

Abstract
This study shows significant effects of protein surface charges on stability and these effects are not eliminated by salt screening. The stability for a variant of protein G B1 domain was studied in the pH-range of 1.5–11 at low, 0.15 M, and 2 M salt. The variant has three mutations, T2Q, N8D, and N37D, to guarantee an intact covalent chain at all pH values. The stability of the protein shows distinct pH dependence with the highest stability close to the isoelectric point. The stability is pH-dependent at all three NaCl concentrations, indicating that interactions involving charged residues are important at all three conditions. We find that 2 M salt stabilizes the protein at low pH (protein net charge is +6 and total number of charges is 6) but not at high pH (net charge is ≤−6 and total number of charges is ≥18). Furthermore, 0.15 M salt slightly decreases the stability of the protein over the pH range. The results show that a net charge of the protein is destabilizing and indicate that proteins contain charges for reasons other than improved stability. Salt seems to reduce the electrostatic contributions to stability under conditions with few total charges, but cannot eliminate electrostatic effects in highly charged systems.
INTRODUCTION
Noncovalent interactions govern the folding of proteins to their native structures. These interactions are of different character and the relative contributions from hydrogen-bonding, van der Waal's, hydrophobic, and electrostatic interactions to the stability of proteins are not resolved. In particular, the importance of electrostatic interactions seems to be a disputable matter. Ionic interactions may affect the protein stability through repulsion of charges of equal sign and attraction of charges of opposite charge, and the net contribution of these interactions in the folded state, relative to unfolded state, governs the stability. In some cases, the stability of proteins has been altered by changing charges on the surface (1,2), whereas, in other cases, surface charges have been removed without affecting the overall stability (3). The stabilizing effects of charge modifications seem to be highly context-dependent (4,5). The seemingly contradictory results may reflect that individual interactions are nonadditive (5) and that highly charged systems behave as saturated with charge (6). Thermophilic proteins often have a higher content of charged residues compared to their mesophilic counterparts (7,8) and it has been claimed that specific ionic interactions stabilize these proteins (9–13). Halophilic proteins are coated with acidic residues that give the protein a high net negative charge. These proteins require a high salt concentration and unfold at low salt concentrations (14–16). This indicates that a high negative charge is destabilizing for the protein but the charges can be shielded by counterions.
A high fraction of charged residues is favorable for proteins in terms of higher solubility, prevention of aggregation, and often has functional importance. For example, charged residues may set up the electrostatic potential for efficient channeling of substrate to the active site through long-range attractive forces (17,18), and is often important in catalysis (19). Furthermore, correct folding of the protein has been shown to be optimized by a high net charge via electrostatic repulsion in unwanted conformations (20). However, a recent study shows that bovine carbonic anhydrase still folds properly and is active despite 18 surface-charge modifications (21). Generally, though, repulsion among charged groups on the surface may lead to decreased stability. At high salt concentration, the contributions from surface charges are reduced and the protein can have more charged residues and still remain stable. In contrast, the dielectric constant for water decreases with increasing temperature, giving higher impact to electrostatic interactions, in regard to both attractive and repulsive forces. Common to both extreme conditions, high salt and high temperature is an increased hydrophobic effect that may enhance the overall stability.
Here we present an investigation of the role of electrostatic interactions in protein stability using a variant of the 56-amino-acid immunoglobulin G (IgG)-binding domain of Streptococcal protein-G (PGB1). Protein surface charges are modulated by pH variations, but the addition of salt may attenuate the effects on protein stability. We have measured protein stability as a function of pH at low, physiological, and high salt concentrations. PGB1 is an excellent model protein. It is highly stable and soluble, it has both α-helical and β-sheet character, it does not contain cysteines, its three-dimensional structure is known (22–24), it has a Trp residue that is buried in the folded state, and its thermodynamic properties are characterized (25). However, wild type (wt) PGB1 contains two classical Asn-Gly deamidation sites that can be removed by mutagenesis. Our PGB1 variant was designed not to deamidate under conditions of elevated pH and temperature, and has the mutations T2Q, N8D, and N37D. With these mutations the protein contains 12 negative and six positive charges at neutral pH (excluded the N- and C-terminals). The T2Q mutation is introduced to prevent processing of the N-terminal methionine (26). Hereafter, our variant is referred to as PGB1-QDD. Nuclear magnetic resonance (NMR) and surface plasmon resonance (SPR) studies show that the structure and function of the wt protein are essentially retained. The temperature denaturation of PGB1-QDD was investigated at three different salt concentrations and a large range of pH values using circular dichroism (CD) spectroscopy and differential scanning calorimetry (DSC). Urea denaturations were monitored by CD and fluorescence spectroscopy. Our findings suggest that the protein is most stable close to its isoelectric point (pI), and that 2 M NaCl does not eliminate the pH dependence of the stability; and also suggest that even at high salt, the pKa values may differ between folded and unfolded proteins.
MATERIALS AND METHODS
Cloning and expression of PGB1-QDD
PCR with overlapping oligonucleotides was used to produce a synthetic full-length gene for PGB1 (56 amino acids) with the following three substitutions: Thr-2→Gln, Asn-8→Asp, and Asn-37→Asp (PGB1-QDD), with NdeI and SacI restriction sites. The PCR band was digested with NdeI and SacI and cloned into a modified Pet3a vector with NdeI and SacI cloning sites. The ligation product was transformed into Escherichia coli ER2566 and plasmids were prepared from single colonies. A plasmid preparation with verified DNA sequence was transformed into E. coli BL21 DES3 PLysS Star. Single colonies were used to inoculate overnight cultures of LB medium with 50 μg/ml ampicillin and 30 μg/ml chloramphenicol. The overnight cultures were diluted 1:100 in the day cultures of 500 mL each in 2.5 L baffled flasks. Protein production was induced by adding 0.4 mM isopropyl-β-D-1-thiogalactosid at an OD600 of 0.6–0.8, and the culture was harvested by centrifugation 3–4 h later. PGB1-QDD labeled with 13C and 15N was produced as above, but the day culture was grown in minimal medium containing 13.7 mM 15NH4Cl, 12.5 mM 13C-glucose, 0.042 M Na2HPO4, 0.22 M KH2PO4, 0.00855 M NaCl, 1 mM MgSO4, 0.1 mM CaCl2, 1 μg/ml Vitamin B1, 18 μM FeCl3, 50 μg/ml ampicillin, and 30 μg/ml chloramphenicol.
