

Abstract
trkB activation results in tyrosine phosphorylation of N-terminal Kir3 residues, decreasing channel activation. To determine the mechanism of this effect, we reconstituted Kir3, trkB, and the mu opioid receptor in Xenopus oocytes. Activation of trkB by BDNF (brain-derived neurotrophic factor) accelerated Kir3 deactivation following termination of mu opioid receptor signaling. Similarly, overexpression of RGS4, a GTPase-activating protein (GAP), accelerated Kir3 deactivation. Blocking GTPase activity with GTPγS also prevented Kir3 deactivation, and the GTPγS effect was not reversed by BDNF treatment. These results suggest that BDNF treatment did not reduce Kir3 affinity for Gβγ but rather acted to accelerate GTPase activity, like RGS4. Tyrosine phosphatase inhibition by peroxyvanadate pretreatment reversibly mimicked the BDNF/trkB effect, indicating that tyrosine phosphorylation of Kir3 may have caused the GTPase acceleration. Tyrosine to phenylalanine substitution in the N-terminal domain of Kir3.4 blocked the BDNF effect, supporting the hypothesis that phosphorylation of these tyrosines was responsible. Like other GAPs, Kir3.4 contains a tyrosine-arginine-glutamine motif that is thought to function by interacting with G protein catalytic domains to facilitate GTP hydrolysis. These data suggest that the N-terminal tyrosine hydroxyls in Kir3 normally mask the GAP activity and that modification by phosphorylation or phenylalanine substitution reveals the GAP domain. Thus, BDNF activation of trkB could inhibit Kir3 by facilitating channel deactivation.
The family of G protein-activated potassium channels (Kir3 or GIRK)1 is a principal effector mediating the actions of a wide range of pertussis toxin-sensitive, G protein-coupled receptors (GPCR) (1). Channel activation provides key regulation of both cardiac and neuronal excitability. The activity of this channel is controlled by a large number of modulators including phosphatidylinositol bisphosphate, Na+, eicosanoids, ATP, Mg2+, and phosphorylation (1–6). For example, in a previous study (7), we found that tyrosine phosphorylation of Kir3 resulted in channel inhibition. Brain-derived neurotrophic factor (BDNF) activation of trkB receptors caused the phosphorylation of specific tyrosine residues in the N-terminal domain of Kir3.1 and Kir3.4, reducing basal channel conductance. Gβγ, released by opioid receptor activation, overcame the inhibition and evoked the original maximal effect. The results suggest that channel phosphorylation regulates the specific interaction with Gβγ, but the mechanism of this effect is unclear. However, the finding may be physiologically significant because tyrosine kinase cascades initiated by neurotrophic factors such as BDNF and nerve growth factor are up-regulated under conditions such as inflammation (8). It is possible that tyrosine phosphorylation of Kir3 may mediate neuronal excitability in conjunction with GPCRs under conditions of inflammation. The goal of the present study was to define the mechanism responsible for BDNF inhibition of G protein regulation of Kir3 functioning.
Recent evidence shows that both arms of the heterotrimeric G protein complex, Gαi and Gβγ, interact with Kir3 (9). Binding of the Gβγsubunit activates Kir3, whereas Gαi binding suppresses basal Kir3 activity and enhances G protein activation (9). This action of Gαi binding resembles the effect of BDNF-induced tyrosine phosphorylation on Kir3. Both reduce basal channel activity without blocking GPCR activation of Kir3, suggesting the possibility of mechanistic similarity. Thus, we explored the hypothesis that the underlying mechanisms were related and that tyrosine phosphorylation controls Gαi regulation of Kir3 response to GPCR activation. Understanding the basis for the modulation of Kir3 by tyrosine phosphorylation would provide additional insight into the processes regulating the activity of this physiologically significant channel and a molecular basis for interaction between GPCR and tyrosine kinase receptor signaling.
