTTCAGw GAATTGCATCCAATCACCCCCAGGGAATTCnGCTAATGTCTCC |
| *Homeobox-binding site described in [46], GCTAATGTCTC: 87-97 + | |
| Cap, M00253, NCANHNNN: 69-76 + (0.965); 87-94 - (0.903); 11-18 - (0.964) | |
| CCAAT box, M00254, NNNRRCCAATSA: 60-71 + (0.949) | |
| CdxA, M00100, 'MTTTATR': 19-25 + (0.913) | |
| CdxA, M00101, AWTWMTR: 19-25 + (0.988) | |
| c-Ets-1(p54), M00032, NCMGGAWGYN: 30-39 + (0.906) | |
| c-Ets-1(p54), M00074, NNACMGGAWRTNN: 24-36 - (0.901) | |
| En-1, M00396, GTANTNN: 66-72 - (0.948) | |
| GATA-1, M00075, SNNGATNNNN: 64-73 - (0.930) | |
| GATA-3, M00077, NNGATARNG: 64-72 - (0.917) | |
| NF-Y, M00185, TRRCCAATSRN: 62-72 + (0.910) | |
| p53, M00272, NGRCWTGYCY: 33-42 + (0.949) | |
| SRY, M00148, AAACWAM: 67-73 + (0.917) | |
| pax6 2.1 (UCSC) | TGGGTCCATTTTCCAGAyGGTTTGTTACTCTTGCTGCmTGATTTrG |
| Cap, M00253, NCANHNNN: 6-13 + (0.921) | |
| CdxA, M00101, AWTWMTR: 9-15 + (0.918) | |
| SRY, M00148, AAACWAM: 21-27 - (0.942) | |
| pax6 2.2 (-) | ATTTTGGTTGCTTTCAGGTwTAATTAACTTT |
| Nkx2-5, M00241, CWTAATTG: 21-28 - (0.902) | |
| pax6 2.3 (UCSC) | ATTGTAATCATTTCAATTATCTTCA |
| Cap, M00253, NCANHNNN: 8-15 + (0.927) | |
| En-1, M00396, GTANTNN: 14-20 - (0.948) | |
| Nkx2-5, M00241, CWTAATTG: 14-21 - (0.930) | |
| pax6 2.4 (-) | GGTTGCTTTCAGGTwTAATTAACTTTGAACAACAAATA |
| Nkx2-5, M00241, CWTAATTG: 16-23 - (0.902) | |
| pax6 3.1 (UCSC) | TTGTAATTACTGCCCTTCATGTGGTCCGGTGCCTTGAACCATCTTTAATTAAAAGCATAATTAAGG |
| AML-1a, M00271, TGTGGT: 20-25 + (1.000) | |
| Cap, M00253, NCANHNNN: 39-46 + (0.910); 55-62 + (0.909); 6-13 - (0.916) | |
| CdxA, M00100, MTTTATR: 56-62 - (0.934) | |
| CdxA, M00101, AWTWMTR: 6-12 + (0.988); 44-50 + (0.913); 47-53 + (0.900); 48-54 + (0.905); 59-65 + (0.903); 60-66 + (0.926); 56-62 - (0.998); 47-53 - (0.913); 44-50 - (0.901); 43-49 - (0.907); 2-8 - (0.949); | |
| En-1, M00396, GTANTNN: 3-9 + (0.912); 4-10 - (0.912) | |
| HSF2 , M00147, NGAANNWTCK: 35-44 + (0.908) | |
| Nkx2-5, M00241, CWTAATTG: 56-63 + (0.935), 58-65 - (0.954) | |
| USF, M00217, NCACGTGN: 17-24 - (0.921) | |
| pax6 3.2 (UCSC) | AAGGCTTGCAGCTGCCTCCAAATCAATAGAyGTCAAAGAAATATGAAAACArTC |
| CdxA, M00101, AWTWMTR: 39-45 + (0.953); 36-42 - (0.925) | |
| SRY, M00148, AAACWAM: 35-41 + (0.961) | |
| Cap, M00253, NCANHNNN: 8-15 + (0.931); 39-46 - (0.940); 8-15 - (0.931) | |
| AP-4, M00175, VDCAGCTGNN: 7-16 - (0.902) | |
| MyoD, M00184, NNCACCTGNY: 7-16 + (0.957) | |
| SRY, M00160, NWWAACAAWANN: 19-30 + (0.928) | |
| pax6 3.3 (UCSC) | GCATAATTAAGGGAAGATCTAAAGAAAGACAATTACCAGATGGTCT |
| Cap, M00253, NCANHNNN: 1-8 + (0.909) | |
| CdxA, M00100, MTTTATR: 2-8 - (0.934) | |
| CdxA, M00101, AWTWMTR: 5-11 + (0.903); 6-12 + (0.926); 32-38 + (0.939); 2-8 - (0.998) | |
| En-1, M00396, GTANTNN: 30-36 - (1.000) | |
| GATA-1, M00075, SNNGATNNNN: 36-45 + (0.936) | |
| GATA-2, M00076, NNNGATRNNN: 36-45 + (0.922) | |
| GATA-3, M00351, ANAGATMWWA: 13-22 + (0.949) | |
| HOXA3, M00395, CNTANNNKN: 29-37 - (0.939) | |
| Msx-1, M00394, CNGTAWNTG: 30-38 - (0.915) | |
| MyoD, M00184, NNCACCTGNY: 35-44 - (0.919) | |
| Nkx2-5, M00241, CWTAATTG: 2-9 + (0.935); 4-11 - (0.954) | |
| SRY, M00148, AAACWAM: 21-27 + (0.961); 25-31 + (0.927) | |
| USF, M00122, NNRNCACGTGNYNN: 33-46 + (0.907); 33-46 - (0.904) |
For each block, the consensus sequence is given followed by the possible binding sites situated in this block: motifs previously described in the literature [47] are marked with an asterisk. The motifs are summarized by their motif name (in bold), by their consensus sequence, if known, as described in the original article, by the sequence of the motif instance in our search, by the positions of the motif instance relative to the consensus sequence of the entire block and by the strand (indicated by a '+' or a '-') on which the motif occurred. Motif hits derived by Transfac are indicated by their matrix accession number, the consensus of this binding site and the instances of this motif in our search. These are further characterized by their positions relative to the consensus sequence of the entire block, by the strand on which the motif occurred and by the corresponding MotifLocator score (in parentheses). The blocks identified by the UCSC genome browser as conserved between mammals and Fugu are marked with 'UCSC', while the blocks detected by our two-step methodology but not present in the UCSC genome browser are indicated with a '-'.
Table 4.
