

Abstract
The heterogenic nature of troponin T (TnT) isoforms in fast skeletal and cardiac muscle suggests important functional differences. Dynamic features of rat cardiac TnT (cTnT) and rat fast skeletal TnT (fsTnT) reconstituted cardiac muscle preparations were captured by fitting the force response of small amplitude (0.5%) muscle length changes to the recruitment-distortion model. The recruitment of force-bearing cross-bridges (XBs) by increases in muscle length was favored by cTnT. The recruitment magnitude was ∼1.5 times greater for cTnT- than for fsTnT-reconstituted muscle fibers. The speed of length-mediated XB recruitment (b) in cTnT-reconstituted muscle fiber was 0.50–0.57 times as fast as fsTnT-reconstituted muscle fibers (3.05 vs. 5.32 s−1 at sarcomere length, SL, of 1.9 μm and 4.16 vs. 8.36 s−1 at SL of 2.2 μm). Due to slowing of b in cTnT-reconstituted muscle fibers, the frequency of minimum stiffness (fmin) was shifted to lower frequencies of muscle length changes (at SL of 1.9 μm, 0.64 Hz, and 1.16 Hz for cTnT- and fsTnT-reconstituted muscle fibers, respectively; at SL of 2.2 μm, 0.79 Hz, and 1.11 Hz for cTnT- and fsTnT-reconstituted muscle fibers, respectively). Our model simulation of the data implicates TnT as a participant in the process by which SL- and XB-regulatory unit cooperative interactions activate thin filaments. Our data suggest that the amino-acid sequence differences in cTnT may confer a heart-specific regulatory role. cTnT may participate in tuning the heart muscle by decreasing the speed of XB recruitment so that the heart beats at a rate commensurate with fmin.
INTRODUCTION
Troponin T (TnT) is essential for the Ca2+-regulated acto-myosin interactions that generate force in striated muscle (1,2). TnT not only bridges between the Ca2+ receptor, troponin C (TnC), and force-bearing cross-bridges (XBs) but also regulates XBs (3,4) by virtue of its unique interaction with tropomyosin (Tm) and other thin-filament regulatory proteins. There are major amino-acid sequence differences between cardiac TnT (cTnT) and fast skeletal muscle TnT (fsTnT) (5), many of which are in regions that are important for establishing the size of the regulated functional unit (Tm1-Tn1-actin7) in the thin filament (6), for modulating kinetic rates of transition between on- and off-states of Tm-Tn (7), and for regulating the rate of XB binding to actin (6). These observations suggest an important heart-specific functional role for cTnT in Ca2+ activation of thin filaments and force generation in cardiac muscle.
In addition to Ca2+ activation of thin filaments, changes in sarcomere length (SL) also regulate force in both cardiac and skeletal muscle through length-dependent activation. A greater effect of length-dependent activation in cardiac muscle compared to fast skeletal muscle has been linked to special features of cardiac thin filaments (1,8). Although various mechanisms have been proposed for length-dependent activation (9–12), the molecular mechanism by which increased SL recruits more XBs in cardiac muscle is not well understood. Previous studies have shown that cardiac troponin I (cTnI) (11) and cTnT (13) may be involved in length-dependent activation of cardiac myofilaments, possibly related to the pivotal role cTnT plays in bridging cTnI and cTnC to the Tm-actin filament.
The dual role of TnT in thin-filament activation by Ca2+ and changes in SL may be expressed in one or more of three myofilament kinetic steps:
Ca2+ binding to the thin-filament regulatory unit (Tm-Tn).
Regulatory unit (RU) switching between on- and off-states.
XB cycling between attached and detached states.
Because of the interactions of thin-filament kinetics with both Ca2+ binding kinetics and XB cycling kinetics, a RU-induced effect on thin-filament kinetics propagates to affect the other kinetic steps (14–16). Changes in RU composition, as occurs with different TnT isoforms, will affect the dynamics of XB recruitment from both Ca2+ activation and SL changes. In cardiac muscle, a cTnT effect on the rate of RU on-/off-transitions could occur either through a direct cTnT-induced effect on Tm-Tm overlapping ends or through direct or indirect effects of cTnT on the actin filament. Furthermore, XBs themselves affect the balance between RU on- and off-states through cooperative activation. This suggests that some aspect of RU-related cooperativity modulates the recruitment of XBs and, therefore, length-dependent activation. We believe that such RU-related mechanisms are likely to be different in cardiac muscle than in fast skeletal muscle, due in part to differences in the primary structure of cTnT.
Accordingly, in this study, we tested the hypothesis that cTnT plays a role in the process by which SL and XB-RU interactions activate cardiac thin filaments. To better determine the effect of specific alterations of cTnT on muscle mechanodynamics, we have used a new mathematical model of myofilament mechanodynamics (14–16). Our data fit well with a model in which cTnT is important for modulating the magnitude of XB recruitment in cardiac muscle. Our results also show that cTnT may participate in tuning the heart muscle by decreasing the speed of XB recruitment so that it is ideal for the heart to beat at a rate commensurate with the frequency of minimum stiffness, fmin.
MATERIALS AND METHODS
PCR cloning of full-length rat fsTnT DNA
A full-length cDNA clone for adult rat fsTnT (class IA α-1) was isolated from adult rat skeletal muscle first-strand cDNA (OriGene Technologies, Rockville, MD). For PCR amplification, we used two oligonucleotide primers whose nucleotide sequences were based on the previously published rat fsTnT sequence α-1 (rat fsTnT class IA α-1, Expasy/Swissprot accession No. P09739).
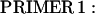 |
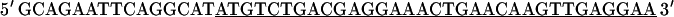 |
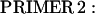 |
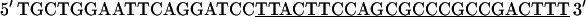 |
PCR cloning of full-length rat cTnC DNA
A full-length rat cTnC DNA clone was isolated from rat heart muscle first-strand cDNA (OriGene). We used two oligonucleotide primers whose nucleotide sequences were based on the previously published mouse cTnC sequence (Expasy/Swissprot accession No. P19123).
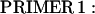 |
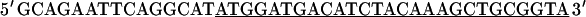 |
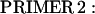 |
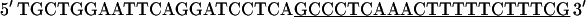 |
PCR cloning of full-length rat cTnI DNA
A full-length rat cTnI DNA clone was isolated from rat heart muscle first-strand cDNA (OriGene). We used two oligonucleotide primers whose nucleotide sequences were based on the previously published (17) mouse cTnI sequence (Expasy/Swissprot accession No. P48787).
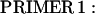 |
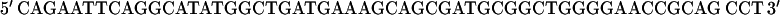 |
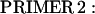 |
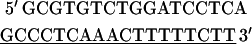 |
PCR-amplified DNA fragments were gel-purified, digested with appropriate restriction enzymes and subcloned into the Nde I-BamH I site of the pSBETa expression vector (Roche, Pleasantan, CA). DNA clones containing proper inserts were sequenced. Adult rat cTnT cDNA clone was a gift from Dr. J.J. Lin (University of Iowa).
Nucleotide sequences of rat fsTnT, rat cTnC, and rat cTnI
The nucleotide sequence from our rat fsTnT (Genbank Accession No. bankit718582 DQ062204) clone matched perfectly with the previously published sequence. To amplify the full-length rat cTnC and rat cTnI clones from the first-strand cDNA, mouse cTnC and mouse cTnI nucleotide sequences were used as templates, respectively. When compared with the nucleotide sequence derived from an annotated rat genomic sequence (NCBI accession No. XM-214266), there were three nucleotide differences in our rat cTnC clone (Genbank Accession No. bankit718648 DQ062205). However, the amino-acid sequences from our rat cTnC clone were found to be identical to the amino-acid sequence derived from an annotated rat genomic sequence. Similarly, we also found four nucleotide changes compared to the published rat cTnI sequence (18), with no changes in amino-acid sequence (Genbank Accession No. bankit719338 DQ062462).
