

Abstract
There is substantial evidence suggesting that angiotensin II plays an important role in elevating blood pressure of spontaneously hypertensive rats, despite normal plasma renin activity, and that converting enzyme inhibitors (captopril) can effectively normalize blood pressure in the spontaneously hypertensive rats. One mechanism by which angiotensin II induces hypertension is via oxidative stress and endothelin, as seen in subpressor angiotensin II—induced hypertension. In fact, it has been shown that antioxidants lower mean arterial pressure in spontaneously hypertensive rats. However, the relationship between angiotensin II, oxidative stress, and endothelin in the spontaneously hypertensive rats is still relatively undefined. This study examines the relationship between mean arterial pressure, plasma renin activity, angiotensin II, oxidative stress, and endothelin in spontaneously hypertensive rats compared with normotensive Wistar Kyoto rats, and the effects of captopril on this association. Untreated spontaneously hypertensive rats had increased plasma angiotensin II levels despite normal plasma renin activity, oxidative stress, and endothelin. Captopril treatment in spontaneously hypertensive rats lowered mean arterial pressure, angiotensin II, oxidative stress, and endothelin, and increased plasma renin activity. In contrast, captopril increased plasma renin activity (suggesting effective captopril treatment) but did not significantly alter mean arterial pressure, angiotensin II, oxidative stress, or endothelin of Wistar Kyoto rats. These results suggest that in spontaneously hypertensive rats, angiotensin II is a primary instigator of hypertension, and that captopril selectively lowers angiotensin II, oxidant stress, and endothelin, which in turn may contribute to the blood pressure-lowering efficacy of captopril in spontaneously hypertensive rats.
Keywords: angiotensin II; angiotensin-converting enzyme inhibitors; captopril; endothelin; oxidative stress; rats, spontaneously hypertensive
Numerous studies have been performed to define the regulatory factors that participate in the pathogenesis of hypertension in spontaneously hypertensive rats (SHR). These studies have demonstrated that despite the presence of normal plasma renin activity (PRA), angiotensin II (Ang II) still appears to play a key role in the pathogenesis of the increased blood pressure. Blood pressure is normalized by administrating renin inhibitors,1 converting enzyme inhibitors,2,3 or Ang II receptor blockers.4-6 However, despite the efficacy of these agents in lowering blood pressure, the mechanisms that lead to the Ang II—dependent increase in blood pressure in the SHR remain undefined.
One mechanism by which Ang II can induce hypertension is via oxidative stress and endothelin (ET). This pathway has been shown to be essential in the maintenance of blood pressure in several models of Ang II—dependent hypertension, including subpressor Ang II—induced hypertension7-11 and 1-kidney Goldblatt hypertension.12,13 Thus, it is tempting to postulate that the normal or modestly elevated levels of Ang II in SHR (which are in fact inappropriately high, considering that blood pressure is elevated) may activate oxidative pathways that promote hypertension. In fact, blood pressure in SHR is decreased by tempol,14,15 a SOD mimetic with antioxidant properties. Despite these advances, the relationship between Ang II, oxidative stress, and ET in the SHR are still relatively obscure. Consequently, the goals of this study were to evaluate whether hypertension in the SHR is associated with elevated Ang II, oxidant stress, and ET compared with 2 normotensive rat strains: (1) Wistar Kyoto (WKY) rats, which are a genetically related normotensive control strain and (2) Sprague Dawley (SD) rats, which are a genetically unrelated normotensive control strain. Also, we wanted to examine the relationship between blood pressure, Ang II, oxidant stress, and ET in SHR and WKY rats in which the renin-angiotensin system has been blocked by captopril.
Methods
All experiments were performed according to the Guidelines for the Care and Use of Laboratory Animals, National Institute of Health, and approved by the Institutional Animal Care and Use Committee, Mayo Clinic. Male SD, WKY, and SHR rats 12 to 13 weeks old (250 to 300 grams; Harlan, Indianapolis, Ind) were placed individually in cages and allowed 6 days to acclimate. All groups were maintained on a standard diet and allowed water ad libitum. At day 1, half of the WKY and SHR rats were started on captopril (100 mg/kg per day added to their drinking water). After 15 days of treatment, we either anesthetized the rats to measure mean arterial pressure (MAP) or collected blood samples from the rats via decapitation for measurement of Ang II, PRA, oxidative stress (thiobarbituric acid reactive substances [TBARS]), and ET.