List of the significant blocks detected in the scl dataset
| Block | Consensus sequence and possible binding sites |
| scl 1.1 (-) | TTGCCAAATTAAAATGAATCATTTGGCCCATAATGGCCGAGGCGCT |
| *Conserved sequence identified in [47], GCCAAAT: 3-9 + | |
| *Putative SKN1 site reported in [47], AATGAATCATTT: 13-24 + | |
| CdxA, M00100, 'MTTTATR': 29-35 - (0.917) | |
| CdxA, M00101, AWTWMTR: 7-13 + (0.901); 8-14 + (0.905); 10-16 + (0.927); 29-35 + (0.927); 29-35 - (0.929); 7-13 - (0.913) | |
| *En-1, M00396, GTANTNN: 30-36 + (0.936) | |
| Cap, M00253, NCANHNNN: 19-26 + (0.932); 10-17 - (0.933) | |
| Pbx-1, M00096, ANCAATCAW:14-22 + (0.941) | |
| AP-1, M00199, NTGASTCAG: 14-22 + (0.913) | |
| *HOXA3, M00395, CNTANNNKN: 29-37 + (0.927) | |
| Tst-1, M00133, NNKGAATTAVAVTDN: 3-17 + (0.901) |
For each block, the consensus sequence is given followed by the possible binding sites situated in this block: motifs previously described in the literature [48] are marked with an asterisk. The motifs are summarized by their motif name (in bold), by their consensus sequence, if known, as described in the original article, by the sequence of the motif instance in our search, by the positions of the motif instance relative to the consensus sequence of the entire block and by the strand (indicated by a '+' or a '-') on which the motif occurred. Motif hits derived by Transfac are indicated by their matrix accession number, the consensus of this binding site and the instances of this motif in our search. These are further characterized by their positions relative to the consensus sequence of the entire block, by the strand on which the motif occurred and by the corresponding MotifLocator score (in parentheses). The blocks identified by the UCSC genome browser as conserved between mammals and Fugu are marked with 'UCSC', while the blocks detected by our two-step methodology but not present in the UCSC genome browser are indicated with a '-'.
Figure 2.
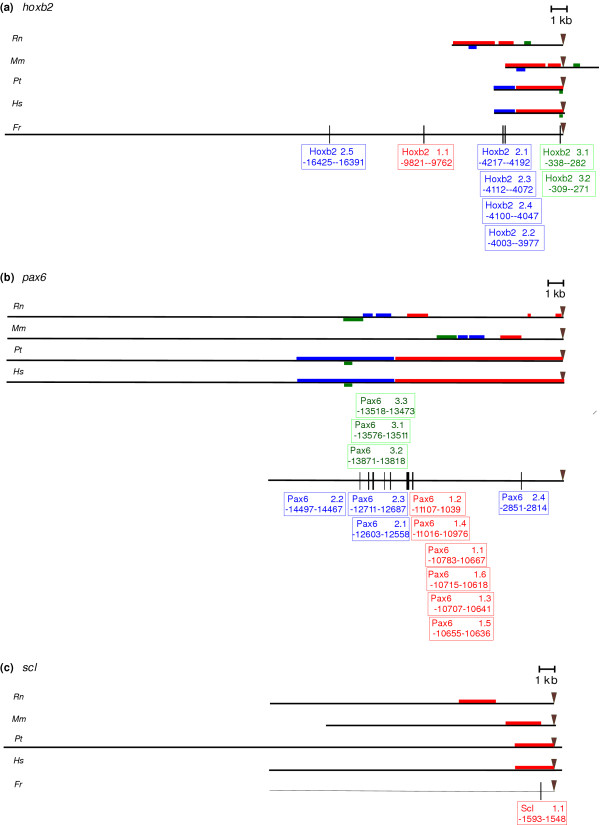
Localization of clusters and conserved blocks in the (a) hoxb2, (b)pax6 and (c)scl datasets. For each dataset, the different orthologous intergenic sequences are shown: Rn,Rattus norvegicus; Mm, Mus musculus; Pt, Pan troglotydes; Hs, Homo sapiens; Fr, Fugu rubripes. Clusters of conserved mammalian subsequences that were subjected to motif detection (that is, clusters containing at least one subsequence per mammalian organism) are represented on the respective mammalian sequences (cluster 1 in red, cluster 2 in blue and cluster 3 in green). The conserved blocks identified using BlockSampler are represented on the Fugu intergenic sequence (in the color of the mammalian cluster it is located in). For each block the localization relative to the start of the Fugu gene is given. The transcription start sites are marked with an inverse triangle.
As a first validation step, we compared our results with the alignments and conserved regions identified by well-established genome browsers, namely the UCSC genome browser [49] and the UCR browser [20] (Table 1).
The UCSC genome browser [50] enables access to current genome assemblies; it offers visualizations of several genomic features, such as cross-species homologies [49,51]. The latter can be viewed as multiple alignments over several species, ranging from closely related mammals to more distantly related species, such as chicken, zebrafish and pufferfish. The multiple alignments were generated with MULTIZ [52]. Of the conserved 22 blocks we identified by aligning intergenic regions of mammals and Fugu, 16 could also be retrieved from the USCS genome browser (Table 1); these are indicated in Tables 2, 3, 4. The remaining six blocks could only be identified using our two-step approach.
The set up of the UCR browser [53] is slightly different from the UCSC browser in that it focuses on the detection of ultra-conserved regions (UCRs) only, that is, regions conserved between human, mouse and Fugu. These regions were identified using sequence alignment strategies (BLAT) applied to complete genome sequences without prior data reduction [20,54]. Although our strategy also identifies regions highly conserved among the species under study, no overlap was detected between our conserved blocks and the UCRs (Table 1); that is, in the regions we studied (up to 40 kb intergenic plus 5' untranslated region), no UCRs were located according to the analysis of Sandelin et al. [20]. The regions the UCR browser identified as ultra-conserved were located much more upstream of the gene compared to the regions we used for our analysis.
To further validate the detected blocks, we tested whether they contain the motifs that were originally reported by Scemama et al. [46], Kammandel et al. [47] and Göttgens et al. [48] for hoxb2, pax6 and scl, respectively (no significant blocks were detected for cfos). The previously described motifs present in the respective blocks are listed in Tables 2, 3, 4 (marked with an asterisk). Of the 17 motifs reported by Scemama et al. [46], 8 were present in the significant hoxb2-blocks (Table 2). Five other motifs were present in non-significant blocks. The latter are blocks with scores that fell below the threshold we chose based on the random analysis (see Materials and methods). The four remaining motifs could not be recovered. All motifs described by Kammandel et al. [47] as conserved among mammalian and Fugu pax6 intergenic regions were recovered by our methodology (Table 3). The conserved block detected in the scl dataset contains three of the five motifs previously identified by Göttgens et al. [48] (Table 4); a fourth motif was picked up in a non-significant block. One motif was not detected in any of the blocks.
Besides these blocks containing known motifs, we identified several blocks (three for hoxb2 and eight for pax6) that correspond to conserved regions not previously described in the literature. To validate these blocks, we checked whether they were enriched for yet undescribed regulatory motifs. Hence, we screened all blocks with the Transfac database of vertebrate transcription factor binding sites [55]. The result of this screening is summarized in Tables 2, 3, 4. As expected [41,56], the conserved blocks we identified contain many potential binding sites; remarkably they tend to be specifically enriched for homeodomain binding sites (in blocks hoxb2 1.1, hoxb2 2.1, hoxb2 2.3, hoxb2 2.4, pax6 1.1, pax6 1.4, pax6 3.1, pax6 3.3 and scl 1.1, homeodomain binding sites were significantly over-represented, with a p value < 10-8). For a more detailed description of both the previously described and the new potential regulatory motifs present in the detected blocks, please refer to the Supplementary website [44].