Expression and purification of recombinant proteins
Recombinant rat fsTnT, rat cTnT, rat cTnC, and rat cTnI (all in pSBETa plasmid DNA) were expressed in BL21 DE3 cells (Novagen, Madison, WI). For all protein preparations, cells from 4 liters were then spun down and sonicated in 50 mM Tris (pH 8.0 at 4°C), 6 M urea, 5 mM EDTA, 0.2 mM PMSF, 5 mM benzamidine-HCl, 10 μM leupeptin, 1 μM pepstatin, 5 μM Bestatin, 2 μM E64, and 1 mM DTT. The insoluble fraction was separated by centrifugation. Rat fsTnT was purified as follows: The supernatant from rat fsTnT culture preparation was used for ammonium sulfate fractionations. The pellet from the 45% ammonium sulfate cut was dissolved in 50 mM Tris (pH 8.0 at 4°C), 6 M urea, 1 mM EDTA, 0.2 mM PMSF, 4 mM benzamidine-HCl, and 1 mM DTT, and then purified by chromatography on a DEAE-fast sepharose (Pharmacia, Basking Ridge, NJ) column. Rat fsTnT was eluted with a 0–0.3M NaCl gradient. Rat cTnT was purified as follows: The pellet from the 70% ammonium sulfate cut was dissolved in 50 mM Tris (pH 8.0 at 4°C), 6 M urea, 1 mM EDTA, 0.2 mM PMSF, 4 mM benzamidine-HCl, and 1 mM DTT, and then purified by chromatography on a DEAE-fast sepharose. Rat cTnT was eluted with a gradient of 0–0.3 M NaCl. Impure fractions were dialyzed against 50 mM Na acetate (pH 5.3 at 4°C), 6 M urea, 1 mM EDTA, 0.2 mM PMSF, 4 mM benzamidine-HCl, and 1 mM DTT and chromatographed on a SP-Sepharose (Pharmacia) column. Rat cTnT was eluted with a gradient of 0–1 M NaCl. Rat cTnI was purified as described previously (17). Rat cTnC was purified as described previously (19). All pure protein fractions were extensively dialyzed against deionized water containing 15 mM β-mercaptoethanol, lyophilized and stored at −80°C.
Reconstitution of recombinant rat TnT isoforms into detergent-skinned rat cardiac muscle fiber bundles
Left ventricular papillary muscle fiber bundles from rat hearts were isolated and dissected, as described previously (13,14). Detergent skinning of muscle fibers were performed overnight at 4°C in the relaxing solution (HR, pCa 9.0) containing 50 mM BES (pH 7.0), 30.83 mM K Propionate, 10 mM NaN3, 20 mM EGTA, 6.29 mM MgCl2, 6.09 mM ATP, 20 mM BDM, 1 mM DTT, 0.1% Triton X-100, and a cocktail of protease inhibitors (4 μM Benzamidine-HCl, 5 μM Bestatin, 2 μM E-64, 10 μM Leupeptin, 1 μM Pepstatin, and 200 μM PMSF). Exchange of rat muscle endogenous troponin complex with rat recombinant troponin complex containing either cTnT-cTnI-cTnC or fsTnT-cTnI-cTnC was based on the method described previously (20), which was modified as follows: The extraction solution containing a mixture of cTnT/fsTnT and cTnI was prepared as cTnT/fsTnT (0.7 mg/ml, w/v) and cTnI (0.7 mg/ml, w/v), which were initially dissolved in 50 mM Tris-HCl (pH 8.0), 6 M urea, 1.0 M KCl, 10 mM DTT, and a cocktail of protease inhibitors. High salt and urea were removed by successive dialysis against the following buffers: 50 mM Tris-HCl (pH 8.0 at 4°C), 4 M urea, 0.7 M KCl, 1 mM DTT, 4 mM benzamidine-HCl and 0.4 mM PMSF and 0.01% NaN3, followed by 50 mM Tris-HCl (pH 8.0 at 4°C), 2 M urea, 0.5 M KCl, 1 mM DTT, 4 mM benzamidine-HCl, and 0.4 mM PMSF and 0.01% NaN3 and then finally against the extraction buffer containing 50 mM BES (pH 7.0 at 20°C), 180 mM KCl, 10 mM BDM, 5 mM EGTA, 6.27 mM MgCl2, 1.0 mM DTT, 4 mM benzamidine-HCl, 0.2 mM PMSF, and 0.01% NaN3. After final dialysis, 5 mM MgATP2- and fresh protease inhibitors were added to the supernatant containing cTnT-cTnI or fsTnT-cTnI. Any undissolved protein was removed by spinning in a microfuge at maximum speed for 15 min. Detergent-skinned muscle fiber bundles were treated with the extraction solution containing cTnT-cTnI or fsTnT-cTnI for ∼3–4 h at room temperature with gentle constant stirring. Muscle fiber bundles were then washed twice with extraction buffer for 15 min. Ca2+-activated maximal tension was measured in pCa 4.3 to determine the residual tension. cTnT-cTnI or fsTnT-cTnI treated muscle fiber bundles were reconstituted overnight (4°C) with cTnC (3 mg/ml) prepared in the relaxation buffer on ice. The composition of relaxation buffer was 50 mM BES, 51.14 mM K propionate, 5.83 mM Na2ATP, 6.87 mM MgCl2, 10 mM EGTA, 5mM NaN3, 1 mM DTT, 10 mM phosphenol pyruvate, 50 μM Leupeptin, 1 μM Pepstatin, 200 μM PMSF, 10 μM oligomycin, and 20 μM A2P5 (ionic strength ∼180 mM). After reconstitution, Ca2+-activated tension and ATPase activity were measured in solutions containing different amounts of free (Ca2+) as described previously (13,14).
Tension-ATPase relation
For simultaneous measurement of tension and ATPase (20°C), we used a system described by Stienen et al. (21) and de Tombe and Stienen (22). Detergent-skinned muscle fiber was attached to a motor and a force transducer using aluminum clips. Sarcomere length (SL) was measured, as previously described (13,22). After 2–3 cycles of full activation and relaxation, the resting SL was readjusted to 1.9–2.2 μm and continuously monitored using a He-Ne laser diffraction system. Using this approach, we found that the resting SL remained stable throughout the experiment. Near-UV light (340 mm) was projected through the muscle chamber just below the muscle fiber, then split via a beam splitter (50:50) and detected at 340 nm (sensitive to change in NADH) and 400 nm (insensitive to NADH). The light intensity at 400 nm served as a reference signal. An analog divider and log amplifier produced a signal proportional to the amount of ATP consumed in the muscle chamber solution. ATPase activity of the skinned muscle fiber bundle was measured as follows: ATP regeneration from ADP was coupled to the breakdown of phosphenol pyruvate to pyruvate and ATP catalyzed by pyruvate kinase, which was linked to the synthesis of lactate catalyzed by lactate dehydrogenase. The breakdown of NADH (which is proportional to the amount of ATP consumed), was measured online by UV absorbance at 340 nm. Maximum activation buffer (pCa 4.3) contained 31 mM potassium propionate, 5.95 mM Na2ATP, 6.61 mM MgCl2, 10 mM EGTA, 10.11 mM CaCl2, 50 mM BES, pH 7.0, 10 mM NaN3, 10 μM leupeptin, 1 μM pepstatin, 10 μM oligomycin, 100 μM PMSF, 0.9 mM NADH, 10 mM phosphenol pyruvate, 4 mg/ml pyruvate kinase (500 U/mg), 0.24 mg/ml lactate dehydrogenase (870 U/mg), and 20 μM A2P5 and the ionic strength of the buffer was 180 mM. The composition of different pCa (−log of free Ca2+-concentration) solution was calculated using the methods described by Fabiato and Fabiato (23).