In pilot studies, we confirmed the well-established notion that anesthesia increases the PRA and Ang II levels in both groups of rats. However, we also found that anesthesia affected these parameters differently in the SHR versus normotensive rats. That is, anesthesia increased PRA by similar magnitudes in both groups of rats: PRA was 10.5±2.5 versus 22.7±3.6 ng/mL per hour in the decapitated and anesthetized SD rats, respectively; and 9.2±1.3 versus 26.4±6.6 ng/mL per hour in the decapitated and anesthetized SHR, respectively. In contrast, anesthesia increased plasma Ang II values to a much greater extent in the SHR than in the SD rats: Ang II levels were 16.5±3.6 versus 34.5±8.8 pg/mL in the decapitated and anesthetized SD rats, respectively; and 38.4±6.2 versus 836±205 pg/mL in the decapitated and anesthetized SHR, respectively. These pilot studies raised the possibility that Ang II formation may be exceptionally responsive to diverse stimuli in the anesthetized SHR (independent of PRA). Thus, in the remaining experiments, we collected the blood samples to measure Ang II, ET, etc, from separate groups of rats in which all manipulations and stresses (animal handling) had been minimized until they were decapitated.
In the acute experiments, half of the rats in each group were anesthetized with Inactin (100 mg/kg body weight, intraperitoneal), then placed on a heating table to maintain body temperature (37°C), and polyethylene cannulas (PE-50 tubing) were inserted into their right femoral artery for measuring MAP. The remaining rats were placed in restraining cones and decapitated by guillotine. Blood was immediately collected in EDTA tubes on ice. All samples from both procedures were centrifuged and plasma was stored at -80°C until assayed. Ang II was measured by enzyme immunoassay kit (Cayman Chemical), PRA was measured by radioimmunoassay (RIA) kit (Dupont NEN), ET was measured by RIA kit (Peninsula Laboratories), and TBARS was measured by a standard colorimetric assay.
Data Analysis
Values are reported as mean±SEM and the significance of differences between groups was assessed using unpaired t test assuming equal variances. P<0.05 indicated a significant difference.
Results
Figure 1 summarizes the basal data obtained in SD, WKY, and SHR. The top panels illustrate MAP, Ang II, and ET, whereas PRA and TBARS are depicted in the bottom panels. The MAP of SHR (160±4.0 mm Hg) was significantly higher than that of WKY and SD rats (105±9.2 and 109±5.5 mm Hg, respectively). The increased blood pressure in the SHR was associated with significantly increased circulating levels of Ang II and ET compared with WKY and SD: the SHR, WKY, and SD had Ang II levels of 40.6±5.3,23.5±3.7, and 15.9±2.1 pg/mL, respectively; and ET levels of 45.3±6.8, 17.6±4.9, and 29.0±3.4 pg/mL, respectively. In contrast, PRA and plasma concentration of TBARS were not different between the 3 groups of rats.
Figure 1.
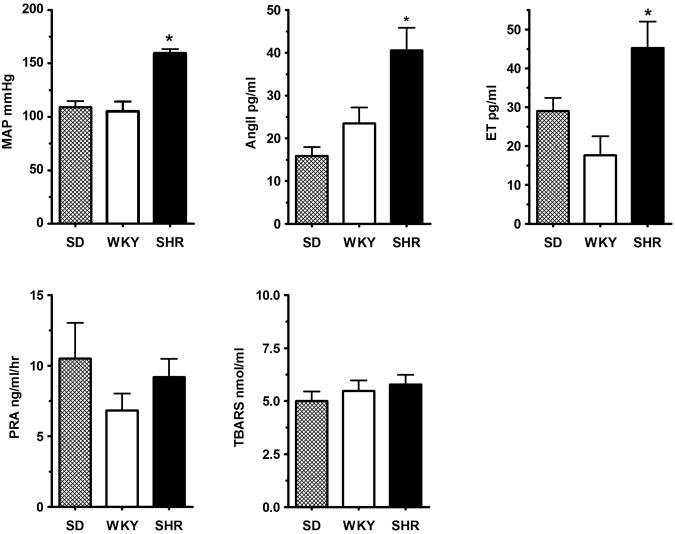
Top panels show MAP (left), circulating levels of Ang II (center), and ET (right) in SD (genetically-unrelated controls; n=11), WKY (genetically related controls; n=5), and SHR (n=7). Bottom panels show PRA (left) and TBARS (right) in SD (n=4), WKY (n=6), and SHR (n=7). The SHR had higher MAP with parallel increases in Ang II and ET levels when compared with either SD or WKY (n=8, 6, and 8, for the SD, WKY, and SHR, respectively). In contrast, PRA and plasma TBARS levels in the SHR were similar to those of SD and WKY rats. *P<0.05 between the SHR values (n=7) and those from the SD (n=4) and WKY rats (n=6).