Besides these well-described benchmark datasets, we applied our method to six additional datasets, differing in composition from the benchmark datasets. They all contained a combination of four mammalian sequences (rat, mouse, human, chimp or dog) to be used in the data reduction step and an additional set of sequences originating from more distantly related orthologs (chicken, Fugu, Tetraodon nigroviridis and zebrafish in different combinations) added in the motif detection step. Four of the six additional datasets were derived from genes functioning in developmental regulation, including three homeobox genes (GSH1, Meis2, HOXB5) and one encoding the zinc finger protein EGR3. Besides these regulators involved in development, two genes, PCDH8 and HIV-EP1, were included, which are, according to our knowledge, unrelated to development. PCDH8 is believed to function as a calcium-dependent cell-adhesion protein and HIV-EP1 binds to enhancer elements present in several viral promoters and in a number of cellular promoters such as those of the class I MHC, interleukin-2 receptor, and interferon-beta genes. In the additional datasets involved in development, we detected several strongly conserved blocks: GSH1 contained four blocks that are conserved among human, chimp, mouse, rat and pufferfish (Fugu and Tetraodon); in Meis2, two blocks were recovered that are retained in all organisms under study except for Fugu; and in HOXB5, six strongly conserved blocks were detected in mammals and pufferfish, while the motif seems to have been lost in chicken. In EGR3, two blocks were found conserved in mammals and fish. In the non-developmental related datasets, only in PCDH8 was one large block detected, conserved in human, chimp, mouse, rat, chicken, Tetraodon and Fugu, but not in zebrafish. This shows that conserved regions might also exist in genes not involved in development, although a possible involvement of this additional gene in developmental processes cannot be ruled out. Detailed results of these analyses can be found in Additional data file 1 and in [44]. Because the motifs in these additional datasets have not been studied as extensively as those of the benchmark datasets, we cannot guarantee all detected blocks are biologically functional.
Evaluation of the developed procedure
To compare the performance of our newly developed two-step strategy to that of other frequently used algorithms, we evaluated to what extent MotifSampler [39], MAVID [10] and 'Threaded Blockset Aligner' (TBA) [52] could recover known motifs in our benchmark sets.
First, we studied the performance of the alignment algorithms MAVID and TBA in detecting conserved regions within our four benchmark datasets. Since MAVID and TBA were originally developed to perform multiple alignments on long sequences, we applied these algorithms to the initial full-length benchmark datasets, that is, the complete mammalian and Fugu intergenics. We evaluated to what extent motifs or conserved regions described in original articles were correctly aligned using either MAVID or TBA. The results are summarized in Table 5 (MAVID and TBA columns) and in [44].
Table 5.
Comparison of two-step procedure with other methodologies
| Gene | Number of motifs | Two-step BS | BS | Two-step MS | MS | MAVID | TBA |
| cfos | 2 | 0 | 0 | 0 | 0 | 0 | 0 |
| hoxb2 | 17 | 8 (+5) | 13 | 2 | 1 | 0 | 0 |
| pax6 | 6 | 6 | 1* | 0 | 0 | 6 | 6 |
| scl | 5 | 3 (+1) | 1 | 0 | 0 | 0 | 0 |
| Total | 30 | 17 (+6) | 15 | 2 | 1 | 6 | 6 |
Number of motifs: the number of motifs reported by Blanchette and Tompa [26] in cfos, Scemama et al. [46] in hoxb2, Kammandel et al. [47] in pax6 and Göttgens et al. [48] in scl. Two-step BS: the number of previously described motifs detected by the two-step procedure, combining data reduction and motif detection using BlockSampler. The numbers in parentheses are the number of motifs present in non-significant blocks. BS: the number of previously described motifs detected by BlockSampler in initial full-length datasets. Two-step MS: the number of previously described motifs detected by combining data reduction and motif detection using MotifSampler. MS: the number of previously described motifs detected by MotifSampler in initial full-length datasets. MAVID: the number of previously described motifs detected (correctly aligned) by MAVID. TBA: the number of previously described motifs detected by TBA. *Only part of a motif was detected.
MAVID alignment of all three cfos datasets (mammalian orthologs combined with each of the three Fugu paralogs) could not recover either of the two motifs previously described by Blanchette and Tompa [26] (Table 5). This is in line with our results showing the overall low homology between the cfos mammalian and Fugu orthologs. The MAVID alignment of most of the hoxb2 blocks containing previously described motifs shows that a conserved region in the mammalian intergenic sequences is broken up into small conserved parts interrupted by gaps when aligned to the longer Fugu sequence, resulting in an incorrect alignment of the regulatory motifs: previously reported motifs were not recovered in the MAVID alignment (Table 5). Our method performs better because the most heterogeneous sequence is only aligned in a second step, using a highly flexible local alignment procedure (BlockSampler). Regarding pax6, most of the blocks containing previously described motifs were correctly aligned by MAVID and all the motifs described by Kammandel et al. [47] could be correctly retrieved over all the orthologs under study (Table 5). This dataset is probably relatively well suited for MAVID because the mammalian sequences are only twice as large as the pufferfish pax6 intergenic region (Table 6). Although the lengths of the intergenic regions in the scl dataset (Table 6) are in the same order of magnitude (ranging from 16.5 to 40 kb), MAVID did not succeed in identifying any of the motifs previously described by Göttgens et al. [48] (Figure 3, Table 5).
Table 6.
Base pair lengths of the intergenic sequences for each benchmark dataset
| Gene | Hs | Mm | Rn | Pt | Fr |
| cfos | 40,154 | 33,157 | 40,132 | 40,154 | 3,606* |
| 3,606† | |||||
| 1,244‡ | |||||
| hoxb2 | 4,973 | 6,744 | 7,640 | 4,878 | 39,219 |
| pax6 | 40,102 | 40,000 | 40,000 | 40,000 | 21,204 |
| scl | 20,981 | 16,471 | 20,343 | 39,999 | 20,155 |
The Fugu cfos intergenic sequences are derived from *SINFRUG00000132418, †SINFRUG00000132419 and ‡SINFRUG00000143787. The Ensemble IDs (+ 1 Genebank accession number) are given in [56]. Fr,Fugu rubripes; Hs, Homo sapiens; Mm, Mus musculus; Pt, Pan troglotydes; Rn, Rattus norvegicus.
Figure 3.
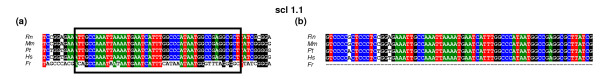
Comparison of two-step strategy with MAVID for the scl data set (a) Conserved block: alignment of the different scl orthologs. The conserved block as identified by BlockSampler - is marked with a boxed area. (b) Visualization of the MAVID alignment of the corresponding region. The dashed line denotes a gap in the alignment. Rn, Rattus norvegicus; Mm, Mus musculus; Pt, Pan troglotydes; Hs, Homo sapiens; Fr, Fugu rubripes.
Although TBA has been shown to outperform MAVID in aligning more divergent sequences [52], applying this alignment tool to the benchmark datasets generated similar results as MAVID: all known pax6-regulating motifs were detected, while motifs present in the other benchmark datasets were not recovered (Table 5, TBA column).
Besides detecting the blocks with previously described motifs, our two-step methodology also discovered blocks (block pax6 2.4, for instance) that could not be recovered when aligning the intergenic sequences with MAVID or TBA [44,57].
Overall, based on our benchmark analysis, the two-step method performs better than MAVID or TBA in identifying conserved blocks in distantly related orthologs: the proposed method is able to recover in our benchmark sets all the known motifs identified by MAVID and TBA but, in addition, finds several previously described motifs ignored by these algorithms (Table 5, two-step BS, MAVID and TBA columns). Using the two-step procedure, first selecting strongly conserved orthologous sequences, clearly facilitates alignment with the more divergent (lower overall similarity) sequence.