Dynamic force-length relationship
Dynamic force-length relationship (FLR) was determined at maximal Ca2+ activation (pCa 4.3) as described previously (14). Briefly, this protocol was designed to provide force and muscle-length information at all frequencies between 0.1 and 40 Hz. Muscle fiber length, LM, was commanded to change according to a constant amplitude (0.5% of LM) sinusoid of continuously varying frequency (chirp). Two chirps were delivered over two sequential time periods. In the first period of 40 s duration, chirp frequencies varied between 0.1 and 4 Hz to emphasize low frequency behavior. In the second time period of 5 s duration, chirp frequencies varied between 1 and 40 Hz to emphasize higher frequency behavior. Measured force changes, ΔF(t), during the FLR protocol were maximally 10% of the Fs baseline. Baseline trends and wander were removed (14) from the ΔF(t) record by fitting a fourth-order polynomial in time to the ΔF(t) signal. The frequency content of the fourth-order polynomial was in a range below the frequency composition of the ΔL(t) signal. Examples of data obtained with this protocol are shown later, in Fig. 6.
FIGURE 6.

Frequency dependency of force response to controlled sinusoidal oscillations in detergent-skinned rat cardiac muscle fiber bundles reconstituted with recombinant rat cTnT and fsTnT. Model-predicted force from the recruitment-distortion model (14) accurately reproduced force measurements for both example tracings shown in panels A and C. Measured force is shown by light shaded trace and the model-predicted force is shown by black trace. The minimal force amplitude response is shown by the arrow in both panels A and C. (A) Force recordings from the double-chirp sinusoidal muscle length change protocol in the cTnT+cTnI+cTnC reconstituted muscle fibers at SL of 2.2 μm. (B) Two chirps were delivered over two sequential time periods: in one period of 40-s duration, chirp frequencies varied between 0.1 and 4 Hz to emphasize low frequency behavior and in a second time period of 5-s duration, chirp frequencies varied between 1 and 40 Hz to emphasize higher frequency behavior (14). Identical chirps were delivered to both muscle fibers shown in panels A and C. (A) Force recordings from the double-chirp sinusoidal muscle length change protocol in the fsTnT+cTnI+cTnC reconstituted muscle fibers at SL of 2.2 μm. There was a small divergence between model prediction and force measurement at high frequencies. Our attempt in modifying the recruitment-distortion model to account for the small high frequency effect, led to no change in the values of model-estimated parameters. The frequency of minimal stiffness, fmin, is 0.79 Hz for cTnT-cTnI-cTnC reconstituted fibers (A) and 1.11 Hz for fsTnT+cTnI+cTnC reconstituted fibers (C).
Estimating parameters of dynamic FLR
Parameters of the dynamic FLR were derived by fitting a recruitment-distortion model (14) to the measurement of ΔF(t) obtained in the dynamic FLR protocol. The recruitment-distortion model is given by the following differential equations:
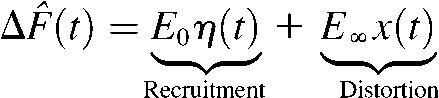 |
(1) |
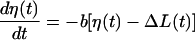 |
(2) |
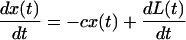 |
(3) |
In these equations, 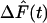 is the model-predicted variation in force in response to measured variation in muscle length, ΔL(t) (through recruitment dynamics; see Eq. 2), and in response to the first time derivative of muscle length,
is the model-predicted variation in force in response to measured variation in muscle length, ΔL(t) (through recruitment dynamics; see Eq. 2), and in response to the first time derivative of muscle length, 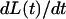 (through distortion dynamics; see Eq. 3). The value η(t) is the recruitment variable; it describes the incremental addition of XBs acting in parallel to produce force. The value x(t) is the distortion variable; it describes the average distortion of internal stretch with the elastic regions of XBs. Parameter E0 is the slope of the static FLR. Parameter E∞ is instantaneous stiffness, as estimated from the initial force response to a sudden stretch. Parameter b is the rate constant governing recruitment dynamics. Parameter c is the rate constant governing distortion dynamics. This model of cardiac muscle dynamic FLR has undergone extensive validation; the model was shown to fit the data well, leaving very little residual error (R2 > 0.98), and parameters of the model (E0, b, E∞, c) were estimated with <1% error (14). Fitting of data in the present study was as described previously (14).
(through distortion dynamics; see Eq. 3). The value η(t) is the recruitment variable; it describes the incremental addition of XBs acting in parallel to produce force. The value x(t) is the distortion variable; it describes the average distortion of internal stretch with the elastic regions of XBs. Parameter E0 is the slope of the static FLR. Parameter E∞ is instantaneous stiffness, as estimated from the initial force response to a sudden stretch. Parameter b is the rate constant governing recruitment dynamics. Parameter c is the rate constant governing distortion dynamics. This model of cardiac muscle dynamic FLR has undergone extensive validation; the model was shown to fit the data well, leaving very little residual error (R2 > 0.98), and parameters of the model (E0, b, E∞, c) were estimated with <1% error (14). Fitting of data in the present study was as described previously (14).
Measurement of rate of tension redevelopment (ktr)
The ktr measurements were made at maximal Ca2+ activation (pCa 4.3). A large slack-release protocol (24) was used to disengage force-generating XBs from the thin filaments, which were isometrically activated. The rate constant of tension redevelopment (ktr) was determined by fitting the rise of tension to the following equation: F = Fobs(1−e−ktr.t) + F0, where F is force at time t, Fobs is observed steady-state force, and ktr is the rate constant of tension redevelopment. In all cases, tension redevelopment in cardiac muscle fibers was well fitted with the monoexponential equation (R2 > 0.97).
Polyacrylamide gel electrophoresis
Protein samples for gel electrophoresis and Western blot analysis were prepared and run on 12.5% SDS-polyacrylamide gels, as previously described (25,26). For Western blot analysis, proteins were transferred onto the PVDF membrane and probed using an anti-mouse primary antibody against either rabbit fsTnT or rat cTnT, as previously described (26).
Data analysis
Data from the normalized pCa-tension measurements were fitted to the Hill equation by using a nonlinear least-square regression procedure to obtain the pCa50 (−log of free Ca2+-concentration required for half-maximal activation) and the Hill coefficient (n). pCa50 and n were determined separately from each muscle fiber experiment and the values averaged. Statistical differences were analyzed by one-way ANOVA, with the criteria for significance set at p < 0.05. Data are expressed as mean ± SE.
RESULTS
Exchange of rat fsTnT into detergent-skinned rat cardiac muscle fiber bundles
Control untreated, cTnT+cTnI+cTnC and fsTnT+cTnI+cTnC reconstituted muscle fibers were solubilized in the gel-loading buffer (20,25) and separated on 12.5% SDS gels. Western blot analysis was performed with the anti-cTnT antibody (Fig. 1 A) to demonstrate the removal of endogenous native cTnT in fsTnT+cTnI+cTnC reconstituted muscle fibers. No immunoreactivity is evident in lane 4 corresponding to the endogenous cTnT band, which demonstrated that most of the endogenous cTnT was replaced by fsTnT (Fig. 1 A). In Fig. 1 B, Western blot analysis was performed with the anti-fsTnT antibody to demonstrate the incorporation of fsTnT in fsTnT+cTnI+cTnC reconstituted muscle fibers. Lane 3 in Fig. 1 B shows that the recombinant fsTnT isoform was incorporated into fsTnT+cTnI+cTnC reconstituted muscle fibers.
FIGURE 1.