To test this hypothesis, we reconstituted the mu opioid receptor (MOR, a GPCR), Kir3, and trkB in Xenopus oocytes and investigated channel activation and deactivation kinetics. When current traces were fit to a simple exponential, trkB accelerated deactivation of Kir3. The kinetics resembled GTPase-activating protein (GAP)-mediated acceleration of channel deactivation, suggesting that trkB might stimulate GAP activity. Sequence alignment showed that Kir3 contains two signature GAP residues (glutamine and arginine) near both N-terminal domain tyrosines. These residues have been shown to promote catalytic activity of the GTPase domain (10–15). Phenylalanine substitution of the two N-terminal tyrosine residues within these tyrosine-arginine-glutamine sequences resulted in Kir3 with constitutively faster kinetics of deactivation. Further, BDNF treatment no longer accelerated deactivation kinetics of the mutant channels. These results suggest that phosphorylation of Kir3 unmasks a GAP domain embedded in the sequence of Kir3 itself, promoting GAP activity.
EXPERIMENTAL PROCEDURES
cRNA Preparation and Injection
Plasmid vectors containing cDNA for the following GenBank accession numbers were obtained from Drs. Lei Yu, Cesar Lebarca, Henry Lester, John Adelman, Mark Bothwell, and Nathan Dascal, respectively: MOR (L13069), Kir3.1 (U01071), Kir3.4 (X83584), trkB (M55293), and RGS4 (AF117211). Kir3.4 was point-mutated by PCR-based site-directed mutagenesis to create the functional homomers Kir3.4(S143T) and Kir3.4(S143T/Y32F/Y53F) (7). cRNA was synthesized from linearized plasmid vectors using RNA polymerases provided by the commercially available mMessage mMachine kit (Ambion Corporation, Austin, TX). The cRNA was subsequently micro-injected into stage V and VI oocytes harvested from mature anesthetized Xenopus laevis frogs. Oocytes were maintained in high salt buffer (ND96: 96 mm NaCl, 2 mm KCl, 1 mmCaCl2, 1 mm MgCl2, 5 mm HEPES, 2.5 mm sodium pyruvate, 50 μg/ml gentamicin, pH 7.5).
Two-electrode Voltage Clamp Technique
Following 3–5 days of expression, the two-electrode voltage clamp technique was used to assess coupling efficiency of MOR to Kir3 in oocytes pretreated in BDNF or vehicle. Briefly, oocyte membranes were voltage-clamped at −80 mV by a microelectrode containing 3 m KCl (resistance between 0.4 and 2 mΩ) connected to a feedback amplifier (Axon Instruments, Inc.). A second microelectrode delivered current necessary to maintain the preset voltage. High potassium buffer (HK+: 2 mm NaCl, 96 mm KCl, 1 mm CaCl2, 1 mm MgCl2, 5 mm HEPES, pH 7.5) and the MOR-agonist [d-Ala2, N-MePhe4-Gly-ol5]enkephalin (DAMGO, diluted in high potassium buffer) were sequentially washed onto the oocytes, and changes in current were recorded and analyzed using pCLAMP 6 software.
Pharmacological Agents
DAMGO was obtained from Sigma and Peninsula Labs (San Carlos, CA). Naloxone was from Sigma. Sodium orthovanadate (Sigma) was activated in 3% hydrogen peroxide for 4–5 h prior to use. BDNF was a gift from AMGEN Corporation. All reagents were dissolved in water and then diluted in ND96 or high potassium buffer.
Data Analysis
Kinetics of Kir3 deactivation were calculated as reported previously by measuring time constant to reversal (τoff, 16, 17). Exponentials were fit to the naloxone reversal phases of the DAMGO-induced Kir3 currents. Cursors were positioned at ~20 and 80% of the maximal equilibrium conductance from current initiation and termination, respectively. Current traces between these points were fit to exponential Equation 1.
| (Eq. 1) |
Time constants exceeding two standard deviations of the mean were excluded from the analysis. The membrane time constant (τoff) was used to estimate kinetics of Kir3 deactivation. Statistical significance was determined using an unpaired Student’s t test, in which a probability of p < 0.05 was considered statistically significant.