Purification of PGB1-QDD
The cell pellet was resuspended in H2O (120 ml to pellet from a 5.4-liter culture), poured into 150 mL boiling buffer A (10 mM Tris/HCl and 1 mM ethylenedinitrilotetraacetic acid (EDTA), pH 7.5), heated to 80°C and then directly cooled on ice. The solution was centrifuged at 15,000 g for 10 min, and the supernatant was rocked on ice with 80 ml DEAE cellulose. The cellulose was packed in a column, washed with buffer A, and eluted using a linear NaCl gradient from 0 to 400 mM in buffer A. Fractions containing PGB1-QDD were pooled and lyophilized, dissolved in 20–25 mL Millipore water (Millipore, Billerica, MA), and separated on a 3.4 × 180 cm Sephadex (Sigma-Aldrich, St. Louis, MO) G50 superfine gel filtration column using 50 mM ammonium acetate, pH 6.5, as running buffer. Fractions containing PGB1-QDD were pooled, lyophilized, and desalted on a Sephadex G25 superfine gel filtration column in water. Typical yield of pure protein was 100–150 mg/liter of culture in both rich and minimal medium.
Nuclear magnetic resonance spectroscopy
The two-dimensional 15N-1H heteronuclear single quantum correlation (HSQC) and two-dimensional 13C-1H HSQC spectra were collected on a 500 MHz Varian UNITY PLUS spectrometer (Varian, Cary, NC) to determine the chemical shifts of 1H-, 15N-, and 13C-resonances of α-carbons. One-thousand-twenty-four points were collected in ω2 and 128 points in ω1, at 25°C for 1.5 mM 13C- and 15N-labeled PGB1-QDD in 93% H2O, 7% D2O, and 10 mM NaN3, pH 5.0. The number of transients was four in both experiments, and 10 mM 2,2-Dimethyl-2-silapentane-5-sulfonate sodium salt (DSS) was used as a reference for the 1H shifts and 13C and 15N were indirectly referenced from the 1H shifts. The NMRPIPE program suite (27) was used to process NMR data with zero-filling to generate data sets with dimensions 256 × 1024 points in ω1 and ω2. The two-dimensional 13C-1H HSQC spectrum was used to determine both α- and β-structural elements. The shifts from the two-dimensional 15N-1H HSQC spectrum were compared to the shifts of wt PGB1 (23).
Thermal denaturation by differential scanning calorimetry
Differential scanning calorimetry (DSC) measurements were performed on a VP-DSC Microcalorimeter (Microcal, Northampton, MA). The temperature was increased from 5°C to 110°C, then decreased to 5°C. Another upward and downward scan was done to see if the denaturation was reversible. The scan rate was 60°C/h. The protein (0.3–0.6 mM) was dissolved in 5 mM phosphate pH 7.5 or in 5 mM sodium acetate pH 4.5. At each pH the protein was studied at three different NaCl concentrations; 0, 0.15, and 2 M, with two replicates for each condition. The protein sample was dialyzed against the same buffer to minimize the difference between the reference buffer and the sample solvent. Protein concentration was determined individually for each condition by amino acid analysis after acid hydrolysis (purchased from the Center for Amino Acid Analysis, Uppsala University, Uppsala, Sweden).
Thermal denaturation by circular dichroism spectroscopy
The circular dichroism (CD) signal at 218 nm was monitored during thermal unfolding of PGB1-QDD from 4°C to 92°C at a scan rate of 1°C/min and response 16 s. The protein (50–60 μM) was dissolved in buffer containing 2 mM Tris, 2 mM N-cyclohexyl-3-aminopropanesulfonic acid (CAPS), 2 mM 2-morpholinoethanesulfonic acid monohydrate, and 0, 0.150, or 2 M NaCl. The pH value was adjusted to a set of values ranging from 1.5 to 11.5. Quartz cuvettes with 1.0 or 0.2 mm pathlength were used. Before the temperature scan, a far-UV spectrum (250–195 nm, scan rate 10 nm/min, response 16 s) was recorded at 25°C for each sample. All CD measurements were performed using a JASCO (Tokyo, Japan) J-720 spectropolarimeter with a JASCO PTC-343 Peltier type thermostated cell holder.
Urea denaturation monitored by fluorescence and CD spectroscopy
A set of protein samples with urea concentrations ranging from 0 to 10.0 M were prepared by mixing appropriate amounts of protein stock and two solutions containing either 0 or 10 M urea in 2 mM Tris, 2 mM CAPS, and 2 mM NaH2PO4 (pH 2.50, 4.50, 7.50, and 10.0 adjusted with HCl or NaOH) and 0 M, 0.15 M, or 2 M NaCl. The protein concentration was ∼20 μM. CD and fluorescence signals were recorded for each sample at 25°C. The CD signal at 218 nm was obtained using a JASCO J-720 spectropolarimeter and a quartz cuvette with 1 or 2 mm pathlength (resolution 1 nm, band width 1 nm, response time 16 s, accumulation 4, and scan rate 10 nm per min). Fluorescence emission at 338 and 348 nm was recorded using a Perkin Elmer (Boston, MA) LS-50B spectrometer with 3 × 3 mm quartz cuvette (excitation at 295 nm, excitation and emission slits 3 nm, scan rate 20 nm/min, accumulation 4).
Isoelectric focusing
Isoelectric focusing, PhastGel IEF pH 4–6.5 (Amersham Biosciences, Piscataway, NJ), was used to determine the pI. The gel contains 22 μmol (ml × pH unit) of Pharmalyte (Amersham Biosciences) and has a linear pH gradient.