Western blot analysis of rat cardiac muscle fiber bundles reconstituted with recombinant rat cTnT and rat fsTnT proteins. (A) Anti-human cTnT antibody was used to probe for rat cardiac TnT. Lane 1, purified recombinant rat cTnT; lane 2, control untreated muscle fibers; lane 3, cTnT+cTnI+cTnC reconstituted muscle fibers; and lane 4, fsTnT+cTnI+cTnC reconstituted muscle fibers. No immunoreactivity is evident in lane 4 corresponding to the native cTnT. (B) Anti-rabbit fast skeletal TnT antibody was used to probe for the presence of rat fsTnT in reconstituted muscle fibers. Lane 1, purified recombinant rat fsTnT; lane 2, cTnT+cTnI+cTnC reconstituted muscle fibers; and lane 3, fsTnT+cTnI+cTnC reconstituted muscle fibers.
Effect of fsTnT on Ca2+ activation in cardiac muscle fibers
The SL dependencies of Ca2+-activated maximal tension and ATPase activity were measured in control untreated and reconstituted muscle fibers at pCa 4.3. At short SL of 1.9 μm (Fig. 2 A), Ca2+-activated maximal tension (in mN/mm2) was 38 ± 1, 36 ± 2, and 23 ± 2 for control untreated, cTnT+cTnI+cTnC reconstituted, and fsTnT+cTnI+cTnC reconstituted muscle fibers, respectively. At short SL, Ca2+-activated maximal ATPase activity (in pmol/mm3/s) was 220 ± 8, 202 ± 10, and 174 ± 7 for control untreated, cTnT+cTnI+cTnC reconstituted, and fsTnT+cTnI+cTnC reconstituted muscle fibers, respectively (Fig. 2 B). At long SL of 2.2 μm, Ca2+-activated maximal tension (in mN/mm2) was 53 ± 1, 55 ± 1, and 38 ± 2 for control untreated, cTnT+cTnI+cTnC reconstituted, and fsTnT+cTnI+cTnC reconstituted muscle fibers, respectively (Fig. 3 A). At long SL, Ca2+-activated maximal ATPase activity (in pmol/mm3/s) was 222 ± 6, 215 ± 7, and 144 ± 7 for control untreated, cTnT+cTnI+cTnC reconstituted and fsTnT+cTnI+cTnC reconstituted muscle fibers, respectively (Fig. 3 B). Thus, both Ca2+-activated maximal tension and ATPase activity (Figs. 2 and 3) in cTnT+cTnI+cTnC reconstituted muscle fibers were not significantly different from those of control untreated muscle fibers at both short and long SL. On the other hand, both Ca2+-activated maximal tension and maximal ATPase activity (Figs. 2 and 3) were significantly depressed in fsTnT+cTnI+cTnC reconstituted muscle fibers at both short and long SL. When a second reconstitution of fsTnT+cTnI+cTnC reconstituted fibers was done to replace fsTnT with cTnT (restoring the cTnT+cTnI+cTnC construct), the depression in Ca2+-activated tension, and ATPase activity were released (data not shown). These observations demonstrated that the depression of tension and ATPase activity were due to the impact of fsTnT on cardiac myofilaments and not related to the reconstitution procedure.
FIGURE 2.

Ca2+-activated maximal tension and ATPase activity in detergent-skinned rat cardiac muscle fiber bundles reconstituted with recombinant rat cTnT and fsTnT. After reconstitution, Ca2+-activated maximal tension and ATPase activity (21,22) were measured in activation buffer at pCa 4.3. Control untreated muscle fibers (control), cTnT+cTnI+cTnC reconstituted muscle fibers (cTnT), and fsTnT+cTnI+cTnC reconstituted muscle fibers (fsTnT). (A) Effect on Ca2+-activated maximal tension at SL of 1.9 μm. (B) Effect on Ca2+-activated maximal ATPase activity at SL of 1.9 μm. Residual Ca2+-activated tension (pCa 4.3) measured after treatment with either cTnT+cTnI or fsTnT+cTnI was minimal (∼1.0 mN/mm2). Number of determinations is at least 10 for each. The asterisk symbol (*) indicates a result significantly different from control (P < 0.001).
FIGURE 3.

Ca2+-activated maximal tension and ATPase activity in detergent-skinned rat cardiac muscle fiber bundles reconstituted with recombinant rat cTnT and fsTnT. After reconstitution, Ca2+-activated maximal tension and ATPase activity (21,22) were measured in activation buffer at pCa 4.3. Control untreated muscle fibers (control), cTnT+cTnI+cTnC reconstituted muscle fibers (cTnT), and fsTnT+cTnI+cTnC reconstituted muscle fibers (fsTnT). (A) Effect on Ca2+-activated maximal tension at SL of 2.2 μm. (B) Effect on Ca2+-activated maximal tension and ATPase activity at SL of 2.2 μm. Residual Ca2+-activated tension (pCa 4.3) measured after treatment with either cTnT+cTnI or fsTnT+cTnI was minimal (∼1.0 mN/mm2). Number of determinations is at least 10 for each. The asterisk symbol (*) indicates a result significantly different from control (P < 0.001).
Furthermore, the steepness of pCa-tension relationships in cTnT+cTnI+cTnC and fsTnT+cTnI+cTnC reconstituted muscle fibers were similar to those of control untreated muscle fibers (Fig. 4 and Table 1). Similar results were observed with pCa-ATPase relations (data not shown). A lack of full complement of Tn would have likely resulted in significant changes in the Hill coefficient value (n), myofilament Ca2+ sensitivity (pCa50), and Ca2+-activated maximal tension (27). These observations demonstrated that both cTnT and fsTnT reconstitution was complete. Data shown in Fig. 4 and Table 1 also demonstrate that the fsTnT isoform did not affect cardiac myofilament Ca2+ sensitivity and cooperativity. Two independent previous studies have shown that modifications of cTnT affect Ca2+-activated maximal tension and ATPase activity in cardiac muscle. For example, our own work demonstrated previously that a modification of the N-terminal region of rat cTnT depressed maximal activation in cardiac myofilaments without affecting n- and pCa50 values (25). Similarly, Communal et al. (28) showed that the deletion of the N-terminus of rat cTnT depressed force and ATPase activity, with no effect on n- and pCa50 values.
FIGURE 4.

Normalized pCa-tension relationship in detergent-skinned rat cardiac muscle fiber bundles reconstituted with recombinant rat cTnT and fsTnT. (A) pCa-tension relations at SL of 1.9 μm. Control untreated muscle fibers (○), cTnT+cTnI+cTnC reconstituted muscle fibers (•), and fsTnT+cTnI+cTnC reconstituted muscle fibers (▴). (B) pCa-tensions relation at SL of 2.2 μm. Control untreated muscle fibers (○), cTnT+cTnI+cTnC reconstituted muscle fibers (•), and fsTnT+cTnI+cTnC reconstituted muscle fibers (▴). pCa50 and Hill coefficient values (n) are listed in Table 1. Standard error bars are smaller than symbols in some cases. Number of determinations is at least 10 for each.
TABLE 1.
Normalized pCa-tension relationship in control untreated, cTnT+cTnI+cTnC, and fsTnT+cTnI+cTnC reconstituted cardiac muscle fiber bundles at short (1.9 μm) and long (2.2 μm) SL
| Control | cTnT+cTnI+cTnC | fsTnT+cTnI+cTnC | |
|---|---|---|---|
| SL 1.9 μm | |||
| pCa50 | 5.50 ± 0.03 | 5.49 ± 0.02 | 5.45 ± 0.02 |
| n | 4.3 ± 0.2 | 4.8 ± 0.2 | 4.2 ± 0.2 |
| SL 2.2 μm | |||
| pCa50 | 5.61 ± 0.03 | 5.63 ± 0.02 | 5.63 ± 0.02 |
| n | 4.2 ± 0.2 | 4.3 ± 0.2 | 4.4 ± 0.2 |
Values are means ± SE. Data from the normalized pCa-tension measurements were fitted to the Hill equation by using a nonlinear least-square regression procedure to derive pCa50 and the Hill coefficient (n) values. pCa50 and n were determined separately from each muscle fiber experiment and the values averaged. Number of determinations is at least 10 for each.