We also tested the performance of MotifSampler as an example of a probabilistic motif detection procedure on the unreduced dataset. In this case, only one previously described motif was detected (Table 5, MS column). This was to be expected as in unreduced datasets the signal to noise ratio is too high for standard motif detection procedures to give reliable and interpretable results.
Our two-step procedure includes two adaptations over previous existing methods: first, it allows for a data reduction step; and secondly, we developed a motif detection procedure specifically adapted to the purpose of detecting large conserved blocks (BlockSampler). To assess the relative contribution of each of these adaptations to the overall result, we set up the following experiment: to study the specific influence of the data reduction step, we compared the results of applying BlockSampler to both the unreduced benchmark datasets and the datasets obtained after data reduction. Table 5 (BS and two-step BS columns) shows the results of this comparison. Overall, the results seem comparable: application of BlockSampler to the complete intergenic sequences results in recovery of 15 of the 30 previously reported motifs (in all four datasets), while the two-step method identified 17. Thus, at first sight, there does not seem to be a major contribution from the data reduction step. A closer look at Table 5, however, shows that the positive contribution of the data reduction (increasing the signal-to-noise ratio) is strongly dependent on the lengths of the intergenic sequences to be aligned. A major positive effect is observed for the large pax6 and scl datasets, whereas for the hoxb2 set, in which the sequences under study are rather short, the data reduction does not offer a clear advantage. To assess the specific improvements of using BlockSampler instead of standard motif detection approaches, we compared the results of BlockSampler to those of MotifSampler when both were applied to the reduced datasets. A reduced dataset thus consists of a subcluster of mammalian sequences (Figure 4) and a complete Fugu ortholog. The performance of MotifSampler was far below that of BlockSampler: MotifSampler only detected two previously described motifs (Table 5, two-step MS column), both in the hoxb2 set, while BlockSampler recovered 17 previously described motifs (Table 5, two-step BS column). Moreover, because MotifSampler searches for short motifs (default eight nucleotides (nt)), it detects many false positive hits. These results show that independent of the data reduction step, BlockSampler is clearly more suited for detecting large conserved blocks than MotifSampler.
Figure 4.
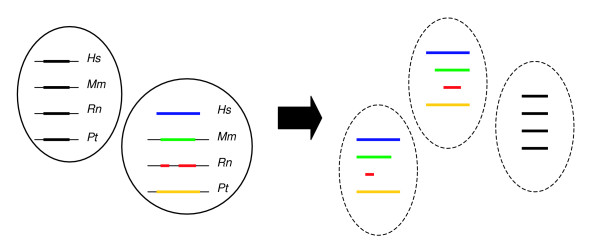
Schematic representation of subclusters, that is, clusters of conserved orthologous sequences that contain one region in each ortholog. See text for details. Rn, Rattus norvegicus; Mm, Mus musculus; Pt, Pan troglotydes; Hs, Homo sapiens.
Discussion
We developed a two-step methodology to search for regions (motifs) conserved over different phylogenetic lineages in long intergenic sequences of heterogeneous size. In a first step, an alignment method is used to select conserved subsequences in intergenic orthologous sequences of comparable size of closely related vertebrate genomes, since these are expected to be enriched for regulatory motifs [21,41]. The combination of this preselected dataset of conserved sequences and the full-length intergenic sequence of a more distant ortholog, which is more likely to differ in size and overall homology, is subjected to probabilistic motif detection. The preselection step facilitates motif detection by enhancing the signal-to-noise ratio in the dataset. For the second motif detection step we used an extension of a Gibbs sampling based algorithm [39] with a higher performance in detecting large conserved blocks within a set of orthologous sequences. Using the strategy mentioned above, we could combine the advantages of alignment methods, which have been shown to be very suitable for aligning long, highly conserved intergenic sequences, and the probabilistic algorithms for motif detection that usually are more appropriate when looking for smaller regions of conservation (lower degree of similarity).
We applied this two-step methodology to four well-studied datasets for which functional phylogenetically conserved motifs had been extensively described. Our approach identified most of the previously described motifs. In addition, we detected several blocks not previously described in the literature or not present in any of the two genome browsers (UCSC and UCR) we compared our results with. Because highly conserved blocks most probably consist of consecutive transcription factor binding sites [21,41,56], we screened the conserved blocks with the Transfac motif database [55]. These blocks contained abundant copies of homeodomain binding sites. This is not unexpected since most of the genes we were studying function in the regulation of development [21,58]. These blocks most probably contain, besides the motifs obtained with the Transfac screening, many more motifs not yet annotated in Transfac. Alternatively, they might have other, not yet characterized biological functions, for example, transcripts of unknown function [59].
Some previously described motifs were missed, however, because of the strong selection criteria we used: since regulatory elements tend to be grouped [21,41,56,60], we assumed that the sequences surrounding a regulatory motif are also conserved (due to the presence of other binding sites). Motifs located in a variable context will probably go undetected.
By applying our method to additional datasets with configurations different from the benchmark dataset we could demonstrate that our methodology is more generally applicable.
Comparing the performance of the two-step procedure with that of MAVID and TBA, as representatives of multiple alignment methods, and MotifSampler, as an example of a motif detection method, showed that our approach outperformed these alternative methods when the intergenic sequences became either too long or too heterogeneous in size.
Additionally, we studied the marginal contribution of the data reduction step and the improved method for motif detection on the final performance of the two-step procedure: overall, BlockSampler performed better than the related algorithm MotifSampler, both on long sequences and on intergenic regions reduced in size. The data reduction step seemed essential when the length of the intergenic sequences to be compared becomes excessive.
Although our two-step procedure has proven successful, there is still room for improvement, for instance by taking into account the phylogenetic relationships between the sequences under study in the second motif detection step. The contribution of finding a motif in an ortholog to the global motif score could be weighted according to its phylogenetic distance from the other sequences in which the motif is also present. Indeed, this way we would account for the specific composition of a dataset because closely related orthologs are less informative than further related ones. If one wanted to relax the assumption of conserved order of motifs in the first data reduction step, it would suffice to replace AVID in this step with a more local aligner such as BLAT [54]. Also, our motif detection algorithm could be extended for more advanced background models [61].
Conclusion
We developed a two-step approach that combines the advantages of both motif detection and multiple alignment algorithms. It has shown to be well suited for identifying conserved regions in intergenic sequences from distantly related orthologs that show a low overall homology and that are heterogeneous in size. The strength of our approach lies in the combination of data reduction and improved motif detection: the first data reduction step is essential when it concerns long intergenic sequences. BlockSampler, the algorithm used in the second motif detection step, has been shown to be optimally suited to identify large conserved regions among orthologous sequences. Applying our method to benchmark sets showed that, although it recovered most of the motifs/blocks previously described in these datasets, some were missed due to the assumptions underlying our analysis and the stringent selection criteria applied. These results indicate that, given the chosen criteria, our method offers a fully automated analysis flow that is highly specific for detecting motifs conserved over different vertebrate lineages in complete intergenic sequences.
Materials and methods
Benchmark datasets
The benchmark datasets were generated as follows. First, a set of orthologous genes was defined using the Ensembl genome browser version 23 [62]. In this study, the benchmark datasets included genes from human (Homo sapiens), mouse (Mus musculus), rat (Rattus norvegicus), chimp (Pan troglodytes) and pufferfish (Fugu rubripes). Regarding the cfos dataset, Ensembl identified three Fugu paralogs - SINFRUG00000132418, SINFRUG00000132419 and SINFRUG00000143787 - that were all included in the analysis. The additional datasets EGR3, GSH1, HIV-EP1, HOXB5, Meis2, PCDH8 contain multiple distantly related orthologs (see [44]).