Surface plasmon resonance
The affinity of PGB1-QDD for human IgG was investigated by surface plasmon resonance (SPR) using the Biacore 3000 apparatus (Biacore, Uppsala, Sweden). IgG (Sigma-Aldrich) was immobilized to the carboxymethylated dextran matrix on the sensor chip via amine coupling. IgG was dissolved in sodium acetate buffer pH 5.0. Equal volumes of 0.1 M N-hydroxysuccinimide and 0.4 M N-ethyl-N′-(dimethylaminopropyl)carbodiimide were first mixed and 70 μl of the mixture was allowed to flow over the chip to activate the surface for 8 min (flow rate = 10 μl/min). IgG coupling was performed at three different concentrations, i.e., 50, 60, and 75 μg/ml, in three flow channels while one channel was kept as a reference. Unreacted N-hydroxysuccinimide ester groups were deactivated by 70 μl of 1.0 M ethanolamine hydrochloride (pH 8.5). The quantity of 50 μl glycine, pH 2.7, was flown over the chip to remove noncovalently-bound material. The association and dissociation of PGB1-QDD to IgG was studied at various concentrations ranging from 4 nM to 60 nM in10 mM phosphate, 0.15 M NaCl, and 0.005% TWEEN 20, at pH 7.5, 6.0, 4.5, and 3.0 (buffer sterile-filtered through 0.22 μm filter and degassed). Three replicates were performed for each condition and each replicate involved kinetic measurements on all the three channels with different IgG concentrations.
DATA ANALYSIS
Least-squares data analysis and error analysis
All least-square analyses were carried out with the Levenberg-Marquardt minimization algorithm using MatLab 6.5 (The MathWorks, Natick, MA). An error analysis of the curve fits was done using the Monte Carlo method. Simulated data sets were generated using the Bootstrap method (28), where data points were drawn randomly from the pool of data points with equal probability and allowing duplicates to simulate errors within each measurement. The Bootstrap method was also employed to draw complete curves to simulate errors between the different curves for the global parameters. Errors in protein concentrations contribute significantly to the errors and were given random values from a Gaussian distribution with a standard deviation estimated to 10% in the Monte Carlo analysis. Multimethod global analysis (29) was carried out for denaturation data to obtain a global ΔCp value.
Denaturation
Assuming a two-state unfolding, N ⇄ U, the fractions native (FN) and unfolded (FU) protein are
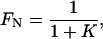 |
(1) |
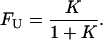 |
(2) |
The relation between equilibrium constant and the standard free energy change ΔG° is
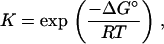 |
(3) |
and the Gibbs-Helmholtz equation describes the temperature-dependence of ΔG°,
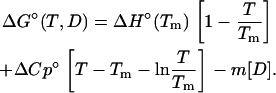 |
(4) |
R is the molar gas constant, T is the temperature, and [D] is the denaturant concentration. ΔG° is zero at the transition midpoint, Tm, or at the urea concentration at the transition midpoint, Cm. Cm = ΔG°/m.
For denaturation with fluorescence, CD, or DSC, the baselines before (YN) and after (YU) the actual unfolding were assumed to be straight lines,
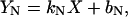 |
(5) |
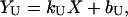 |
(6) |
where kN and kU are the slopes, bN and bU are intercepts, and X is the temperature in Kelvin or urea concentration in M. The value Y is the measured signal ellipticity for CD, and Cp for DSC.
For thermal or urea denaturation measured with CD or fluorescence,
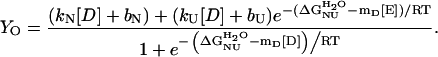 |
(7) |
For the DSC denaturation data,
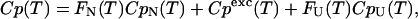 |
(8) |
and the excess heat capacity, 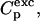 going from folded to unfolded state is
going from folded to unfolded state is
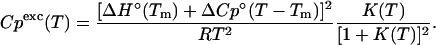 |
(9) |
From the heat capacity change, the enthalpy can be derived as
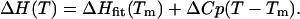 |
(10) |
Surface plasmon resonance
Dissociation was assumed to follow the first-order reaction
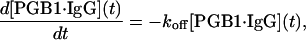 |
(11) |
where koff is the dissociation rate constant. By integrating Eq. 11 the decrease in complex concentration as a function of time can be described as
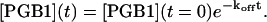 |
(12) |
If assuming that the instrumental response R(t) is linear with complex concentration, Eq. 12 can be used to fit dissociation data as
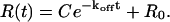 |
(13) |
R0 is the baseline value that is approached as t goes to infinity and C is R(0) − R0.
The association phase of the SPR measurement is affected both by the association and dissociation rate constants and follows a more complex kinetics,
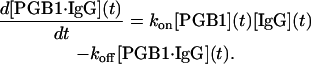 |
(14) |
The value kon is the association rate constant. Since there is a constant flow of PGB1-QDD over the cell the consumption of the substrate is assumed to be negligible resulting in a constant [PGB1]. The total concentration of IgG is the sum of the uncomplexed IgG on the chip and the IgG in complex with PGB1-QDD. Using the fact that the complex concentration at time zero = 0 and a linear instrument response the solution to Eq. 14 becomes
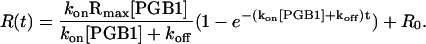 |
(15) |
Equation 15 is fitted to association data where Rmax is the instrument response if all the immobilized IgG-molecules were in complex with PGB1. The koff and kon values were globally fitted to Eqs. 13 and 15 from 60 measurements for each pH, with different concentrations of PGB1-QDD flown over the channels.
The equilibrium constant for the complex formation is defined as
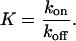 |
(16) |
RESULTS
Nuclear magnetic resonance
The 15N-HSQC spectrum (Fig. 1 a) shows well-resolved 1H-15N cross-peaks for the backbone of PGB1-QDD. All backbone residues are assigned (unpublished data), except for Glu-27, which did not give rise to a peak. The Hα chemical shifts are sensitive to secondary structure and often, α-helical protons are found at lower and β-strand protons at higher chemical shifts than the water resonance. From the 13C-HSQC spectrum (Fig. 1 b), it is therefore clear that PGB1-QDD contains both α- and β-structures consistent with the fold of the wt PGB1 (23). The chemical-shift differences between wt PGB1 and PGB1-QDD are displayed in Fig. 2. Overall, the chemical shift differences are small, except for resonances that are close in space to the sites of mutation. This indicates that PGB1-QDD is structurally similar to wt PGB1 (23).