Effect of fsTnT on the relationship between steady-state isometric tension and ATPase activity
We determined the relationship between steady-state isometric tension and the rate of ATP hydrolysis in muscle fibers reconstituted with cTnT+cTnI+cTnC and fsTnT+cTnI+cTnC. Tension-ATPase relationships were linear (Fig. 5). At long SL (Fig. 5 B), the slopes of the tension-ATPase relationship (tension cost) of cTnT+cTnI+cTnC and fsTnT+cTnI+cTnC reconstituted muscle fibers were not significantly different from those of control untreated muscle fibers (Table 2). The slope of steady-state isometric tension and the rate of ATP hydrolysis has been proposed as a measure of the rate of XB detachment (23). Therefore, our data suggest that the rate of XB detachment in cardiac myofilaments was not altered by the presence of fsTnT at long SL. In contrast, there was a 38% increase in the slope of tension-ATPase relationship in fsTnT+cTnI+cTnC reconstituted muscle fibers at short SL (Fig. 5 A and Table 2), indicating that there was an increase in the rate of XB detachment rate at short SL.
FIGURE 5.

Relationship between steady-state isometric tension and ATPase activity in detergent-skinned rat cardiac muscle fiber bundles reconstituted with recombinant rat cTnT and fsTnT. Steady-state isometric tension and ATPase were measured simultaneously as described before (21,22). Equal variance across the sample was confirmed, which permitted us to bin the data as follows. Tension and ATPase data were pooled in 10% wide steady-state bands and the average values for each band were calculated. Averaged data were fitted using a linear regression analysis. Departure from linearity was found to be nonsignificant in all cases. (A) Tension-ATPase relations at SL of 1.9 μm. Mean slope values from linear fits of at least 10 muscles are listed in Table 2. Control untreated muscle fibers (○), R2 = 0.97; cTnT+cTnI+cTnC reconstituted muscle fibers (•), R2 = 0.97; and fsTnT+cTnI+cTnC reconstituted muscle fibers (▴), R2 = 0.96. (B) Tension-ATPase relations at SL of 2.2 μm. Control untreated muscle fibers (○), R2 = 0.99; cTnT+cTnI+cTnC reconstituted muscle fibers (•), R2 = 0.98; and fsTnT+cTnI+cTnC reconstituted muscle fibers (▴), R2 = 0.99.
TABLE 2.
Effect of activating Ca2+ on the slope of tension-ATPase relationship (tension cost) and XB distortion rate constant (c) at SL of 1.9 and 2.2 μm in control untreated, cTnT+cTnI+cTnC, and fsTnT+cTnI+cTnC reconstituted cardiac muscle fiber bundles
| Control | cTnT+cTnI+cTnC | fsTnT+cTnI+cTnC | |
|---|---|---|---|
| SL 1.9 μm | |||
| Tension cost | 6.26 ± 0.24 | 6.05 ± 0.22 | 8.38 ± 0.49† |
| c (s−1) | 52.75 ± 4.56 | 53.37 ± 4.28 | 94.93 ± 6.76† |
| SL 2.2 μm | |||
| Tension cost | 4.47 ± 0.10 | 4.04 ± 0.15 | 4.09 ± 0.18 |
| c (s−1) | 40.4 ± 0.2 | 31.21 ± 0.4* | 35.53 ± 0.3* |
Values are means ± SE. Tension and ATPase activities were measured simultaneously at different Ca2+ activations as described previously (13,21,22). Tension cost (in pmol/mN/mm/s) is determined from the slopes of tension-ATPase relationships (Fig. 5). The rate constant of XB distortion (c) was determined at maximal Ca2+ activation (pCa 4.3) as described previously (14). Number of determinations is at least 10 for each.
p < 0.01.
p < 0.001.
Effect of TnT isoform on the rate of tension redevelopment (ktr)
SL-dependent effects on maximal ktr values in reconstituted muscle fibers were measured at pCa 4.3. fsTnT significantly increased ktr values in reconstituted cardiac muscle fibers at both short and long SL (Table 3). At short SL, the ktr for fsTnT+cTnI+cTnC reconstituted muscle fiber was 37% higher than in cTnT+cTnI+cTnC reconstituted muscle fibers. Similarly, at long SL, the ktr for fsTnT+cTnI+cTnC reconstituted muscle fiber was 32% higher than in cTnT+cTnI+cTnC reconstituted muscle fibers. Furthermore, the maximum ktr values at short SL were significantly higher than those at long SL for all three groups of muscle fibers tested (Table 3). The ktr values increased by 38% from long to short SL in control untreated, by 29% for cTnT+cTnI+cTnC, and by 34% for fsTnT+cTnI+cTnC reconstituted muscle fibers. In contrast to our observations, studies on rat slow-twitch and rabbit fast-twitch skeletal muscle fibers demonstrated that ktr values at short SL were significantly lower than those measured at longer SL (29). This difference may be related to the differences in the fiber types used in our study. Some support for our study comes from a recent study in which the SL-dependent effect on ktr was measured in rat cardiac trabeculae preparations (30). During submaximal activation (30), ktr values measured at short SL were significantly faster than those measured at long SL. However, they observed a small but statistically insignificant increase in ktr values during maximal Ca2+ activation at short SL (30).
TABLE 3.
Effect of activating Ca2+ on rate constants of tension redevelopment (ktr) and XB recruitment (b) at SL of 1.9 and 2.2 μm in control untreated, cTnT+cTnI+cTnC, and fsTnT+cTnI+cTnC reconstituted cardiac muscle fiber bundles
| Control | cTnT+cTnI+cTnC | fsTnT+cTnI+cTnC | |
|---|---|---|---|
| SL 1.9 μm | |||
| ktr (s−1) | 8.82 ± 0.31 | 7.87 ± 0.47 | 10.78 ± 0.51* |
| b (s−1) | 2.24 ± 0.22 | 3.05 ± 0.52 | 5.32 ± 0.58* |
| SL 2.2 μm | |||
| ktr (s−1) | 6.39 ± 0.18 | 6.10 ± 0.14 | 8.04 ± 0.33* |
| b (s−1) | 4.67 ± 0.27 | 4.16 ± 0.25 | 8.36 ± 0.38* |
Values are means ± SE. The rate constant of monoexponential tension redevelopment (ktr) was determined as described previously (13,24). The rate constant of XB recruitment (b) was determined as described previously (14). The values b and ktr were measured at maximal Ca2+ activation (pCa 4.3). Number of determinations is at least 10 for each.
p < 0.01.
Effect of TnT on XB recruitment and distortion dynamics
Results from the double-chirp, dynamic FLR protocol for muscle fibers reconstituted with cTnT+cTnI+cTnC and fsTnT+cTnI+cTnC are shown in Fig. 6, A and C, respectively. In both examples, the force amplitude first declined to a minimum (see arrows in Fig. 6) and then rose as the frequency of muscle length perturbation increased. Standard errors of the parameter estimates (E0, b, E∞, c) were generally <1% of the estimated parameter value. Both the magnitude and the speed of length-mediated XB recruitment were strongly affected by the isoform of TnT. E0 (XB recruitment magnitude) was ∼1.5 times greater for cTnT+cTnI+cTnC than for fsTnT+cTnI+cTnC reconstituted muscle fibers (82 vs. 56 mN/mm2 per μm at short SL and 186 vs. 126 mN/mm2 per μm at long SL). Thus, the length-induced increase in E0 was favored in muscle fibers reconstituted with cTnT over those fibers reconstituted with fsTnT. The value b in cTnT+cTnI+cTnC reconstituted muscle fiber was only 0.57–0.50 times as fast as that in fsTnT+cTnI+cTnC reconstituted muscle fibers (3.05 vs. 5.32 s−1 at short SL and 4.16 vs. 8.36 s−1 at long SL).