Figure 2 summarizes the data obtained from the untreated and captopril-treated WKY and SHR. The top panels again illustrate MAP, Ang II, and ET; whereas the bottom panels show PRA and TBARS. Captopril treatment significantly lowered blood pressure in the SHR but not in the WKY: MAP was 160±4.0 versus 101.0±6.6 mm Hg in the untreated and captopril-treated SHR, respectively; and 105±9.2 versus 88±1.4 mm Hg in the untreated and captopril-treated WKY, respectively. The effect of captopril on the MAP of SHR and WKY was associated with parallel changes in circulating levels of Ang II and ET. That is, captopril decreased Ang II (from 40.6±5.3 to 21.9±2.3 pg/mL) and ET (from 45.3±6.8 to 18.2±2.0 pg/mL) in the SHR, but it did not affect either parameter in the WKY rats (Ang II was 23.5±3.7 versus 24.6±3.6 pg/mL and ET was 17.6±4.9 versus 18.8±2.2 pg/mL, in the untreated and captopril-treated, respectively). Although the basal levels of TBARS were not elevated in the SHR, captopril still induced a small but significant decrease in their levels in the SHR (from 5.8±0.5 to 4.6±0.3 nmol/mL). Captopril had no effect on TBARS in the WKY rats (5.5±0.5 versus 5.3±0.6 nmol/mL in the untreated and captopriltreated WKY rats, respectively). Finally, captopril treatment increased PRA in both groups (indirectly verifying inhibition of angiotensin-converting enzyme in both groups): PRA increased from 9.2±1.3 to 23.4±5.7 ng/mL per hour in the SHR, and from 6.8±1.2 to 14.5±1.3 ng/mL per hour in the WKY.
Figure 2.
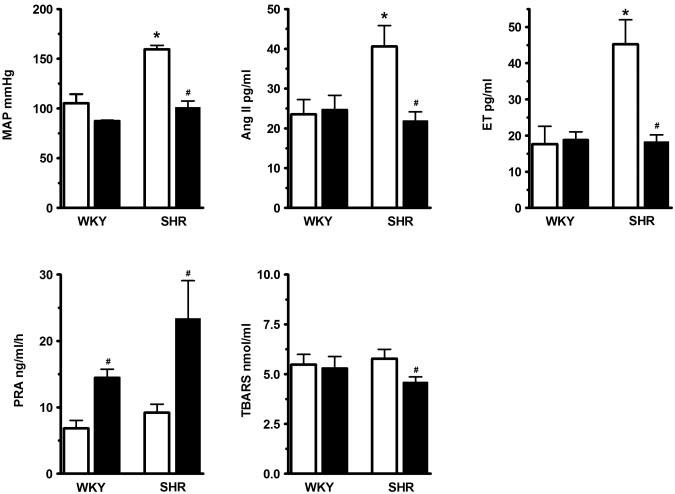
Top panels show MAP (left), plasma Ang II (center), and ET (right) in WKY and SHR that were either left untreated (□ open bars) or treated for 15 days with captopril (■ closed bars). Bottom panels show PRA (left) and TBARS (right) in untreated and captopril-treated WKY and SHR. Captopril treatment in SHR caused analogous decreases in MAP, Ang II, and ET in SHR, but had no discernable effects on these parameters in WKY. Likewise, captopril decreased TBARS only in the SHR. However, it increased PRA in both groups (confirming angiotensin-converting enzyme inhibition). P<0.05 from WKY rats; #P<0.05 from the corresponding untreated WKY or SHR rats. Number of animals (for untreated and captopril-treated WKY and SHR, respectively). MAP: n=5, 6, 7, and 7; Ang II: n=6, 6, 7, and 7; ET: n=6, 6,8, and 12; PRA: n=6, 6, 7, and 7; TBARS: n=5, 6, 7, and 7.