FIGURE 1.

(a) Two-dimensional 15N-1H HSQC with assignments. (b) Hα-Cα region of the two-dimensional 13C-1H HSQC spectrum. Hα shifts are found both to the left and right of the water resonance consistent with both α-helical and β-sheet structure. Glycines show negative peaks and are observed at a lower 13C-chemical shift.
FIGURE 2.

Backbone chemical shift differences between wt PGB1 (23) and PGB1-QDD. (a) The protein is depicted with gray shading; darker tones represent a larger shift difference. The mutated residues are also marked in the structure. The figure was generated using the PDB accession code 1PGB (22) and the program MOLMOL (47). (b) Chemical shift differences calculated as Δδ(1H, 15N) = |Δδ1H| + 0.2|Δδ15N|. The highest shift differences are observed for by A23 and T25, followed by the mutated residues.
Thermal denaturation
CD and DSC were used to monitor the protein folding equilibrium as a function of temperature at pH values ranging from 1.5 to 11 at three different salt concentrations. Solutions with no-added, 0.15, and 2 M NaCl are referred to as low, physiological, and high salt, respectively. The normalized data with fitted curves are presented in Fig. 3 a. The reproducibility within CD and DSC data and the agreement between the two methods is good, and therefore both data sets are fitted using a global ΔCp. The Tm values obtained from the fits are displayed in Fig. 4 a. At low pH, the temperature at the transition midpoint, Tm, has increased with the addition of 2 M salt, an effect that cannot be seen with 0.150 M salt (Fig. 4 a). Hence, a high concentration of salt is required to shield repulsive ionic interactions and to stabilize the positively charged protein at low pH. Around pH 4.5, close to the pI of the protein, there is no stability enhancement with the addition of salt. Rather, under these conditions of no or low net charge, the protein appears to be most stable at low salt. At low and physiological salt, the protein is most stable at a pH close to pI, while at high salt the stability is less sensitive to pH around this value. Around pH 7.5, where the protein is negatively charged, there is no clear difference in Tm between the low and high salt concentrations; whereas, the protein at physiological salt is somewhat destabilized compared to low and high salt conditions (Fig. 4). Noteworthy is that, at high pH where the protein carries significant negative charge, there is no difference in Tm between high and low salt concentrations but the physiological salt concentration yields lower Tm.
FIGURE 3.

The pH-dependent stability. Experimental data points (⋄) and fitted models (solid lines) are color-coded according to pH as indicated by the scale to the right. For the DSC data the fraction unfolded, FU, is also displayed as a solid line. (a) Temperature denaturations of PGB1-QDD studied with CD (top two rows) and DSC (third row) at different pH values and three NaCl concentrations: (left) low salt; (middle) 0.15 M; and (right) 2 M. The top row shows temperature-denaturation curves at low pH values up to the pH with maximum stability at the respective salt concentration. The middle row shows data at high pH values down to the pH with maximum stability. (b) Urea denaturations studied with CD and fluorescence at different pH and the same three NaCl concentrations.
FIGURE 4.

(a) Temperature at the denaturation midpoint, Tm, as a function of pH for the three different NaCl concentrations. Symbol key: □, low salt; ⋄, 0.15 M NaCl; and ▵, 2M NaCl. Open symbols are for CD data and solid symbols are for DSC data. (b) Urea concentration at the denaturation midpoint, Cm, as a function of pH for the three different NaCl concentrations. Symbol key: □, low salt; ⋄, 0.15 M NaCl; and ▵, 2M NaCl.
As can be seen from the CD data, the stability toward thermal denaturation is clearly pH-dependent at all three salt concentrations (Fig. 3 a). The pH variation in thermal stability is smallest at high salt concentration (Fig. 4 a), and this is particularly noticeable at low pH. The addition of 0.15 M salt yields marginal effects on the pH dependence, but the physiological salt curve is shifted to somewhat lower Tm over the whole pH range.
As for DSC data at pH 4.5, the protein shows highest stability at low salt concentration followed by high salt concentration, whereas at physiological salt concentration it shows the lowest stability. These observations are in agreement with the data from CD spectroscopy. At pH 7.5 there is no difference between low and physiological salt concentrations but high salt yields a clear increase in the stability of the protein.
The temperature at the transition midpoint, Tm, measured by DSC and CD are in good agreement, suggesting that the protein undergoes a cooperative unfolding event involving both tertiary and secondary structure. The unfolding measured by both CD and DSC is reversible as long as the temperature does not exceed 95°C. For DSC scans above this temperature, the reversibility is lost. The reversibility has been shown previously for wt PGB1 (25). The value Tm, measured by DSC, generally lies slightly lower than the value measured by CD. However, CD could, in this respect, be more reliable due to the much lower protein concentration. High protein concentrations could give false Tm due to interactions between protein molecules or due to screening effects from counterions of the protein itself (30).
Urea denaturation
PGB1-QDD shows pH- and salt-dependent stability toward urea denaturation. We have analyzed the stability toward urea denaturation at three different salt concentrations (low, 0.15 M, and 2 M NaCl) and four different pH values (2.5, 4.5, 7.5, and 10.0). The normalized data sets with the normalized fitted curves are shown in Fig. 3 b. In all cases, the CD signal at 218 nm and the tryptophan fluorescence at 343 nm show a transition toward lower intensity with increasing urea concentration. Exactly the same samples were analyzed both by CD and fluorescence. When fitted separately, the curves obtained by fluorescence and CD yield the same Cm values, indicating a two-state cooperative unfolding involving both tertiary and secondary structure. Therefore, the denaturation curves by the two methods were analyzed globally. At low salt the protein is most stable at pH 4.5, followed by pH 7.5 and pH 2.5, while at pH 10 the protein is least stable. In the presence of high salt, the protein is slightly more stable at pH 2.5 than at pH 7.5. At pH 2.5 and 4.5, the Cm values are higher at 2 M NaCl than at low salt. The difference between the two salt concentrations is small at pH 7.5 and 10. At high pH, temperature denaturation yields a significantly lower Tm at physiological compared to the other salt concentrations. Therefore, we added a urea denaturation experiment at this condition, and again found a lower stability compared to both low and 2 M salt.