RESULTS
trkB Modulation of Kir3 Activation
We found previously that tyrosine kinase phosphorylation of Kir3 modulates its conductance (7). To investigate how tyrosine phosphorylation cascades initiated by trkB might regulate Kir3 activation by G protein receptors, we coexpressed cRNA for Kir3.1, Kir3.4, trkB, and MOR in Xenopus oocytes. Oocytes were pretreated in either vehicle (ND96) or 0.2 μg/ml BDNF. Plasma membranes were clamped at −80 mV in two-electrode voltage clamp configuration. Perfusion of 96 mm potassium buffer (HK+) revealed an inwardly rectifying basal current (Fig. 1A). Pretreatment in BDNF suppressed basal current to 77 ± 8% (p < 0.05, n = 35) of vehicle controls in agreement with previous studies (Fig. 1B). Basal current was potentiated when the MOR agonist (DAMGO, 1 μm) was perfused into the recording chamber. BDNF had no effect on the amplitude of the DAMGO-induced response (104 ± 1% of vehicle-treated controls, n = 35, Fig. 1B). MOR antagonist perfusion (naloxone, 1 μm) returned current back to basal amplitude, and ND96 perfusion returned the basal current to baseline (Fig. 1A). The rate of channel deactivation during naloxone perfusion appeared to increase after BDNF treatment. To quantify channel deactivation kinetics, we fit the portion of the trace between naloxone and final ND96 applications to a simple exponential. The time constant of deactivation (τoff) was calculated using pCLAMP software as described under “Experimental Procedures.” BDNF significantly accelerated channel deactivation rate; τoff was reduced to 71 ± 3% of vehicle control (p < 0.05, n = 35, Fig. 1, A and B). Channel activation rate, as measured by τon, was not significantly accelerated in BDNF-treated oocytes (data not shown).
Fig. 1. trkB accelerates Kir3 deactivation.
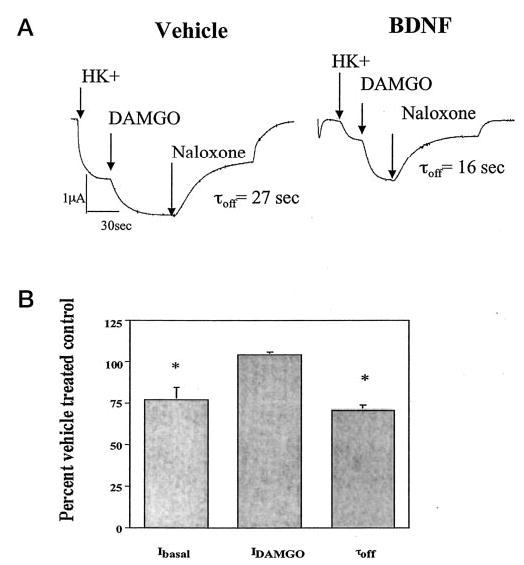
A, Xenopus oocytes were injected with cRNA for MOR, Kir3, and trkB and incubated at 19 °C for 2–4 days. On recording day, oocytes were pretreated in vehicle or 0.2 μg/ml BDNF for 6–12 min. Oocytes were voltage-clamped in two-electrode configuration at −80 mV. At the first arrow, high (96 mm) potassium buffer (HK+) was perfused into the recording chamber until the current stabilized. Then, as noted by each subsequent arrow on the trace, 1 μm DAMGO in HK+, 1 μm naloxone in HK+, and vehicle (ND96) were sequentially perfused into the chamber. pCLAMP6 software was used to fit a simple exponential equation to the naloxone reversal phase of the trace, and time constant of deactivation (τoff) was determined as described under “Experimental Procedures.” B, basal current, DAMGO-induced current, and time constant data were compiled from multiple batches of oocytes injected with Kir3.1/3.4 as potassium channel subunits. BDNF-treated oocytes were normalized to vehicle-treated controls. Data are expressed as percent of vehicle-treated controls following a 6–12-min preincubation in BDNF. trkB activation suppressed basal channel conductance and accelerated channel deactivation without affecting DAMGO-induced current amplitude.
The decrease in the τoff parameter was not a consequence of the reduced basal current amplitude; a regression analysis showed no correlation between DAMGO current amplitude and τoff (r2 < 0.1). Oocytes lacking the trkB receptor did not show significant acceleration of Kir3 deactivation or decrease in basal channel conductance following BDNF treatment (data not shown). Thus, trkB activation by BDNF reduced the basal channel conductance and accelerated the deactivation kinetics of Kir3 without reducing the maximal DAMGO-evoked effect. Because Kir3 conductance is controlled predominately by binding to free Gβγ, these results suggest that tyrosine phosphorylation affected the interaction between channel and Gβγ.