Discussion
The results of this study show that the increased blood pressure of the SHR is associated with a modest increase in baseline levels of circulating Ang II (in the absence of an increase in PRA), along with a greatly exaggerated increase in circulating Ang II in response to anesthesia (which is dissociated from the PRA response). This suggests that the basal Ang II levels and Ang II—generating activity are inappropriately high (independently of PRA), because this system should be shutdown in the presence of high blood pressure. Surprisingly, TBARS were not elevated in the untreated SHR but ET was, suggesting that the Ang II—dependent increases in blood pressure and ET in the SHR are not dependent on a measurable increase in circulating TBARS. However, the blood pressure-lowering effects of a converting enzyme inhibitor in the SHR is associated with decreases in circulating levels of Ang II, ET, and TBARS (despite the normal basal levels of the latter). Despite its effectiveness at inhibiting converting enzyme, captopril did not decrease any of these parameters in the normotensive WKY, further suggesting that the Ang II system is abnormally sustained in SHR.
It is well known that the SHR is a genetic model of hypertension that is characterized by increased peripheral resistance and normal PRA levels. Despite the fact that PRA levels in the SHR are normal, administration of an angiotensin-converting enzyme inhibitor such as captopril was very effective in treating high blood pressure, suggesting that Ang II plays a prominent role in the pathogenesis of hypertension in the SHR. One potential explanation for this may be that normal PRA coinciding with high blood pressure may actually be inappropriately high; thus, by blocking this system, the imbalance between high blood pressure and “normal” PRA is rectified. A second explanation may relate to the enhanced vascular responses to Ang II that have been reported in SHR.16 These exaggerated responses to Ang II would in turn increase peripheral resistance and ultimately blood pressure. Our findings suggest another potential mechanism: basal levels of Ang II are also increased (despite the normal PRA), and Ang II generation is greatly enhanced in response to stimuli (ie, anesthesia) independent of PRA. The modest but sustained increase in basal Ang II (despite the hypertension) may further increase peripheral resistance, whereas the exaggerated generation of Ang II may augment vascular responses to stress stimuli and also help maintain elevated Ang II levels.
The reasons for the increased basal levels of Ang II and its exaggerated increase in response to anesthesia are unknown, but its presence leads further credence to its importance in maintaining the hypertension in SHR. In this respect, it is interesting to note a few similarities with the hypertensive responses that occur as a result of chronically infusing a subpressor dose of Ang II (SP-Ang II). In this model, Ang II levels are sustained independently of blood pressure and sodium intake. As a result, there is autopotentiation of the vascular responses to Ang II, with a resultant increase in vascular reactivity, exaggerated increases in blood pressure in response to stress17 and a progressive increase in peripheral resistance. These abnormalities mimic those that are present in the SHR. Because of these similarities and the fact that they can be reversed by converting enzyme inhibitors and Ang II receptor blockers, it is tempting to speculate that they may be the result of the sustained low-level increase in Ang II that we observed in the SHR and perhaps to an exaggerated response to stress.
Another factor that appears to be implicated in the development of hypertension in SHR is oxidant stress. This view is derived from studies that reported that oxidative stress is increased in SHR, and that antioxidants can lower their blood pressure.14,15 The SHR in our current study did not have elevated circulating levels of TBARS, but the levels were significantly reduced by captopril. The reason for the discrepancy between our study and previous ones is not clear but may be because of the different markers used between studies or the tissue in which they were measured (we only measured circulating levels). However, the fact that they were decreased by captopril suggests Ang II was playing a role in maintaining their production. However, we recognize that because captopril has other effects such as blocking the degradation of kinins, which will also decrease blood pressure, it cannot be ruled out that these factors may have contributed in part to the decrease in oxidant stress. It is apparent, however, that there was some dissociation between the levels of TBARS and both Ang II and blood pressure levels, and that decreasing Ang II (either directly or indirectly) lowers circulating TBARS.