Free energy of unfolding (ΔG°)
The free energy difference between unfolded and native state, ΔG°, at 25°C was extrapolated from high temperature for thermal denaturation and from high urea concentration for the solvent denaturations (Eq. 4, Fig. 5 a). Based on the parameter values from fitting of thermal denaturations at pH 4.5 and 7.5, curves for ΔG° were calculated as a function of temperature (Fig. 5, b and c). The values at 25°C as extrapolated from the urea denaturations are included for comparison.
FIGURE 5.

(a) ΔG° at 25°C as a function of pH. Temperature denaturations are extrapolated from Tm to 25°C and urea denaturations are extrapolated from Cm to 0 M urea (Eq. 4). Symbol key: □, low salt; ⋄, 0.15 M NaCl; and ▵, 2M NaCl. Open symbols are for temperature denaturations; solid symbols are for urea denaturations. (b) ΔG° at pH 4.5 (solid lines) and 7.5 (dashed lines), and low salt as a function of temperature. (c) ΔG° at pH 4.5 (solid lines) and 7.5 (dashed lines), and 2 M salt as a function of temperature. In b and c, urea denaturation data points (▪ in b and ▴ in c) are added for comparison.
Isoelectric point (pI)
The pI of PGB1-QDD was determined by isoelectric focusing at low salt concentration and found to be 4.3 ± 0.1 (data not shown). The pI, based on model pKa values for individual amino acids (31), is 4.2. This corresponds to a protein where all electrostatic interactions are shielded. Hence, there is no difference in pI between the model and experiment. Wt PGB1 is reported to have a pI of 4.0 determined by isoelectric focusing (25). In this case, the calculated pI is 4.4 based on model pKa values and 4.2 based on measured pKa values (32). We find that pI is, within error, the same for wt and PGB1-QDD despite the introduction of two negative charges.
Surface plasmon resonance
The IgG binding of PGB1-QDD was measured using SPR technology. The affinity was determined at three different pH values; 4.5, 6.0, and 7.5 at 0.15 M NaCl as summarized in Table 1. Examples of data are shown in Fig. 6. The rate constants for association and dissociation were obtained by global fitting to all data at each pH. The association rate constant was found to be largely pH-independent in the range studied. However, the dissociation rate constant at pH 7.5 is one order-of-magnitude higher compared to the other two pH values. The affinity we obtain of PGB1-QDD for IgG at pH 7.5 is only half an order-of-magnitude lower compared to reported values for whole protein G (33), and at lower pH the affinities are comparable. Hence, the affinity differences between the pH values are larger than between our PGB1 fragment and full length Protein G. At pH 7.5, our protein carries two extra negative charges compared to the wild type, which may affect the kinetic rate constants through electrostatic repulsion. The dissociation data at pH 4.5 are generally of much lower quality compared to the other pH values, perhaps due to aggregation problems, instability of IgG or instrument limitations. No binding was observed at pH 3.0.
TABLE 1.
Association, dissociation, and equilibrium constants for the binding of PGB1-QDD to IgG at different values of pH
| pH | 10log kon (s−1) | 10log koff (s−1) | 10log K |
|---|---|---|---|
| 4.5 | 5.31 ± 0.02 | −3.23 ± 0.02 | 8.54 |
| 6.0 | 5.32 ± 0.01 | −3.04 ± 0.02 | 8.36 |
| 7.5 | 5.40 ± 0.01 | −2.34 ± 0.01 | 7.74 |
FIGURE 6.

(a) Association of PGB1-QDD to IgG at pH 6.0 for two different concentrations of PGB1-QDD (16 nM, ○, and 60 nM, ♦) with the fitted curves using global kon and koff as solid lines. Only data every 10 s is shown. (b) Association of PGB1-QDD to IgG at pH 7.5 for two different concentrations of PGB1-QDD (16 nM □, and 60 nM ▾) with the fitted curves using global kon and koff as solid lines. Only data every 10 s are shown. (c) Dissociation of PGB1-QDD from IgG at pH 6.0 and 7.5 for two different concentrations of PGB1-QDD, with the same symbols as for association and the fitted curves using global koff as solid lines. Only data every 50 s are shown.
DISCUSSION
The results of this study indicate that a high concentration of salt does not eliminate the influence of surface charges of a protein. In 2 M salt there is still a strong pH dependence of the stability of PGB1-QDD. Based on fitted parameters from thermal denaturation, we have generated contour plots (Fig. 7) to illustrate the pH- and temperature-dependence of ΔG° for the protein denaturation at three salt concentrations. In these plots, the ΔG° = 0 contour line corresponds to a situation where native and denatured states are equally populated. The pH dependences of ΔG° are comparable between low and physiological salt. Although the curve is somewhat flatter at 2 M salt, the effects are surprising small. In a situation with complete shielding of surface charges, horizontal ΔG° lines would appear. Based on thermodynamic arguments, a pH-dependence of protein stability reflects differences in pKa-values of ionizable groups between folded and unfolded proteins. Our findings hence suggest that such a difference remains at high salt especially for carboxyl groups.
FIGURE 7.

ΔG° contour maps calculated using fitted parameters from temperature denaturations at (a) low, (b) physiological, and (c) high salt concentrations.