trkB Acceleration of Kir3 Deactivation Is Mediated by Enhanced GAP Activity
The kinetic profile we describe for Kir3 deactivation resembles GAP-mediated acceleration of deactivation without concomitant decrease in DAMGO current amplitude or increase in τon (18). To illustrate the kinetic similarity, we injected 10 ng of cRNA for the GAP RGS4 into oocytes expressing Kir3, MOR, and trkB 12–18 h before recording. When traces were fit to a simple exponential, Kir3 deactivation (as measured by τoff) was accelerated from 39 ± 3 s (Kir3.1/3.4, n = 28) to 27 ± 3 s (Kir3.1/3.4, n = 36) when oocytes not expressing RGS4 were pretreated in BDNF (Fig. 2). Injection of RGS4 accelerated channel deactivation to 12 ± 2 s (τoff, n = 7). BDNF treatment reduced τoff to 7 ± 1 s (n = 11). These results show that trkB modulation of deactivation kinetics is similar to the effect of the GAP RGS4, leading to the speculation that both operate by similar mechanisms.
Fig. 2. trkB acceleration of Kir3 deactivation recapitulates RGS4 kinetics.
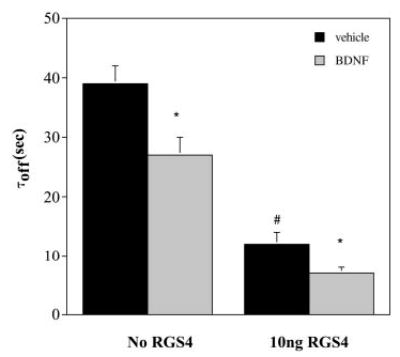
Oocytes expressing Kir3.1, Kir3.4, trkB, and MOR were injected with 10 ng of RGS4 12–16 h prior to recording. RGS4 accelerated Kir3 deactivation, an effect enhanced by trkB activation. There was no statistical difference in the time constant parameter between batches. *, p < 0.05 of BDNF effect compared with vehicle-treated control. #, p < 0.05 for RGS4-expressing oocytes compared with oocytes not injected with exogenous RGS4.
To test the hypothesis that trkB activation increased GAP activity (rather than decreasing Gβγ binding affinity for Kir3) we inhibited GTP hydrolysis by injecting GTPγS, a non-hydro-lyzable analogue of GTP (19). If Kir3 deactivation rate was controlled solely by Gβγdissociation and not Gβ γsequestration by Gαi·GDP, then BDNF-accelerated deactivation could result from a reduction in Gβγ affinity for the channel. Alternatively, if the deactivation rate was controlled by Gβγ sequestration by Gαi·GDP, then the accelerated deactivation rate would result from a BDNF-induced increased GTPase activity that would be blocked by GTPγS. In oocytes expressing MOR, trkB, and Kir3, GTPγS virtually eliminated Kir3 deactivation whether or not RGS4 was injected (Fig. 3, a and b). Pretreatment with BDNF did not accelerate deactivation kinetics of GTPγS-injected oocytes (Fig. 3c). Because BDNF pretreatment did not even partially recover deactivation in the presence of GTPγS, trkB-mediated acceleration of deactivation kinetics was probably not a result of a reduction in Gβγ affinity for the channel. These results suggest that trkB accelerates channel deactivation by facilitating GTP hydrolysis and subsequent Gβγ sequestration by Gαi.
Fig. 3. GTPγS blocks RGS4 and trkB acceleration of Kir3 deactivation.
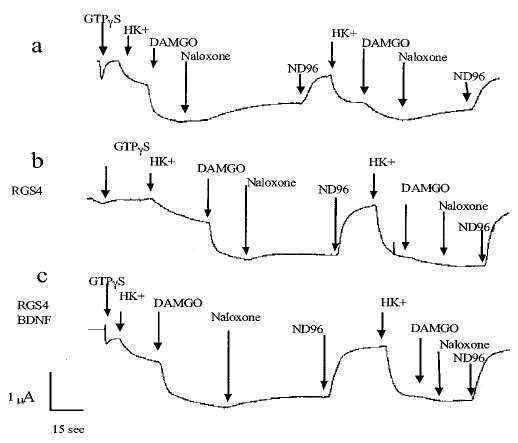
a, a representative oocyte expressing Kir3.1, Kir3.4, MOR, and trkB was injected with 120 pmol of GTPγS. DAMGO-induced increase in current did not significantly reverse during naloxone perfusion, and subsequent applications of HK+ and then DAMGO showed that the activation of Kir3 did not reverse. b, a representative oocyte also expressing RGS4 showed a similar response. c, BDNF pretreatment of a representative oocyte had no effect on deactivation kinetics. RGS4 (10 ng) was injected 12–16 h before recording. Membranes were clamped at −80 mV and 120 pmol of GTPγS was injected. High potassium buffer, DAMGO, naloxone, and vehicle were sequentially perfused as described. After current returned to baseline, the perfusion sequence was repeated. GTPγS injection significantly blocked channel deactivation.