Both the infinite frequency (E∞) and Ca2+-activated steady-state tension (Fss) measure the number of parallel XBs (15). At long SL, E∞ for cTnT+cTnI+cTnC reconstituted fibers was 827 mN/mm2/μm, whereas for fsTnT+cTnI+cTnC fibers, E∞ was 558 mN/mm2 per μm. Thus, the ratio, E∞/Fss, was not different between cTnT and fsTnT reconstituted muscle fibers indicating that fsTnT had no effect on force per XB. The E∞/Fss ratio also remained unaffected at short SL (data not shown). Therefore, the depression in maximal tension and ATPase activity in fsTnT+cTnI+cTnC reconstituted muscle fibers was likely be due to a decrease in the number of XBs. Whether the stabilization of cardiac thin filaments in the submaximally activated state involves altered fsTnT-Tm (25) or fsTnT-TnI interactions remains to be explored. At short SL, the value c of muscle fibers reconstituted with cTnT+cTnI+cTnC was much lower than that for fibers reconstituted with fsTnT+cTnI+cTnC (53.4 vs. 94.9 s−1). However at long SL, c of fibers reconstituted with cTnT+cTnI+cTnC was not different than that for fibers reconstituted with fsTnT+cTnI+cTnC (31.2 vs. 35.5 s−1).
TnT-induced changes in the recruitment-distortion model parameters
Just as b was slower in muscle fibers reconstituted with cTnT+cTnI+cTnC than those reconstituted with fsTnT+cTnI+cTnC, ktr was also slower at both short and long SL (Table 3). Directional similarity in the estimated values of b and ktr suggests that TnT modulates b. At short SL, ktr was faster and b was slower for all three groups of muscle fibers tested in this study. The mechanism of recruiting XBs in ktr experiments differs significantly from those in the dynamic FLR measured using small changes in muscle length. The value ktr is an approximation of a single rate constant for force redevelopment for a given SL under the experimental condition where most XBs have been mechanically broken. On the other hand, b was estimated by fitting small changes in force around a steady-state force with small changes in muscle length. The XB recruitment rate constant embraces the entire myofilament system, which includes the thin-filament overlap, length-dependent XB attachment, and amplification of XB attachment by cooperativity (14). The question of how various XB recruitment mechanisms interact and how they are affected by different SLs remains open. Although the directionality of changes in ktr versus b and c versus tension cost were similar, the magnitude of changes in both ktr and c were higher in fsTnT reconstituted muscle fibers at short SL (Table 3).
DISCUSSION
We have documented a new functional role for cTnT in cardiac thin filaments. Our study demonstrates that cardiac muscle fibers reconstituted with cTnT+cTnI+cTnC differ from those reconstituted with fsTnT+cTnI+cTnC in the magnitude and the speed of dynamic length-mediated recruitment of XBs. There are many mechanisms for recruitment of XBs into the force-bearing state in cardiac muscle. Most obviously, Ca2+ activation is an important XB recruitment mechanism. Just as important is the SL of muscle at any given Ca2+ in recruiting XBs. In this study, we use an incremental measure of length-mediated XB recruitment with our assessment of E0 and b from the dynamic FLR. XB recruitment from the noncycling pool into the cycling pool (force generating XBs) is aided by the transition of RU from the nonpermissive to the permissive states (31). Ca2+ bound to RU is obligatory for transition of RU from the nonpermissive to permissive states. However, Ca2+ binding only initiates the process, and the amount of subsequent nonpermissive to permissive RU transition that occurs depends on both SL and cooperative interactions between XB and RU states. Our data implicates TnT as a participant in the mechanism by which both SL and XB-RU cooperative interactions aid in recruiting cycling XBs during thin-filament activation.
XB recruitment magnitude effects in muscle activation have typically been related to so-called length-dependent activation in cardiac muscle. The differential effect of cTnT on the rate constant of XB recruitment (b) and the magnitude of XB recruitment (E0) suggests that cTnT participates importantly in the mechanism of this fundamental cardiac muscle functional property. Most importantly, cTnT slows b in cardiac muscle fibers. The significance of slowing b may be explained by considering the frequency dependence of cardiac muscle stiffness. Frequency-dependent cardiac muscle stiffness, σ(jω), was calculated from the recruitment-distortion model by Fourier transformation of Eqs. 1–3 to give
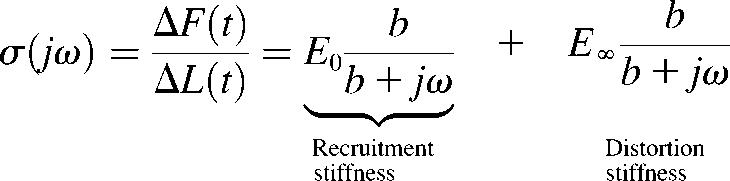 , ,
|
(4) |
where j = √−1, and ω is angular frequency, E0 is the slope of the static FLR, E∞ is instantaneous stiffness, b is the rate constant governing XB recruitment dynamics, and c is the rate constant governing XB distortion dynamics. In this complex stiffness formulation, recruitment dynamics take on a low-pass filter character and distortion dynamics take on a high-pass filter character. When b is much less than c, recruitment and distortion dynamic processes separate such that the recruitment dynamics dominate at low frequencies and the distortion dynamics dominate at high frequencies. At lower frequencies, the speed of muscle length change is not sufficient to cause an appreciable XB distortion because b ≪ c. The transition of dominance from recruitment to distortion may result in stiffness at the frequency of transition that is less than at any other frequency, i.e., a frequency of minimum stiffness, fmin (Fig. 7). Stiffness drops to a minimum at fmin because the speed of muscle-length change outpaces the speed of XB recruitment. Therefore, fmin occurs at lower frequencies when b decreases, as in the case of cTnT reconstituted muscle fibers (Figs. 6 and 7).
FIGURE 7.

Stiffness magnitude spectra of detergent-skinned rat cardiac muscle fiber bundles reconstituted with recombinant rat cTnT and fsTnT. Dynamic FLR was determined at full activation (pCa 4.3) after muscle fiber bundles were reconstituted with recombinant proteins. Dashed line represents muscle fiber bundles reconstituted with cTnT+cTnI+cTnC and solid lines represent muscle fiber bundles reconstituted with fsTnT+cTnI+cTnC. (A) Dynamic stiffness measurements at SL of 1.9 μm. The frequency of minimal stiffness, fmin, is 0.64 Hz for cTnT-cTnI-cTnC reconstituted muscle fibers and 1.16 Hz for fsTnT+cTnI+cTnC reconstituted muscle fibers. (B) Dynamic stiffness measurements at SL of 2.2 μm. The frequency of minimal stiffness, fmin, is 0.79 Hz for cTnT-cTnI-cTnC reconstituted muscle fibers and 1.11 Hz for fsTnT+cTnI+cTnC reconstituted muscle fibers.