Subsequently, the intergenic regions of these orthologs were selected using the Ensembl mart database release 21.1. The region upstream of the transcription start (as defined by Ensembl) was limited to 40 kb. Additionally, the 5' untranslated region was included. Lengths of the respective intergenics are given in Table 6; the benchmark datasets, cfos, hoxb2, pax6 and scl can be found in [44]. The rat cfos ortholog ENSRNOG00000008015, Fugu hoxb2 ortholog SINFRUG00000136637, chimp pax6 ortholog ENSPTRG00000003474, and scl chimp ENSPTRG00000003474 contain long N-stretches, probably as a result of incomplete preliminary annotation.
Remarkably, where Fugu is known to have a very compact genome [23], the Fugu hoxb2 mentioned above is very long compared to the mammalian hoxb2 intergenic sequences (Table 6). This is probably due to the presence of a pseudogene (SINFRUG00000157209) in the intergenic region of SINFRUG00000136637 at circa 5.9 kb from the transcription start site of hoxb2, which was not yet annotated in the release version 23 of ENSEMBL.
All intergenic sequences were selected as described above, except the intergenic sequence of the Fugu scl ortholog. Because the putative scl ortholog annotated by ENSEMBL (SINFRUG00000145588) did not contain motifs shown to be present in the Fugu scl ortholog by Göttgens et al. [48], we used the Genbank Fugu scl sequence [Genbank: AJ131019]. This sequence (referring to a cosmid sequence of circa 33 kb) was also used in the original study of Barton et al. [63]. To delineate the intergenic region of scl, we aligned the coding sequence from the scl homolog SINFRUG00000145588 with the AJ131019 sequence using 'blast 2 sequences' [64]. The coding region was located from positions 20,156 to 22,165; we then selected the upstream region (from positions 1 to 20,155).
A two-step procedure for phylogenetic footprinting
A schematic representation of the developed two-step procedure is given in Figure 1.
Step 1: data reduction
In this step, a dataset consisting of the complete intergenic sequences of comparable size originating from orthologs of closely related organisms is reduced to a dataset of preselected sequences conserved among all/most compared orthologs. First, related vertebrate intergenic regions of comparable size (in this study these sequences corresponded to the mammalian human, chimp, rat, mouse and dog sequences) are aligned using the pairwise alignment algorithm AVID (using default parameters) [65]. For each ortholog, sequences corresponding to the significantly conserved regions of the pairwise alignment are selected using VISTA [66]. Significance of the alignment is defined by two parameters (VISTA parameters): the window length (L), the region for which the percent identity is calculated; and the conservation level (C) in the selected window, the minimal percent identity of the aligned region to be considered as significantly conserved. The parameter settings were adapted to the evolutionary distance of the compared organisms. The closer the organisms were related, the higher the threshold on the degree of conservation chosen. The conservation parameters used were: for human-mouse comparison, 85% over 200 nt; human-rat, 85% over 200 nt; human-chimp, 85% over 350 nt; human-dog, 80% over 200 nt; mouse-rat, 85% over 350 nt; mouse-chimp, 85% over 200 nt; mouse-dog, 80% over 200 nt; rat-chimp, 85% over 200 nt; rat-dog, 80% over 200 nt; and chimp-dog, 80% over 200 nt.
To identify orthologous regions conserved in multiple related vertebrate sequences of comparable size (that is, multiple alignment), homologies between all preselected sequences were determined (using AVID with default parameters). Subsequently, multiple conserved regions were identified using the graph based clustering TribeMCL [67]. We chose TribeMCL as this is a well-known graph-based clustering algorithm that was originally designed to recover transitivity relations between biological sequences (that is, orthologous proteins). Each resulting cluster corresponds to a region conserved in multiple sequences and consists of a set of preselected sequences originating from the different related orthologs of comparable size that mutually show a minimal degree of conservation. Several runs of TribeMCL were performed for each dataset, using different values of clustering parameters I and P (see [44]). The parameter I did not seem to have a major influence on the size of the clusters and, therefore, was set at 4. For the P value, three different values were tested per dataset and the parameter that resulted in small tightly linked clusters was chosen as these clusters correspond to strongly conserved regions. The parameters of choice for the benchmark datasets were: for cfos, I = 4 and P = 0; for hoxb2, I = 4 and P = -10; for pax6, I = 4 and P = 0; and for scl, I = 4 and P = -10. Concerning the additional datasets, the parameter setting of choice was I = 4 and P = 0 for EGR3, HIV-EP1, HOXB5, Meis2 and PCHD8 and I = 4 and P = -10 for GSH1.
Some clusters contain different subsequences derived from the intergenic sequence of a single organism that match one larger sequence of another organism; for example, two subsequences in rat that match one larger sequence in human. To minimize the noise in the datasets used for motif detection, such clusters are split into subclusters. Subclusters contain only a single subsequence of each ortholog (paralog; Figure 4). A subcluster is tagged by a profile containing the IDs of the different subsequences composing this subcluster. The input dataset for motif detection (Figure 1) thus consists of the mammalian subsequences in a subcluster together with the intergenic region of the corresponding Fugu ortholog.
Step 2: Motif detection
To find motifs conserved in the preselected intergenic sequences of orthologous genes, we developed BlockSampler as an extension of MotifSampler [39]. In contrast to the previous version of MotifSampler, which could only handle a single background model, in BlockSampler each orthologous intergenic sequence in the input dataset is scored with its appropriate species-specific background model. Previous studies have shown that using the correct species-specific higher order background model improves the reliability of the results [37,68]. In this study we used species-specific third-order background models.
The current implementation also allows selecting a user-defined core ortholog. This is the sequence of interest in which the motif should be present (in our case the sequence of heterogeneous length - the Fugu sequence). The idea behind this is that we are interested in motifs present in this core sequence that are supported by their presence in the preselected conserved orthologous regions. In this study, the most divergent Fugu orthologs were chosen as core sequences. The Gibbs sampling procedure searches for a common motif that has exactly one occurrence in the core sequence and no or one occurrence in the remainder of the sequences. After short motif seeds are identified, these are extended using a simple protocol to find larger conserved blocks: if the consensus score over a 5 nt region adjacent to the current motif exceeds a given threshold, the motif is extended with one nucleotide (in that direction). The larger a conserved block, the higher the confidence in the motif.
BlockSampler was run 100 times for each input set (subcluster plus Fugu ortholog) and corresponding random sets using default parameters; searching plus strand only (s = 0), prior set to 0.2, initial motif length of 8 nt. Only the threshold of the consensus score (default 1.0) was augmented to 1.2, selecting stronger conserved blocks. This generated 100 conserved blocks for each input set. To avoid redundancy, blocks overlapping more than 80% were merged. Concerning the benchmark datasets that consisted of only one distantly related ortholog, namely Fugu, we then selected those blocks that were conserved among all vertebrates under study. When studying more diverse datasets containing multiple distantly related species (with regard to mammals), we relaxed this requirement by allowing a block to be absent from one of the orthologs under study.
To account for the fact that short blocks are more likely to have a higher degree of conservation than long bocks, consensus scores [39] were compensated for their length. Blocks were then ranked according to this normalized consensus score (Csad), calculated using the formula Csad = (L/L+E)Cs, where L is the length of the conserved block, E is an empirical factor (set to 5) and Cs the consensus score.