It is apparent from our data that the salt effects are quite different for positively (low pH) and negatively (high pH) charged protein (Figs. 4, 5, and 7). At low pH, high salt concentration stabilizes the protein significantly, but no effect is seen at physiological salt. Hence a high concentration of salt is required to shield repulsive ionic interactions when the protein net charge is +6. At an approximate pH of 7.5, where the protein has a net charge of −6, there is no clear difference in Tm between the low and high salt concentrations, while the protein at physiological salt is somewhat destabilized compared to low and high salt conditions (Fig. 4). At pH 7.5 the protein has the same absolute net charge but opposite sign as compared to pH 2.5, but there are more total charges at pH 7.5 (18 compared to 6 at pH 2.5) and hence the charges are closer in space. This might be the reason why salt is unable to stabilize the protein at pH 7.5. It has been shown that the interactions between charged groups that are very close to each other in the folded protein such as ion pairs are insensitive to salt (34,35). Moreover, Dominy et al. (36) saw that thermophilic and hyperthermophilic proteins with mostly favorable electrostatic interactions on their surface, were destabilized by the addition of salt (36). At an approximate pH of 10, Tm is markedly reduced at all salt concentrations. Here, the protein has a net charge ∼−9. At high pH there is no difference in Tm between high and low salt concentrations, but the physiological salt concentration yields lower Tm, indicating an uneven shielding of charges in the folded and unfolded states. There is also a possibility that our results reflect salt effects on other types of interactions, e.g., hydrophobic effects, and/or that short- and long-range electrostatic effects are opposing and screened to different extents. There are studies showing that long-range interactions are shielded at a low ionic strength, while short-range interactions persist at a high salt concentration (34,36–38). If electrostatic repulsion was the driving force for protein unfolding at high pH, one could expect the addition of 2 M salt to shield at least long-range repulsions and contribute to increased stability of the protein. On the other hand, it has been shown that highly charged systems do not always behave as expected from an electrostatic point of view (6), and similar effects could be responsible for our unexpected behaviors at high pH.
To compare urea and thermal denaturation, and the salt effects at different pH values, ΔG° was calculated from the fitted parameters (Fig. 5). The ΔG° from urea denaturation is extrapolated from high to low urea concentration, and ΔG° from thermal denaturation is extrapolated from high to low temperature. Despite this, the values from the two methods fall into the same range. This gives an indication that the extrapolations are valuable and are useful when comparing different conditions. Both Cm and Tm are increased by the addition of 2 M salt at low pH, while this is not the case at high pH (Fig. 4). At pH 4.5 where the protein shows the highest stability, salt screening changes Cm and Tm in opposite directions. This may be due to differences in solution conditions, high temperature, and no denaturant versus room temperature and high urea concentration. The intermediate salt concentration seems to destabilize the protein at high pH in both methods. These curves show that ΔG° does not vary much over the temperature range. Previously it was reported that PGB1 has a low ΔCp that makes the protein stable over a wide range of temperatures (25). There is no distinct difference for ΔG° between the different salt concentrations.
According to the Höfmeister series, the salt, sodium chloride, used in this study is neither a strong salting-in nor a strong salting-out agent, and therefore the nature of the electrolyte, as such, should not contribute to the stability (39). Studies have shown that denaturing salts such as guanidinium chloride more efficiently shield electrostatic interactions in a protein than sodium chloride (40,41). However, salts in general are thought to increase the hydrophobic interaction and this might be one of the reasons why the stability is enhanced by the addition of salt at low pH. One explanation why such an effect is not seen at the high pH is due to the high net charge (−9) and the associated charge repulsion. Short-range repulsion is increased at high pH compared to low pH, and may account for the inability of salt to shield the charges. The increased hydrophobic interactions are not enough to overcome the large unfavorable electrostatic repulsions. The different stability effects at low and high pH with added salt could be because the Na+ and Cl− ions are not equally polarizable. The chloride ion is more polarizable, and might therefore more effectively shield charges than the sodium ion.
An alternative possibility is that all charges might not be completely surface-exposed and that the pH-dependent stability remaining in high salt might derive from desolvation penalties (42,43). Lysines are extended residues and likely to be in contact with the solvent, accounting for complete shielding at low pH. Carboxyl residues, on the other hand, are not as extended, and there may be structural effects on desolvation of charges giving reduced stability of the protein at high pH.
Around the isoelectric point of the protein, a minimum of net electrostatic contributions to protein stability are expected. Our results show that PGB1-QDD has maximum stability around the isoelectric point which is slightly reduced by salt, in agreement with the previous study by Dominy et al. (36). This implies that a net charge of the protein is destabilizing. Many proteins are negatively charged at neutral pH and have a pI around 4, where they also show the highest stability. However, most proteins function under physiological conditions, and at this pH and salt concentration, at least PGB1-QDD is largely destabilized. Hence, stability and function do not correlate. Why are proteins not created with a pI closer to the pH where they function? Stability may disfavor functionality and a too stable protein does not function as well in the cell. Many protein functions like catalysis and binding are dependent on protein charges. Another reason could be that the protein has a higher tendency to aggregate and form amyloid (44) around its pI. Avoidance of aggregation is especially critical in a cellular environment with high protein concentration; this may be the reason why most proteins are negatively charged so they repel each other and DNA. A sacrifice in stability can be compensated for by a decrease in aggregation. Moreover, the proteins in vivo must be accessible for proteases to allow for spatial and temporal control of cellular activities.
The stability of the protein cannot only be described by the net charge of the protein even though the protein shows its highest stability close to the pI. As mentioned in the Introduction, halophilic proteins have a higher content of negatively charged residues and unfold in the presence of low salt. In this study we show that such negative charges cannot be randomly introduced to create a halophilic protein. At high pH, PGB1-QDD is a highly negatively charged system that does not show greater stability with added salt. It seems that halophilicity is structure-dependent and not just charge-dependent.