When complex stiffness is calculated using the dynamic FLR parameters of the cTnT+cTnI+cTnC reconstituted muscle fibers, fmin is well defined with values of 0.64 and 0.79 Hz at short and long SL, respectively (Fig. 7). The minimum in the stiffness magnitude at fmin is clearly identified, i.e., the minimum stiffness magnitude at fmin is much less than that at zero frequency. However, when b is almost doubled to the value it possessed in the fsTnT+cTnI+cTnC reconstituted fiber, fmin is increased by 80% to 1.16 Hz at short SL and 40% to 1.11 Hz at long SL (Fig. 7). Assuming, as others have (32–34), that fmin and minimum frequency stiffness are important features of heart muscle that tunes the dynamics of muscle contraction to heart rate, our results suggest that cTnT is an important protein in achieving that tuning.
The sense in which this tuning may occur is as follows. When muscle shortens, force decreases according to the speed of shortening and the stiffness of the muscle. If the muscle shortens quickly, as during high frequency length change, the high stiffness of the muscle at these high frequencies (→ E∞) causes muscle force to drop to very low values by the end of the shortening period (ΔF → E∞ΔL). If the muscle shortens slowly, as during low frequency length change, the modest stiffness of the muscle at these low frequencies (→ E0) causes muscle force to drop modestly to low values by the end of the shortening period (ΔF → E0ΔL). However, if the muscle shortens at speeds commensurate with fmin, the minimum stiffness at fmin (→ Emin) causes force to drop by an increment (ΔF → EminΔL) that is less than during rapid shortening or slow shortening. Because the amount of work done by the muscle during shortening is determined by the area under the force-length trajectory, the trajectory in which force is maintained highest during shortening is the one in which most work is performed. This will be the trajectory associated with shortening at speeds commensurate with fmin. Because cTnT participates in determining fmin, it participates in setting the requirements for tuned operation of the cardiac system. cTnT exerts this effect on cardiac thin-filament tuning by its effect on the speed of XB recruitment (b).
SL-dependence of rate of tension redevelopment (ktr) and XB detachment rate constant in cardiac muscle
When maximally activated (pCa 4.3), there was a small but significant increase in ktr at short SL, compared to long SL, in all three groups of muscle fibers tested. At short SL, ktr increased by 38%, 29%, and 34% in control, cTnT+cTnI+cTnC, and fsTnT+cTnT+cTnI reconstituted muscle fibers, respectively (Table 3). A recent study by Adhikari et al. (30) demonstrated a small, but statistically insignificant 14% increase in maximal ktr at short SL in detergent-skinned rat cardiac trabeculae preparations. However, in the study of Adhikari et al., measurements made during submaximal activation at short SL showed significantly decreased force and significantly increased ktr. To account for this decreased force and increased ktr at short SL, Adhikari et al. (30) hypothesized that the XB detachment rate constant, g, increased at short SL due to an increased XB radial strain (35,36). In the two-state model, ktr = fapp + gapp and force is proportional to fapp/(fapp + gapp). Therefore, an increase in g may explain why maximal ktr increases and force drops at short SL in cardiac muscle fibers. Interestingly, our data demonstrate that both the tension cost (which is a measure of g) and the XB detachment rate constant (c, which is proportional to g) increase significantly at short SL (Table 2). Note that the model estimated c agrees well with the directionality of the trend in the experimentally obtained value of the tension cost. Although the tension cost and c increased at short SL in both control untreated as well as reconstituted muscle fiber bundles, an increase in the tension cost and c were more pronounced in fsTnT+cTnI+cTnC reconstituted muscle fibers. The mechanism by which changes in SL and TnT isoforms impact XB recruitment is not well understood. The small increase in ktr as shown in this study and Adhikari et al. (30) contrasts with previous data from rat slow-twitch and rabbit fast-twitch skeletal muscle fiber studies, which demonstrated a small but significant decrease in maximal ktr at short SL (29).
The molecular mechanism by which RU impacts XB detachment rate is unknown. TnT interacts strongly with both TnI and Tm, which bind directly to actin to regulate different XB states. Parts of TnT may also interact directly with actin monomers in a functional unit (37,38). Therefore, TnT has the ability to alter XB cycling either directly or indirectly through its effect on TnI, Tm, and actin (39,40). For example, qualitative changes in cardiac RU have been shown to affect length-dependent activation (slow skeletal TnI in the heart, PKA phosphorylation of cTnI, mutations in cTnT), which suggest that altered protein-protein interactions within the thin filament alter length-dependent activation (11,13,41). For example, we recently showed that the tension cost increased at both short and long SL in a mutant cTnT in which the amino-acid residue 160 was deleted (42). Our data show that cTnT differentially modulates XB recruitment compared to the effect of fsTnT and may tune the heart muscle by decreasing the speed of XB recruitment so that the heart beats at a rate commensurate with the frequency of minimum stiffness (fmin). A link between tuning of cardiac muscle by a thin-filament protein and heart rate has significant implications for cardiomyopathy in humans, where mutations in thin-filament proteins are known to be causal.
Acknowledgments
This work was supported by National Institutes of Health grant No. HL-075643 (to M.C.).
References
- 1.Gordon, A. M., E. Homsher, and A. M. Regnier. 2000. Regulation of contraction in striated muscle. Physiol. Rev. 80:853–924. [DOI] [PubMed] [Google Scholar]
- 2.Potter, J. D., Z. Sheng, B. S. Pan, and J. Zhao. 1995. A direct regulatory role for troponin T and a dual role for troponin C in the Ca2+ regulation of muscle contraction. J. Biol. Chem. 270:2557–2562. [DOI] [PubMed] [Google Scholar]
- 3.Tobacman, L. S., M. Nihli, C. Butters, M. Heller, V. Hatch, R. Craig, W. Lehman, and E. Homsher. 2002. The troponin tail domain promotes a conformational state of the thin filament that suppresses myosin activity. J. Biol. Chem. 277:27636–27642. [DOI] [PubMed] [Google Scholar]
- 4.Maytum, R., M. A. Geeves, and S. S. Lehrer. 2002. A modulatory role for the troponin T tail domain in thin filament regulation. J. Biol. Chem. 277:29774–29780. [DOI] [PubMed] [Google Scholar]
- 5.Perry, S. V. 1998. Troponin T: genetics, properties and function. J. Muscle Res. Cell Motil. 19:575–602. [DOI] [PubMed] [Google Scholar]
- 6.Schaertl, S., S. S. Lehrer, and M. A. Geeves. 1995. Separation and characterization of the two functional regions of troponin involved in muscle thin filament regulation. Biochemistry. 34:15890–15894. [DOI] [PubMed] [Google Scholar]
- 7.Razumova, M. V., A. E. Bukatina, and K. B. Campbell. 2000. Different myofilament nearest-neighbor interactions have distinctive effects on contractile behavior. Biophys. J. 78:3120–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allen, D. G., and J. C. Kentish. 1985. The cellular basis of the length-tension relation in cardiac muscle. J. Mol. Cell. Cardiol. 17:821–840. [DOI] [PubMed] [Google Scholar]
- 9.Irving, T. C., J. P. Konhilas, D. Perry, R. Fischetti, and P. P. de Tombe. 2000. Myofilament lattice spacing as a function of sarcomere length in isolated rat myocardium. Am. J. Physiol. Heart Circ. Physiol. 279:H2568–H2573. [DOI] [PubMed] [Google Scholar]