As mentioned, there appears to be numerous similarities between SHR and the SP-Ang II—induced hypertension.18 In addition to the inappropriately high Ang II levels, the responsiveness to antioxidant therapy, the autopotentiation to Ang II, and the increase in peripheral resistance and pressor responses, both models also have increased levels of ET. We found that this increase in ET in the SHR was blocked by captopril and thus followed the same pattern as Ang II. Thus, it is tempting to suggest that the reason for the elevated ET levels in the SHR may also be because of the sustained Ang II levels. This would be similar to the SP-Ang II hypertensive model in which Ang II is associated with an increase in ET that contributes to the hypertension.8,10 The increase in blood pressure was prevented by bosentan (a mixed ET receptor blocker). Interestingly, those studies found that the increase in ET was secondary to the increase in oxidant stress (TBARS), whereas in our present study, the increase in ET was independent of an increase in TBARS. The reason for this discrepancy is not clear but may be caused by different pathways of Ang II—induced ET in these hypertensive models or caused by an increase in oxidant stress that we did not detect in the circulating levels of TBARS.
In conclusion, our results suggest that in the SHR, there is an abnormal elevation in Ang II (which is dissociated from PRA) and an exaggerated increase in circulating Ang II in response to stimuli such as anesthesia. It appears that this sustained increase in Ang II is likely an essential factor in the maintenance of oxidant stress and the increase in ET, all of which interact to cause autopotentiation of vascular responses, enhanced pressor responses, and increased peripheral resistance, with the consequent sustained increase in blood pressure.
Perspectives
There is substantial evidence showing that Ang II plays a leading role in the development of several forms of experimental hypertension. Ang II—induced changes in oxidant stress and ET are vital components implicated in the maintenance of hypertension. Our previous studies in SP-Ang II—induced hypertension delineated the contribution of each component (Ang II, oxidative stress, and ET) to the maintenance of blood pressure. Whereas our current study did not evaluate the precise contribution of each component in SHR, it does imply, when taken together with previous studies,8,10,17 that all of these components play a considerable role in the pathogenesis of hypertension. Captopril treatment decreased MAP and circulating levels of all of these components in SHR but not WKY, suggesting that the system is dysfunctional during hypertension. These studies also underscore the notion that an abnormal relationship between Ang II, oxidant stress, or ET, whether they are activated simultaneously or independently, can have a profound effect on blood pressure regulation. Although these associations are thought to be important in the pathogenesis of human hypertension, their interactions have not been clearly defined. Further assessment of these interactions between Ang II, oxidant stress, and ET may further help us define the mechanisms of hypertension in humans and design more effective alternative (or combination) antihypertensive therapies.
Acknowledgments
This work was supported in part by National Institutes of Health grant HL16496 (J.C.R.). L.A.J. was supported a Clinician-Scientist Award from the National Kidney Foundation and National Institutes of Health grant DK02943. M.C.O. was supported by a postdoctoral fellowship grant from the American Heart Association (0020640Z), the Spanish Society for Liver Study (AEEH), and the Spanish Ministerio de Educacion y Cultura (PF0029000129).
References
- 1.Wood JM. Current Advances in ACE Inhibition 2. Churchill Livingstone; London: 1991. [Google Scholar]
- 2.Antonaccio MJ, High JP, Rubin B, Schaeffer T. Contribution of the kidneys but not adrenal glands to the acute antihypertensive effects of captopril in spontaneously hypertensive rats. Clin Sci (Lond) 1979;57:127s–130s. doi: 10.1042/cs057127s. [DOI] [PubMed] [Google Scholar]