This investigation would not be possible with wt PGB1, because basic pH catalyses deamidation of Asn residues that precede glycine, and wt PGB1 has two such sites. Deamidation is a spontaneous event leading to both normal and iso-Asp (45,46), and with two sites the protein will evolve to a mixture of nine different forms. Each form may have its own thermodynamic parameters for unfolding, and data interpretation for such mixtures could be highly complex. Our results show that PGB1-QDD is functional at several values of pH and has the same structure as wt PGB1, and hence comparisons between the two variants are valid. Due to introduced negative charges at positions 8 and 37, our protein will have a higher net charge than wt PGB1 at high and intermediate values of pH and we find that this affects the stability. The Tm for wt PGB1 has been determined before to 87.5°C at pH 5.4, where it showed the highest melting temperature (25). PGB1-QDD shows a Tm of 74°C at this pH and a somewhat higher Tm of 78°C at pH 4.5. This shows that the introduced charges alter the Coulombic network and the addition of counterions is not enough to shield the unfavorable interactions.
Acknowledgments
We thank Dr. Ingemar André for assistance with NMR experiments and analysis and for valuable discussions.
This work was supported by the Swedish Research Council (to S.L.).
References
- 1.Grimsley, G., K. Shaw, L. Fee, R. Alston, B. Huyghues-Despointes, R. Thurlkill, J. Scholtz, and C. Pace. 1999. Increasing protein stability by altering long-range Coulombic interactions. Protein Sci. 8:1843–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akke, M., and S. Forsen. 1990. Protein stability and electrostatic interactions between solvent-exposed charged side chains. Proteins. 8:23–29. [DOI] [PubMed] [Google Scholar]
- 3.Loladze, V. V., and G. I. Makhatadze. 2002. Removal of surface charge-charge interactions from ubiquitin leaves the protein folded and very stable. Protein Sci. 11:174–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makhatadze, G. I., V. V. Loladze, D. N. Ermolenko, X. Chen, and S. T. Thomas. 2003. Contribution of surface salt bridges to protein stability: guidelines for protein engineering. J. Mol. Biol. 327:1135–1148. [DOI] [PubMed] [Google Scholar]
- 5.Wunderlich, M., A. Martin, and F. X. Schmid. 2005. Stabilization of the cold shock protein CspB from Bacillus subtilis by evolutionary optimization of Coulombic interactions. J. Mol. Biol. 347:1063–1076. [DOI] [PubMed] [Google Scholar]
- 6.Andre, I., T. Kesvatera, B. Jonsson, K. S. Akerfeldt, and S. Linse. 2004. The role of electrostatic interactions in calmodulin-peptide complex formation. Biophys. J. 87:1929–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cambillau, C., and J.-M. Claverie. 2000. Structural and genomic correlates of hyperthermostability. J. Biol. Chem. 275:32383–32386. [DOI] [PubMed] [Google Scholar]
- 8.Suhre, K., and J.-M. Claverie. 2003. Genomic correlates of hyperthermostability, an update. J. Biol. Chem. 278:17198–17202. [DOI] [PubMed] [Google Scholar]
- 9.Xiao, L., and B. Honig. 1999. Electrostatic contributions to the stability of hyperthermophilic proteins. J. Mol. Biol. 289:1435–1444. [DOI] [PubMed] [Google Scholar]
- 10.Kumar, S., and R. Nussinov. 2001. How do thermophilic proteins deal with heat? Cell. Mol. Life Sci. 58:1216–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karshikoff, A., and R. Ladenstein. 2001. Ion pairs and the thermotolerance of proteins from hyperthermophiles: a “traffic rule” for hot roads. Trends Biochem. Sci. 26:550–556. [DOI] [PubMed] [Google Scholar]
- 12.Sanchez-Ruiz, J. M., and G. I. Makhatadze. 2001. To charge or not to charge? Trends Biotechnol. 19:132–135. [DOI] [PubMed] [Google Scholar]
- 13.Szilagyi, A., and P. Zavodszky. 2000. Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: results of a comprehensive survey. Structure. 8:493–504. [DOI] [PubMed] [Google Scholar]
- 14.Elcock, A. H., and J. A. McCammon. 1998. Electrostatic contributions to the stability of halophilic proteins. J. Mol. Biol. 280:731–748. [DOI] [PubMed] [Google Scholar]
- 15.Rao, J., and P. Argos. 1981. Structural stability of halophilic proteins. Biochemistry. 20:6536–6543. [DOI] [PubMed] [Google Scholar]
- 16.Jaenicke, R., and P. Zavodszky. 1990. Proteins under extreme physical conditions. FEBS Lett. 268:344–349. [DOI] [PubMed] [Google Scholar]
- 17.Kangas, E., and B. Tidor. 2001. Electrostatic complementarity at ligand binding sites: application to chorismate mutase. J. Phys. Chem. B. 105:880–888. [Google Scholar]
- 18.Ciriolo, M. R., A. Battistoni, M. Falconi, G. Filomeni, and G. Rotilio. 2001. Role of the electrostatic loop of Cu,Zn superoxide dismutase in the copper uptake process. Eur. J. Biochem. 268:737–742. [DOI] [PubMed] [Google Scholar]
- 19.Tougu, V., and T. Kesvatera. 1996. Role of ionic interactions in cholinesterase catalysis. Biochim. Biophys. Acta. 1298:12–30. [DOI] [PubMed] [Google Scholar]
- 20.Otzen, D. E., O. Kristensen, and M. Oliveberg. 2000. Designed protein tetramer zipped together with a hydrophobic Alzheimer homology: a structural clue to amyloid assembly. Proc. Natl. Acad. Sci. USA. 97:9907–9912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gudiksen, K. L., I. Gitlin, J. Yang, A. R. Urbach, D. T. Moustakas, and G. M. Whitesides. 2005. Eliminating positively charged lysine ɛ-NH3+ groups on the surface of carbonic anhydrase has no significant influence on its folding from sodium dodecyl sulfate. J. Am. Chem. Soc. 127:4707–4714. [DOI] [PubMed] [Google Scholar]