To assess the relative individual contributions of the data reduction and motif detection steps to the final result, we applied BlockSampler on the full-length benchmark datasets. We used the same parameter setting as described above but, because of the longer sequence length in the full datasets, we increased the number of runs (1,000 runs for each benchmark dataset). Blocks were selected as described above. The best scoring 10% of the remaining blocks were searched for known motifs.
Randomization
To set a threshold on the adapted consensus score of the blocks (blocks with a score above the threshold are considered relevant), we compared block scores of the genuine set with those of corresponding random sets. For each genuine dataset, 100 random sets were generated. A corresponding random set contains, besides the different homologous regions of the genuine subcluster under study, a random Fugu intergenic sequence. This additional random sequence was not orthologous with the mammalian sequences and thus is unlikely to contain the same motifs. In each random set, motifs were identified using the same procedure as described for the genuine set. For each random set the best scoring motif was selected, that is, the block with the highest normalized consensus score. This resulted in a group of the best scoring 100 false positive motifs. These scores were approximately normally distributed. As a threshold, we choose the 90th percentile of the best scoring random motifs.
Motif validation
For each block we detected, a BLAT search against the human genome (May 2004 assembly) was performed [54,69]. This linked to the UCSC genome browser [51], where alignments between multiple vertebrate organisms were generated using MULTIZ [52]. Subsequently, we checked in the UCR browser [20] whether UCRs were identified in the intergenic regions under study.
To assess whether known transcription factor binding sites are located in the detected blocks, we compared the consensus sequence of each block with motifs described in the literature. In addition, we scanned the block consensus sequence with the Transfac 6.0 public database of vertebrate transcription factor binding site profiles [55]. This scanning was performed using MotifLocator [70-72] with a 0th order vertebrate background model. Hits with a score >0.9 were regarded as potential binding sites. The binding sites are indicated by the Transfac factor name [55].
To calculate the statistical over-representation of homeodomain binding sites, 100 sequences were selected randomly from the Fugu genome and screened to make sure they differed from the genes under study. These random sequences were screened with matrix models from homeodomain binding sites (obtained from TRANSFAC 8.2) using MotifLocator, as described above. We calculated the chi-square statistic with Yates correction of the 2 × 2 contingency table test for the set of homeodomain binding sites [73]. Homeobox binding sites were significantly over-represented in a certain block at a p value of 10-8.
Performance evaluation
To evaluate our newly developed procedure, we compared its performance to that of two algorithms often used for phylogenetic footprinting, namely the motif detection algorithm MotifSampler [39] and the multiple alignment procedures MAVID [10,65] and TBA [52]. These three algorithms were applied to the benchmark datasets and the resulting motifs (conserved in all organisms under study) were compared to those detected by the two-step procedure. We aligned the full-length initial datasets (Table 6) [44] using the online MAVID version at [74] with the default parameter setting [9].
Besides MAVID, we used TBA as it has been shown to outperform MAVID [52]. All the necessary tools were obtained from the Miller Lab website [75]. To generate a multiple alignment using TBA, we first pairwise aligned the initial datasets using blastz. We used the evolutionary tree ((human chimp)(rat mouse) Fugu); the additional blastz parameter file (latest version) was obtained from the E Margulies ftp site [76]. The final multiple alignment was obtained by running the TBA executable.
We applied MotifSampler both on the reduced datasets (subcluster + complete Fugu intergenic sequence [44]) and on the complete intergenic sequences (initial datasets). For the reduced sets we performed 100 MotifSampler runs, while for the complete datasets MotifSampler was run 1,000 times, each time using the standard parameter settings of the algorithm: the algorithm searches for only one motif (n = 1) of 8 nt (w = 8) on both strands (s = 1) and the prior probability of 1 motif copy (p) is 0.5. A third order vertebrate background model was used.
Additional data files
The following additional data are available with the online version of this paper. Additional data file 1 contains the list of significant blocks detected in the six additional datasets and, for each block, the results of the Transfac screening. Additional data file 2 contains the stand-alone version of BlockSampler. Additional data file 3 contains the corresponding BlockSampler help file.
Supplementary Material
The list of significant blocks detected in the six additional datasets and, for each block, the results of the Transfac screening
The stand-alone version of BlockSampler
The corresponding BlockSampler help file
Acknowledgments
Acknowledgements
R Van Hellemont is a fellow of the IWT. This work is partially supported by: IWT project GBOU-SQUAD-20160; Research Council KULeuven GOA Mefisto-666, GOA-Ambiorics, EF/05/007 SymBioSys, IDO genetic networks; FWO projects G.0115.01, G.0413.03 and G.0318.05; IUAP V-22 (2002-2006). The authors would like to thank Tine Blomme for help with the identification of orthologs.
Contributor Information
Ruth Van Hellemont, Email: Ruth.vanhellemont@esat.kuleuven.be.
Pieter Monsieurs, Email: Pieter.monsieurs@esat.kuleuven.be.
Gert Thijs, Email: Gert.thijs@esat.kuleuven.be.
Bart De Moor, Email: Bart.demoor@esat.kuleuven.be.
Yves Van de Peer, Email: Yves.vandepeer@psb.ugent.be.
Kathleen Marchal, Email: Kathleen.Marchal@biw.kuleuven.be.
References
- Tagle DA, Koop BF, Goodman M, Slightom JL, Hess DL, Jones RT. Embryonic epsilon and gamma globin genes of a prosimian primate (Galago crassicaudatus). Nucleotide and amino acid sequences, developmental regulation and phylogenetic footprints. J Mol Biol. 1988;203:439–455. doi: 10.1016/0022-2836(88)90011-3. [DOI] [PubMed] [Google Scholar]
- Fickett JW, Wasserman WW. Discovery and modeling of transcriptional regulatory regions. Curr Opin Biotechnol. 2000;11:19–24. doi: 10.1016/S0958-1669(99)00049-X. [DOI] [PubMed] [Google Scholar]
- Levy S, Hannenhalli S, Workman C. Enrichment of regulatory signals in conserved non-coding genomic sequence. Bioinformatics. 2001;17:871–877. doi: 10.1093/bioinformatics/17.10.871. [DOI] [PubMed] [Google Scholar]
- Boffelli D, McAuliffe J, Ovcharenko D, Lewis KD, Ovcharenko I, Pachter L, Rubin EM. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science. 2003;299:1391–1394. doi: 10.1126/science.1081331. [DOI] [PubMed] [Google Scholar]