Tyrosine Phosphorylation of Kir3 Is the Mechanism of trkB-mediated GAP Activation
Previously, we identified tyrosine residues in the Kir3 N-terminal tail that were phosphorylated by trkB and modulated Kir3 gating parameters (7). To investigate whether phosphorylation state of Kir3 controlled channel deactivation, we pretreated oocytes with the phosphatase inhibitor peroxyvanadate (100 μm) before voltage clamp experimentation. Peroxyvanadate pretreatment reduced τoff from 39 ± 3 s (n = 4) to 23 ± 5 s (n = 4). A 3-min perfusion with ND96 returned τoff to 34 ± 2 s (n = 4), approaching vehicle-treated controls (Fig. 4, A and C). Conversely, when oocytes were pretreated first in vehicle followed by a 3-min treatment in peroxyvanadate, channel deactivation was accelerated (Fig. 4B). These results suggest that constitutive phosphorylation of Kir3 may result in enhanced GTP hydrolysis.
Fig. 4. Phosphatase inhibition accelerates Kir3 deactivation.
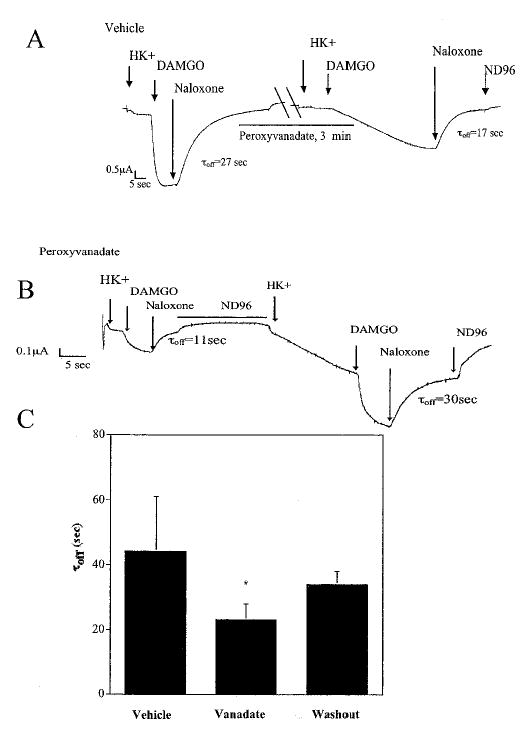
Oocytes expressing MOR, trkB, Kir3.1, and Kir3.4 were pretreated 6–10 min in vehicle of 100 μm sodium orthovanadate preactivated in 3% hydrogen peroxide for 4–5 h. Membranes were clamped at −80 mV and sequentially perfused with high potassium buffer, DAMGO, and naloxone (see arrows above trace). A, peroxyvanadate treatment accelerated channel deactivation. B, conversely, in peroxyvanadate-pretreated oocytes, naloxone reversal was initially accelerated. After 3 min of ND96 perfusion, channel 1 deactivation returned to control levels. C, these results were replicated in five oocytes, and data were summarized. *, p < 0.05 compared with vehicle-treated control.
We hypothesized that BDNF/trkB-induced phosphorylation of specific N-terminal tyrosine residues accelerated Kir3 deactivation kinetics. Deactivation kinetics were compared for Kir3.4(S143T) (a variant able to form functional homomers as described) (20) and Kir3.4(S143T/Y32F/Y53F) (a homomeric channel with both N-terminal tyrosine residues point-mutated to phenylalanines) (7). trkB activation accelerated channel deactivation in oocytes expressing Kir3.4(S143T) (vehicle, τoff = 26 ± 4 s, n = 18 and BDNF, τoff = 17 ± 1 s, n = 17). However, trkB no longer accelerated deactivation in the double tyrosine to phenylalanine mutant (vehicle, τoff = 15 ± 1 s, n = 30 and BDNF, τoff = 15 ± 1 s, n = 40, Fig. 5A). These results suggest that trkB-mediated phosphorylation of these tyrosine residues is important in facilitating GAP activity of the channel.