- 10.Konhilas, J. P., T. C. Irving, and P. P. de Tombe. 2002. Myofilament calcium sensitivity in skinned rat cardiac trabeculae: role of interfilament spacing. Circ. Res. 90:59–65. [DOI] [PubMed] [Google Scholar]
- 11.Konhilas, J. P., T. C. Irving, B. M. Wolska, E. E. Jweied, A. F. Martin, R. J. Solaro, and P. P. de Tombe. 2003. Troponin I in the murine myocardium: influence on length-dependent activation and interfilament spacing. J. Physiol. 547:951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martyn, D. A., B. B. Adhikari, M. Regnier, J. Gu, S. Xu, and L. C. Yu. 2004. Response of equatorial x-ray reflections and stiffness to altered sarcomere length and myofilament lattice spacing in relaxed skinned cardiac muscle. Biophys. J. 86:1002–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandra, M., V. L. Rundell, J. C. Tardiff, L. A. Leinwand, P. P. de Tombe, and R. J. Solaro. 2001. Ca2+ activation of myofilaments from transgenic mouse hearts expressing R92Q mutant cardiac troponin T. Am. J. Physiol. Heart Circ. Physiol. 280:H705–H713. [DOI] [PubMed] [Google Scholar]
- 14.Campbell, K. B., M. Chandra, R. D. Kirkpatrick, B. K. Slinker, and W. C. Hunter. 2004. Interpreting cardiac muscle force-length dynamics using a novel functional model. Am. J. Physiol. Heart Circ. Physiol. 286:H1535–H1545. [DOI] [PubMed] [Google Scholar]
- 15.Campbell, K. B., M. V. Razumova, R. D. Kirkpatrick, and B. K. Slinker. 2001. Myofilament kinetics in isometric twitch dynamics. Ann. Biomed. Eng. 29:384–405. [DOI] [PubMed] [Google Scholar]
- 16.Campbell, K. B., M. V. Razumova, R. D. Kirkpatrick, and B. K. Slinker. 2001. Nonlinear myofilament regulatory processes affect frequency-dependent muscle fiber stiffness. Biophys. J. 81:2278–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo, X., J. Wattanapermpool, K. A. Palmiter, A. M. Murphy, and R. J. Solaro. 1994. Mutagenesis of cardiac troponin I: role of the unique NH2-terminal peptide in myofilament activation. J. Biol. Chem. 269:15210–15216. [PubMed] [Google Scholar]
- 18.Murphy, A. M., L. Jones 2nd, H. F. Sims, and A. W. Strauss. 1991. Molecular cloning of rat cardiac troponin I and analysis of troponin I isoform expression in developing rat heart. Biochemistry. 30:707–712. [DOI] [PubMed] [Google Scholar]
- 19.Pan, B. S., and R. G. Johnson. 1996. Interaction of cardiotonic thiadiazinone derivatives with cardiac troponin C. J. Biol. Chem. 271:817–823. [PubMed] [Google Scholar]
- 20.Chandra, M., J. J. Kim, and R. J. Solaro. 1999. An improved method for exchanging troponin subunits in detergent skinned rat cardiac fiber bundles. Biochem. Biophys. Res. Commun. 263:219–223. [DOI] [PubMed] [Google Scholar]
- 21.Stienen, G. J. M., R. Zaremba, and G. Elzinga. 1995. ATP utilization for calcium uptake and force production in skinned muscle fibers of Xenopus laevis. J. Physiol. 482:109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Tombe, P. P., and G. J. M. Steinen. 1995. Protein kinase A does not alter economy of force maintenance in skinned rat cardiac trabeculae. Circ. Res. 76:734–741. [DOI] [PubMed] [Google Scholar]
- 23.Fabiato, A., and F. Fabiato. 1979. Calculator programs for computing the composition of the solutions containing multiple metals and ligands used for experiments in skinned muscle cells. J. Physiol. (Paris). 75:463–505. [PubMed] [Google Scholar]
- 24.Brenner, B., and E. Eisenberg. 1986. Rate of force generation in muscle: correlation with actomyosin ATPase activity in solution. Proc. Natl. Acad. Sci. USA. 83:3542–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chandra, M., D. E. Montgomery, J. J. Kim, and R. J. Solaro. 1999. The N-terminal region of troponin T is essential for the maximal activation of cardiac myofilaments. J. Mol. Cell. Cardiol. 31:867–880. [DOI] [PubMed] [Google Scholar]
- 26.Montgomery, D. E., J. C. Tardiff, and M. Chandra. 2001. Cardiac troponin T mutations: correlation between the type of mutation and the nature of myofilament dysfunction in transgenic mice. J. Physiol. 536:583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moss, R. L., J. D. Allen, and M. L. Greaser. 1986. Effects of partial extraction of troponin complex upon the tension-pCa relation in rabbit skeletal muscle. Further evidence that tension development involves cooperative effects within the thin filament. J. Gen. Physiol. 87:761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Communal, C., M. Sumandea, P. P. de Tombe, J. Narula, R. J. Solaro, and R. J. Hajjar. 2002. Functional consequences of caspase activation in cardiac myocytes. Proc. Natl. Acad. Sci. USA. 99:6252–6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDonald, K. S., M. R. Wolff, and R. L. Moss. 1997. Sarcomere length dependence of the rate of tension redevelopment and submaximal tension in rat and rabbit skinned skeletal muscle fibers. J. Physiol. 501:607–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adhikari, B. B., M. Regnier, A. J. Rivera, K. L. Kreutziger, and D. A. Martyn. 2004. Cardiac length dependence of force and force redevelopment kinetics with altered cross-bridge cycling. Biophys. J. 87:1784–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Campbell, K. B. 1997. Rate constant of muscle force redevelopment reflects cooperative activation as well as cross-bridge kinetics. Biophys. J. 72:254–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steiger, G. J. 1971. Stretch activation and myogenic oscillation of isolated contractile structures of heart muscle. Pflugers Arch. 330:347–361. [DOI] [PubMed] [Google Scholar]
- 33.Shibata, T., W. C. Hunter, and K. Sagawa. 1987. Dynamic stiffness of barium-contractured cardiac muscles with different speeds of contraction. Circ. Res. 60:770–779. [DOI] [PubMed] [Google Scholar]
- 34.Kawai, M., Y. Saeki, and Y. Zhao. 1993. Cross-bridge scheme and the kinetic constants of elementary steps deduced from chemically skinned papillary and trabecular muscles of the ferret. Circ. Res. 73:35–50. [DOI] [PubMed] [Google Scholar]
- 35.Brenner, B., and L. C. Yu. 1985. Equatorial x-ray diffraction from single skinned rabbit psoas fibers at various degrees of activation. Changes in intensities and lattice spacing. Biophys. J. 48:829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goldman, Y. E., and R. M. Simmons. 1986. The stiffness of frog skinned muscle fibers at altered lateral filament spacing. J. Physiol. 378:175–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heeley, D. H., and L. B. Smillie. 1988. Interaction of rabbit skeletal muscle troponin T and F-actin at physiological ionic strength. Biochemistry. 27:8227–8232. [DOI] [PubMed] [Google Scholar]
- 38.Dahiya, R., C. A. Butters, and L. S. Tobacman. 1994. Equilibrium linkage analysis of cardiac thin filament assembly. Implications for the regulation of muscle contraction. J. Biol. Chem. 269:29457–29461. [PubMed] [Google Scholar]
- 39.Greene, L. E., and E. Eisenberg. 1980. Cooperative binding of myosin subfragment-1 to the actin-troponin-tropomyosin complex. Proc. Natl. Acad. Sci. USA. 77:2616–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cassell, M., and L. S. Tobacman. 1996. Opposite effects of myosin subfragment 1 on binding of cardiac troponin and tropomyosin to the thin filament. J. Biol. Chem. 271:12867–12872. [DOI] [PubMed] [Google Scholar]
- 41.Arteaga, G. M., K. A. Palmiter, J. M. Leiden, and R. J. Solaro. 2000. Attenuation of length dependence of calcium activation in myofilaments of transgenic mouse hearts expressing slow skeletal troponin I. J. Physiol. 526:541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chandra, M., M. L. Tschirgi, and J. C. Tardiff. 2005. Increase in tension-dependent ATP consumption induced by cardiac troponin T mutation. Am. J. Physiol. Heart Circ. Physiol. 289:H2112–H2119. [DOI] [PubMed] [Google Scholar]