- Chapman MA, Donaldson IJ, Gilbert J, Grafham D, Rogers J, Green AR, Gottgens B. Analysis of multiple genomic sequence alignments: a web resource, online tools, and lessons learned from analysis of mammalian SCL loci. Genome Res. 2004;14:313–318. doi: 10.1101/gr.1759004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern B, Frech K, Dress A, Werner T. DIALIGN: finding local similarities by multiple sequence alignment. Bioinformatics. 1998;14:290–294. doi: 10.1093/bioinformatics/14.3.290. [DOI] [PubMed] [Google Scholar]
- Morgenstern B. DIALIGN 2: improvement of the segment-to-segment approach to multiple sequence alignment. Bioinformatics. 1999;15:211–218. doi: 10.1093/bioinformatics/15.3.211. [DOI] [PubMed] [Google Scholar]
- Bray N, Pachter L. MAVID multiple alignment server. Nucleic Acids Res. 2003;31:3525–3526. doi: 10.1093/nar/gkg623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray N, Pachter L. MAVID: constrained ancestral alignment of multiple sequences. Genome Res. 2004;14:693–699. doi: 10.1101/gr.1960404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudno M, Do CB, Cooper GM, Kim MF, Davydov E, Green ED, Sidow A, Batzoglou S. LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003;13:721–731. doi: 10.1101/gr.926603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major MB, Jones DA. Identification of a gadd45beta 3' enhancer that mediates SMAD3- and SMAD4-dependent transcriptional induction by transforming growth factor beta. J Biol Chem. 2004;279:5278–5287. doi: 10.1074/jbc.M311517200. [DOI] [PubMed] [Google Scholar]
- Wasserman WW, Fickett JW. Identification of regulatory regions which confer muscle-specific gene expression. J Mol Biol. 1998;278:167–181. doi: 10.1006/jmbi.1998.1700. [DOI] [PubMed] [Google Scholar]
- Abrahams BS, Mak GM, Berry ML, Palmquist DL, Saionz JR, Tay A, Tan YH, Brenner S, Simpson EM, Venkatesh B. Novel vertebrate genes and putative regulatory elements identified at kidney disease and NR2E1/fierce loci. Genomics. 2002;80:45–53. doi: 10.1006/geno.2002.6795. [DOI] [PubMed] [Google Scholar]
- Aparicio S, Morrison A, Gould A, Gilthorpe J, Chaudhuri C, Rigby P, Krumlauf R, Brenner S. Detecting conserved regulatory elements with the model genome of the Japanese puffer fish, Fugu rubripes. Proc Natl Acad Sci USA. 1995;92:1684–1688. doi: 10.1073/pnas.92.5.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagheri-Fam S, Ferraz C, Demaille J, Scherer G, Pfeifer D. Comparative genomics of the SOX9 region in human and Fugu rubripes: conservation of short regulatory sequence elements within large intergenic regions. Genomics. 2001;78:73–82. doi: 10.1006/geno.2001.6648. [DOI] [PubMed] [Google Scholar]
- Montpetit A, Sinnett D. Comparative analysis of the ETV6 gene in vertebrate genomes from pufferfish to human. Oncogene. 2001;20:3437–3442. doi: 10.1038/sj.onc.1204444. [DOI] [PubMed] [Google Scholar]
- Nobrega MA, Ovcharenko I, Afzal V, Rubin EM. Scanning human gene deserts for long-range enhancers. Science. 2003;302:413. doi: 10.1126/science.1088328. [DOI] [PubMed] [Google Scholar]
- Santini S, Boore JL, Meyer A. Evolutionary conservation of regulatory elements in vertebrate Hox gene clusters. Genome Res. 2003;13:1111–1122. doi: 10.1101/gr.700503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelin A, Bailey P, Bruce S, Engstrom PG, Klos JM, Wasserman WW, Ericson J, Lenhard B. Arrays of ultraconserved non-coding regions span the loci of key developmental genes in vertebrate genomes. BMC Genomics. 2004;5:99–107. doi: 10.1186/1471-2164-5-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfe A, Goodson M, Goode DK, Snell P, McEwen GK, Vavouri T, Smith SF, North P, Callaway H, Kelly K, et al. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 2005;3:e7.0116–e7.0130. doi: 10.1371/journal.pbio.0030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio S, Chapman J, Stupka E, Putnam N, Chia JM, Dehal P, Christoffels A, Rash S, Hoon S, Smit A, et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science. 2002;297:1301–1310. doi: 10.1126/science.1072104. [DOI] [PubMed] [Google Scholar]
- Brenner S, Elgar G, Sandford R, Macrae A, Venkatesh B, Aparicio S. Characterization of the pufferfish (Fugu) genome as a compact model vertebrate genome. Nature. 1993;366:265–268. doi: 10.1038/366265a0. [DOI] [PubMed] [Google Scholar]
- Venkatesh B, Gilligan P, Brenner S. Fugu: a compact vertebrate reference genome. FEBS Lett. 2000;476:3–7. doi: 10.1016/S0014-5793(00)01659-8. [DOI] [PubMed] [Google Scholar]
- Elemento O, Tavazoie S. Fast and systematic genome-wide discovery of conserved regulatory elements using a non-alignment based approach. Genome Biol. 2005;6:1–R18. doi: 10.1186/gb-2005-6-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M, Tompa M. Discovery of regulatory elements by a computational method for phylogenetic footprinting. Genome Res. 2002;12:739–748. doi: 10.1101/gr.6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M, Tompa M. FootPrinter: A program designed for phylogenetic footprinting. Nucleic Acids Res. 2003;31:3840–3842. doi: 10.1093/nar/gkg606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. The value of prior knowledge in discovering motifs with MEME. Proc Int Conf Intell Syst Mol Biol. 1995;3:21–29. [PubMed] [Google Scholar]
- Hertz GZ, Hartzell GW, III, Stormo GD. Identification of consensus patterns in unaligned DNA sequences known to be functionally related. Comput Appl Biosci. 1990;6:81–92. doi: 10.1093/bioinformatics/6.2.81. [DOI] [PubMed] [Google Scholar]
- Hertz GZ, Stormo GD. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics. 1999;15:563–577. doi: 10.1093/bioinformatics/15.7.563. [DOI] [PubMed] [Google Scholar]
- Lawrence CE, Altschul SF, Boguski MS, Liu JS, Neuwald AF, Wootton JC. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science. 1993;262:208–214. doi: 10.1126/science.8211139. [DOI] [PubMed] [Google Scholar]
- McCue L, Thompson W, Carmack C, Ryan MP, Liu JS, Derbyshire V, Lawrence CE. Phylogenetic footprinting of transcription factor binding sites in proteobacterial genomes. Nucleic Acids Res. 2001;29:774–782. doi: 10.1093/nar/29.3.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliften PF, Hillier LW, Fulton L, Graves T, Miner T, Gish WR, Waterston RH, Johnston M. Surveying Saccharomyces genomes to identify functional elements by comparative DNA sequence analysis. Genome Res. 2001;11:1175–1186. doi: 10.1101/gr.182901. [DOI] [PubMed] [Google Scholar]
- Hughes JD, Estep PW, Tavazoie S, Church GM. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J Mol Biol. 2000;296:1205–1214. doi: 10.1006/jmbi.2000.3519. [DOI] [PubMed] [Google Scholar]
- Workman CT, Stormo GD. ANN-Spec: a method for discovering transcription factor binding sites with improved specificity. Pac Symp Biocomput. 2000:467–478. doi: 10.1142/9789814447331_0044. [DOI] [PubMed] [Google Scholar]
- Liu X, Brutlag DL, Liu JS. BioProspector: discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. Pac Symp Biocomput. 2001:127–138. [PubMed] [Google Scholar]
- Thijs G, Lescot M, Marchal K, Rombauts S, De Moor B, Rouze P, Moreau Y. A higher-order background model improves the detection of promoter regulatory elements by Gibbs sampling. Bioinformatics. 2001;17:1113–1122. doi: 10.1093/bioinformatics/17.12.1113. [DOI] [PubMed] [Google Scholar]