Fig. 5. Tyrosine phosphorylation in GAP motifs of the Kir3.4 N-terminal tail is the mechanism of trkB-mediated GAP activity.
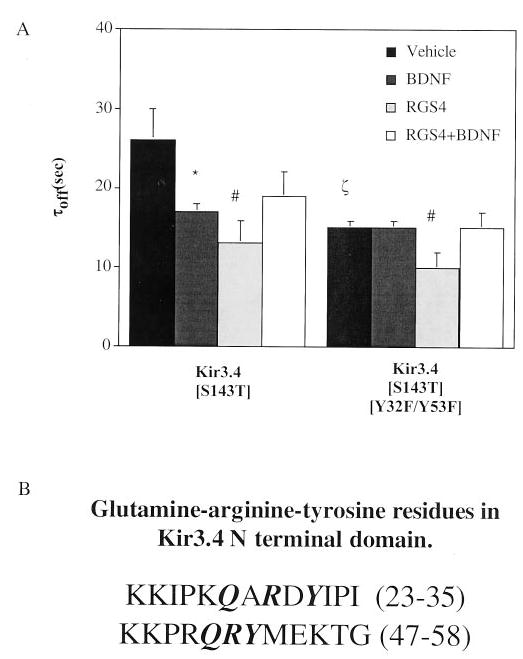
A, when tyrosines 32 and 53 were mutated to phenylalanines in Kir3.4(S143T) homomeric channels, channel deactivation was insensitive to trkB-mediated acceleration. Channel mutants were expressed in Xenopus oocytes, and two-electrode voltage clamp experiments were performed as described following pretreatment in vehicle or BDNF. Overexpression of 10 ng of RGS4 12–16 h prior to two-electrode voltage clamp recordings accelerated channel deactivation independently of Kir3 GAP activity conferred by N-terminal tyrosines. *, p < 0.05 of BDNF effect compared with vehicle-treated control. #, p < 0.05 for RGS4-expressing oocytes compared with oocytes lacking exogenously expressed RGS4. ζ, p < 0.05 for vehicle-treated oocytes expressing Kir3.4(S143T) compared with Kir3.4(S143T/Y32F/Y53F). B, Kir3.4 GAP-like tyrosine-arginine-glutamine motifs in the N terminus.
Interestingly, we found that the Kir3.4(S143T/Y32F/Y53F) mutations constitutively accelerated Kir3.4 kinetics (compare Kir3.4(S143T), τoff = 26 ± 4 s, n = 18 with Kir3.4(S143T/Y32F/Y53F), τoff = 15 ± 1 s, n = 30, Fig. 5A). These results suggest that the hydroxyl group in the tyrosine dampens GAP activity because phenylalanine substitution for tyrosine promotes GAP activity. A direct measure of GTPase activity of Kir3 was not possible because we would not have attained the high level of channel expression required (21).
Kir3 Has Potential GAP Activity
Recently, the crystal structures of G protein·GAP combinations such as Ras-Ras·GAP, Rho-Rho·GAP, and Gαi·RGS have been solved (10–12, 14, 22–24). A sequence alignment of the amino acids in the vicinity of Kir3.4 N-terminal tyrosine residues revealed that these tyrosines are part of a signature consensus sequence recurring in some GAPs to facilitate hydrolysis of GTP by directly inserting these amino acid residues into the catalytic binding pocket (Fig. 5B). RGS4 is also a GAP, but crystal structure data indicate that it does not contribute amino acid residues to the Gαi but instead holds the Gαi in a catalytically favored transition state (24). To determine whether trkB worked cooperatively with RGS4 to accelerate channel deactivation, we investigated the effect of BDNF pretreatment on the τoff parameter in oocytes expressing RGS4, trkB, Kir3 mutants, and MOR. RGS4 accelerated channel deactivation in the Kir3.4(S143T/Y32F/Y53F) mutants (τoff = 10 ± 2 s, n = 20, Fig. 5A), but BDNF had no additional effect (τoff = 15 ± 2 s, n = 18). In fact, time constants resembled BDNF-pretreated oocytes lacking RGS4 (Fig. 5A). Similar acceleration of deactivation was seen in Kir3.4(S143T) mutants (compare RGS4, τoff = 13 ± 3 s, n = 37 with RGS4+BDNF, τoff = 19 ± 3 s, n = 8). These results suggest an independent mechanism of Kir3 GAP activity for RGS4- and trkB-mediated Kir3 phosphorylation.