- 22.Gallagher, T., P. Alexander, P. Bryan, and G. Gilliland. 1994. Two crystal structures of the B1 immunoglobulin-binding domain of streptococcal protein G and comparison with NMR. Biochemistry. 33:4721–4729. [PubMed] [Google Scholar]
- 23.Gronenborn, A., D. Filpula, N. Essig, A. Achari, M. Whitlow, P. Wingfield, and G. Clore. 1991. A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science. 253:657–661. [DOI] [PubMed] [Google Scholar]
- 24.Akerstrom, B., T. Brodin, K. Reis, and L. Bjorck. 1985. Protein G: a powerful tool for binding and detection of monoclonal and polyclonal antibodies. J. Immunol. 135:2589–2592. [PubMed] [Google Scholar]
- 25.Alexander, P., S. Fahnestock, T. Lee, J. Orban, and P. Bryan. 1992. Thermodynamic analysis of the folding of the streptococcal protein G IgG-binding domains B1 and B2: why small proteins tend to have high denaturation temperatures. Biochemistry. 31:3597–3603. [DOI] [PubMed] [Google Scholar]
- 26.Smith, C., J. Withka, and L. Regan. 1994. A thermodynamic scale for the β-sheet forming tendencies of the amino acids. Biochemistry. 33:5510–5517. [DOI] [PubMed] [Google Scholar]
- 27.Delaglio, F., S. Grzesiek, G. Vuister, G. Zhu, J. Pfeifer, and A. Bax. 1995. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 6:277–293. [DOI] [PubMed] [Google Scholar]
- 28.Efron, B., and G. Gong. 1983. A leisurely look at the bootstrap, the jackknife, and cross-validation. Am. Stat. 37:36–48. [Google Scholar]
- 29.Xue, W.-F., J. Carey, and S. Linse. 2004. Multi-method global analysis of thermodynamics and kinetics in reconstitution of monellin. Proteins. 57:586–595. [DOI] [PubMed] [Google Scholar]
- 30.Linse, S., B. Jonsson, and W. Chazin. 1995. The effect of protein concentration on ion binding. Proc. Natl. Acad. Sci. USA. 92:4748–4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nozaki, Y., and C. Tanford. 1967. Examination of titration behavior methods. In Enzymology, Vol. 11. Academic Press, New York. 715–734.
- 32.Khare, D., P. Alexander, J. Antosiewicz, P. Bryan, M. Gilson, and J. Orban. 1997. pKa measurements from nuclear magnetic resonance for the B1 and B2 immunoglobulin G-binding domains of protein G: comparison with calculated values for nuclear magnetic resonance and x-ray structures. Biochemistry. 36:3580–3589. [DOI] [PubMed] [Google Scholar]
- 33.Saha, K., F. Bender, and E. Gizeli. 2003. Comparative study of IgG binding to proteins G and A: nonequilibrium kinetic and binding constant determination with the acoustic waveguide device. Anal. Chem. 75:835–842. [DOI] [PubMed] [Google Scholar]
- 34.Kao, Y.-H., C. A. Fitch, S. Bhattacharya, C. J. Sarkisian, J. T. J. Lecomte, and E. B. Garcia-Moreno. 2000. Salt effects on ionization equilibria of histidines in myoglobin. Biophys. J. 79:1637–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luisi, D., C. Snow, J. Lin, Z. Hendsch, B. Tidor, and D. Raleigh. 2003. Surface salt bridges, double-mutant cycles, and protein stability: an experimental and computational analysis of the interaction of the Asp 23 side chain with the N-terminus of the N-terminal domain of the ribosomal protein l9. Biochemistry. 42:7050–7060. [DOI] [PubMed] [Google Scholar]
- 36.Dominy, B. N., D. Perl, F. X. Schmid, I. Brooks, and L. Charles. 2002. The effects of ionic strength on protein stability: the cold shock protein family. J. Mol. Biol. 319:541–554. [DOI] [PubMed] [Google Scholar]
- 37.Lee, K., C. Fitch, J. Lecomte, and E. B. Garcia-Moreno. 2002. Electrostatic effects in highly charged proteins: salt sensitivity of pKa values of histidines in staphylococcal nuclease. Biochemistry. 41:6556–6567. [DOI] [PubMed] [Google Scholar]
- 38.Lee, K. K., C. A. Fitch, and E. B. Garcia-Moreno. 2002. Distance dependence and salt sensitivity of pairwise, Coulombic interactions in a protein. Protein Sci. 11:1004–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baldwin, R. 1996. How Höfmeister ion interactions affect protein stability. Biophys. J. 71:2056–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ibarra-Molero, B., V. V. Loladze, G. I. Makhatadze, and J. M. Sanchez-Ruiz. 1999. Thermal versus guanidine-induced unfolding of ubiquitin. An analysis in terms of the contributions from charge-charge interactions to protein stability. Biochemistry. 38:8138–8149. [DOI] [PubMed] [Google Scholar]
- 41.Perez-Jimenez, R., R. Godoy-Ruiz, B. Ibarra-Molero, and J. M. Sanchez-Ruiz. 2004. The efficiency of different salts to screen charge interactions in proteins: a Höfmeister effect? Biophys. J. 86:2414–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang, A.-S., and B. Honig. 1993. On the pH dependence of protein stability. J. Mol. Biol. 231:459–474. [DOI] [PubMed] [Google Scholar]
- 43.Yang, A.-S., and B. Honig. 1994. Structural origins of pH and ionic strength effects on protein stability: acid denaturation of sperm whale apomyoglobin. J. Mol. Biol. 237:602–614. [DOI] [PubMed] [Google Scholar]
- 44.Schmittschmitt, J. P., and J. M. Scholtz. 2003. The role of protein stability, solubility, and net charge in amyloid fibril formation. Protein Sci. 12:2374–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reissner, K. J., and D. W. Aswad. 2003. Deamidation and isoaspartate formation in proteins: unwanted alterations or surreptitious signals? Cell. Mol. Life Sci. 60:1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bischoff, R., and H. V. J. Kolbe. 1994. Deamidation of asparagine and glutamine residues in proteins and peptides: structural determinants and analytical methodology. J. Chromatogr. B Biomed. Sci. Appl. 662:261–278. [DOI] [PubMed] [Google Scholar]
- 47.Koradi, R., M. Billeter, and K. Wüthrich. 1996. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14:51–55. [DOI] [PubMed] [Google Scholar]