- 3.Harrap SB, Nicolaci JA, Doyle AE. Persistent effects on blood pressure and renal haemodynamics following chronic angiotensin converting enzyme inhibition with perindopril. Clin Exp Pharmacol Physiol. 1986;13:753–765. doi: 10.1111/j.1440-1681.1986.tb02379.x. [DOI] [PubMed] [Google Scholar]
- 4.Bunkenburg B, Schnell C, Baum HP, Cumin F, Wood JM. Prolonged angiotensin II antagonism in spontaneously hypertensive rats. Hemodynamic and biochemical consequences. Hypertension. 1991;18:278–288. doi: 10.1161/01.hyp.18.3.278. [DOI] [PubMed] [Google Scholar]
- 5.Gillies LK, Lu M, Wang H, Lee RM. AT1 receptor antagonist treatment caused persistent arterial functional changes in young spontaneously hypertensive rats. Hypertension. 1997;30:1471–1478. doi: 10.1161/01.hyp.30.6.1471. [DOI] [PubMed] [Google Scholar]
- 6.Wong PC, Price WA, Jr., Chiu AT, Duncia JV, Carini DJ, Wexler RR, Johnson AL, Timmermans PB. Hypotensive action of DuP 753, an angiotensin II antagonist, in spontaneously hypertensive rats. Nonpeptide angiotensin II receptor antagonists: X. Hypertension. 1990;15:459–468. doi: 10.1161/01.hyp.15.5.459. [DOI] [PubMed] [Google Scholar]
- 7.Reckelhoff JF, Zhang H, Srivastava K, Roberts LJ, 2nd, Morrow JD, Romero JC. Subpressor doses of angiotensin II increase plasma F(2)isoprostanes in rats. Hypertension. 2000;35:476–479. doi: 10.1161/01.hyp.35.1.476. [DOI] [PubMed] [Google Scholar]
- 8.Ortiz MC, Sanabria E, Manriquez MC, Romero JC, Juncos LA. Role of endothelin and isoprostanes in slow pressor responses to angiotensin II. Hypertension. 2001;37:505–510. doi: 10.1161/01.hyp.37.2.505. [DOI] [PubMed] [Google Scholar]
- 9.Ruef J, Moser M, Kubler W, Bode C. Induction of endothelin-1 expression by oxidative stress in vascular smooth muscle cells. Cardiovasc Pathol. 2001;10:311–315. doi: 10.1016/s1054-8807(01)00095-3. [DOI] [PubMed] [Google Scholar]
- 10.Ortiz MC, Manriquez MC, Romero JC, Juncos LA. Antioxidants block angiotensin II-induced increases in blood pressure and endothelin. Hypertension. 2001;38:655–659. doi: 10.1161/01.hyp.38.3.655. [DOI] [PubMed] [Google Scholar]
- 11.Nishiyama A, Seth DM, Navar LG. Renal interstitial fluid concentrations of angiotensins I and II in anesthetized rats. Hypertension. 2002;39:129–134. doi: 10.1161/hy0102.100536. [DOI] [PubMed] [Google Scholar]
- 12.Lerman LO, Nath KA, Rodriguez-Porcel M, Krier JD, Schwartz RS, Napoli C, Romero JC. Increased oxidative stress in experimental renovascular hypertension. Hypertension. 2001;37:541–546. doi: 10.1161/01.hyp.37.2.541. [DOI] [PubMed] [Google Scholar]
- 13.Dobrian AD, Schriver SD, Prewitt RL. Role of angiotensin II and free radicals in blood pressure regulation in a rat model of renal hypertension. Hypertension. 2001;38:361–366. doi: 10.1161/01.hyp.38.3.361. [DOI] [PubMed] [Google Scholar]
- 14.Schnackenberg CG, Welch WJ, Wilcox CS. Normalization of blood pressure and renal vascular resistance in SHR with a membranepermeable superoxide dismutase mimetic: role of nitric oxide. Hypertension. 1998;32:59–64. doi: 10.1161/01.hyp.32.1.59. [DOI] [PubMed] [Google Scholar]
- 15.Schnackenberg CG, Wilcox CS. Two-week administration of tempol attenuates both hypertension and renal excretion of 8-iso prostaglandin f2alpha. Hypertension. 1999;33:424–428. doi: 10.1161/01.hyp.33.1.424. [DOI] [PubMed] [Google Scholar]
- 16.Hollenberg GE. Different reactivity to angiotensin II of peripheral and renal arteries in spontaneously hypertensive rats: effect of acute and chronic angiotensin converting enzyme inhibition. J Hypertens. 1986;4:S480–S482. [PubMed] [Google Scholar]
- 17.Pelaez LI, Manriquez MC, Nath KA, Romero JC, Juncos LA. Low-dose angiotensin II enhances pressor responses without causing sustained hypertension. Hypertension. 2003;42:798–801. doi: 10.1161/01.HYP.0000085782.99773.B6. [DOI] [PubMed] [Google Scholar]
- 18.Reckelhoff JF, Romero JC. Role of oxidative stress in angiotensininduced hypertension. Am J Physiol Regul Integr Comp Physiol. 2003;284:R893–R912. doi: 10.1152/ajpregu.00491.2002. [DOI] [PubMed] [Google Scholar]