- Thijs G, Moreau Y, De Smet F, Mathys J, Lescot M, Rombauts S, Rouze P, De Moor B, Marchal K. INCLUSive: integrated clustering, upstream sequence retrieval and motif sampling. Bioinformatics. 2002;18:331–332. doi: 10.1093/bioinformatics/18.2.331. [DOI] [PubMed] [Google Scholar]
- Thijs G, Marchal K, Lescot M, Rombauts S, De Moor B, Rouze P, Moreau Y. A Gibbs sampling method to detect overrepresented motifs in the upstream regions of coexpressed genes. J Comput Biol. 2002;9:447–464. doi: 10.1089/10665270252935566. [DOI] [PubMed] [Google Scholar]
- Tompa M, Li N, Bailey TL, Church GM, De Moor B, Eskin E, Favorov AV, Frith MC, Fu Y, Kent WJ, et al. Assessing computational tools for the discovery of transcription factor binding sites. Nat Biotechnol. 2005;23:137–144. doi: 10.1038/nbt1053. [DOI] [PubMed] [Google Scholar]
- Pennacchio LA. Insights from human/mouse genome comparisons. Mamm Genome. 2003;14:429–436. doi: 10.1007/s00335-002-4001-1. [DOI] [PubMed] [Google Scholar]
- Elgar G, Sandford R, Aparicio S, Macrae A, Venkatesh B, Brenner S. Small is beautiful: comparative genomics with the pufferfish (Fugu rubripes). Trends Genet. 1996;12:145–150. doi: 10.1016/0168-9525(96)10018-4. [DOI] [PubMed] [Google Scholar]
- Wasserman WW, Krivan W. In silico identification of metazoan transcriptional regulatory regions. Naturwissenschaften. 2003;90:156–166. doi: 10.1007/s00114-003-0409-4. [DOI] [PubMed] [Google Scholar]
- Supplementary Website http://homes.esat.kuleuven.be/~kmarchal/Supplementary_Information_VanHel_2005/SuppWebsite.html
- Marchal K, De Keersmaecker S, Monsieurs P, van Boxel N, Lemmens K, Thijs G, Vanderleyden J, De Moor B. In silico identification and experimental validation of PmrAB targets in Salmonella typhimurium by regulatory motif detection. Genome Biol. 2004;5:R9.1–R9.20. doi: 10.1186/gb-2004-5-2-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scemama JL, Hunter M, McCallum J, Prince V, Stellwag E. Evolutionary divergence of vertebrate Hoxb2 expression patterns and transcriptional regulatory loci. J Exp Zool. 2002;294:285–299. doi: 10.1002/jez.90009. [DOI] [PubMed] [Google Scholar]
- Kammandel B, Chowdhury K, Stoykova A, Aparicio S, Brenner S, Gruss P. Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity. Dev Biol. 1999;205:79–97. doi: 10.1006/dbio.1998.9128. [DOI] [PubMed] [Google Scholar]
- Gottgens B, Barton LM, Chapman MA, Sinclair AM, Knudsen B, Grafham D, Gilbert JG, Rogers J, Bentley DR, Green AR. Transcriptional regulation of the stem cell leukemia gene (SCL) - comparative analysis of five vertebrate SCL loci. Genome Res. 2002;12:749–759. doi: 10.1101/gr.45502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. 10.1101/gr.229102. Article published online before print in May 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UCSC Genome Browser http://genome.ucsc.edu/
- Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, Roskin KM, Schwartz M, Sugnet CW, Thomas DJ, et al. The UCSC Genome Browser Database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UCR Browser http://mordor.cgb.ki.se/UCRbrowse/
- Kent WJ. BLAT - the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. 10.1101/gr.229202. Article published online before March 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingender E, Chen X, Fricke E, Geffers R, Hehl R, Liebich I, Krull M, Matys V, Michael H, Ohnhauser R, et al. The TRANSFAC system on gene expression regulation. Nucleic Acids Res. 2001;29:281–283. doi: 10.1093/nar/29.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies EH, Blanchette M, Haussler D, Green ED. Identification and characterization of multi-species conserved sequences. Genome Res. 2003;13:2507–2518. doi: 10.1101/gr.1602203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ureta-Vidal A, Ettwiller L, Birney E. Comparative genomics: genome-wide analysis in metazoan eukaryotes. Nat Rev Genet. 2003;4:251–262. doi: 10.1038/nrg1043. [DOI] [PubMed] [Google Scholar]
- Boffelli D, Nobrega MA, Rubin EM. Comparative genomics at the vertebrate extremes. Nat Rev Genet. 2004;5:456–465. doi: 10.1038/nrg1350. [DOI] [PubMed] [Google Scholar]
- Encode Project Consortium The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- Thomas JW, Touchman JW, Blakesley RW, Bouffard GG, Beckstrom-Sternberg SM, Margulies EH, Blanchette M, Siepel AC, Thomas PJ, McDowell JC, et al. Comparative analyses of multi-species sequences from targeted genomic regions. Nature. 2003;424:788–793. doi: 10.1038/nature01858. [DOI] [PubMed] [Google Scholar]
- Down TA, Hubbard TJ. NestedMICA: sensitive inference of over-represented motifs in nucleic acid sequence. Nucleic Acids Res. 2005;33:1445–1453. doi: 10.1093/nar/gki282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensembl Genome Browser http://www.ensembl.org
- Barton LM, Göttgens B, Gering M, Gilbert JG, Grafham D, Rogers J, Bentley D, Patient R, Green AR. Regulation of the stem cell leukemia (SCL) gene: a tale of two fishes. Proc Natl Acad Sci USA. 2001;98:6747–6752. doi: 10.1073/pnas.101532998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusova TA, Madden TL. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett. 1999;174:247–250. doi: 10.1016/S0378-1097(99)00149-4. [DOI] [PubMed] [Google Scholar]
- Bray N, Dubchak I, Pachter L. AVID: A global alignment program. Genome Res. 2003;13:97–102. doi: 10.1101/gr.789803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004;32:W273–W279. doi: 10.1093/nar/gkh053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchal K, Thijs G, De Keersmaecker S, Monsieurs P, De Moor B, Vanderleyden J. Genome-specific higher-order background models to improve motif detection. Trends Microbiol. 2003;11:61–66. doi: 10.1016/S0966-842X(02)00030-6. [DOI] [PubMed] [Google Scholar]
- BLAT Search Genome http://genome.ucsc.edu/cgi-bin/hgBlat
- Aerts S, Thijs G, Coessens B, Staes M, Moreau Y, De Moor B. Toucan: deciphering the cis-regulatory logic of coregulated genes. Nucleic Acids Res. 2003;31:1753–1764. doi: 10.1093/nar/gkg268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coessens B, Thijs G, Aerts S, Marchal K, De Smet F, Engelen K, Glenisson P, Moreau Y, Mathys J, De Moor B. INCLUSive: A web portal and service registry for microarray and regulatory sequence analysis. Nucleic Acids Res. 2003;31:3468–3470. doi: 10.1093/nar/gkg615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BioI@SCD Software http://homes.esat.kuleuven.be/~dna/Bioi/Software.html
- Kato M, Hata N, Banerjee N, Futcher B, Zhang MQ. Identifying combinatorial regulation of transcription factors and binding motifs. Genome Biol. 2004;5:R56.1–R56.13. doi: 10.1186/gb-2004-5-8-r56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAVID Multiple Alignment Server http://baboon.math.berkeley.edu/mavid/ [DOI] [PMC free article] [PubMed]
- Miller Lab http://bio.cse.psu.edu/
- E Margulies FTP Site ftp://kronos.nhgri.nih.gov/pub/outgoing/elliott/tba/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The list of significant blocks detected in the six additional datasets and, for each block, the results of the Transfac screening
The stand-alone version of BlockSampler
The corresponding BlockSampler help file