We conclude that Kir3.4 contains critical GAP-like amino acids and probably acts as a GAP for Gαi. GAP activity is kept in check by the tyrosine hydroxyl. Either elimination of the hydroxyl by phenyalanine substitution or phosphorylation of the hydroxyl by tyrosine kinase signaling cascades results in potentiation of GAP activity. These results suggest a novel control mechanism of channel deactivation.
DISCUSSION
The principal finding of this study is that tyrosine phosphorylation of Kir3 by trkB/BDNF reduces channel activation by accelerating GTPase kinetics. Our data did not support the alternative hypothesis that the binding affinity of channel for Gβγ was reduced by channel phosphorylation. The concept that the channel regulates the GTPase activity of the Gαi subunit is novel, although the principle of an effector providing feedback regulation of its activator is well established (15, 25).
Tyrosine phosphorylation has been described previously as a regulator of ion conductance of potassium and other ion channels (7, 26–31). Tyrosine kinase cascades are ubiquitous, and they modulate signaling in cardiac myocytes and neurons among other physiological systems (32, 33). Our study provides a novel link between Kir3 tyrosine phosphorylation and G protein-mediated signaling. To our knowledge, this is the first demonstration that tyrosine phosphorylation of an ion channel regulates intrinsic GTPase accelerating capacity.
Recent evidence shows that Gαi binds directly to the Kir3 itself, suppressing basal Gβγ binding and basal current amplitude (9, 34–38). Peleg and et al. (9) found that Kir3 Gαi interaction decreased Gβγ binding to the channel, and Gαi bound directly to the N-terminal domain residues of Kir3. The tyrosine residues masking Kir3.4 GAP activity in our study are within this domain. Further, the arginine, glutamine, and tyrosine residues in the portion of Kir3 contacting Gαi are residues found in GTPase-activating proteins shown by crystallography to be critical in stabilizing the transition state of Gαi, favoring catalysis (10, 14, 23). Glutamine and arginine residues in GAPs such as Ras·GAP, p50Rho·GAP, cdc42·GAP, and the yeast Ypt/Rab·GAP·Gyp1p are critical in mimicking residues in their respective G proteins to stabilize the transition phase of the Gαi binding pocket (10–12, 14, 23, 39). The contribution of the tyrosine residue is less clear, although some studies imply that a tyrosine hydroxyl on the G protein itself (11, 12, 15, 40) or the GAP in the case of the RGS4·Gαi interaction (24) is part of a network providing polar interaction between the G domain and GAP, facilitating catalysis. Our data support a role for the tyrosine hydroxyl in reducing rather than enhancing catalysis. Nevertheless, without crystal structure data for Kir3, speculations about the contributions of specific residues remain unverified.
The trkB facilitation of Kir3 deactivation kinetically resembles RGS4-mediated return to basal current (16, 18). Our data conform kinetically to the properties of the known GTPase-activating protein RGS4 (18, 21). Like kinetics after trkB activation, the RGS proteins elicit a similar acceleration of return to inactive heterotrimeric G protein state, presumably by mediating direct hydrolysis of GTP and resulting in a return of Gαi to the GDP-bound state (13, 18, 42, 43). Besides, if trkB induced a decrease in the affinity of Gβγ for Kir3, we would have expected trkB/BDNF at least partially to overcome blockage of GTPase activity by enhancing Gβγ dissociation from Kir3, thereby restoring deactivation kinetics in oocytes injected with GTPγS. Because BDNF/trkB did not restore GTPase activity in this experiment, we favor the hypothesis that trkB activation increases GTP hydrolysis.
CONCLUSIONS
This report provides a novel mechanism for Kir3.4 modulating its own kinetic parameters by dual GAP sequences in its N-terminal tail. GAP facilitation by phosphorylation may promote Kir3 deactivation, potentially providing a new mechanism for regulation of channel function. In opioid regulation of inflammatory pain, for example, endogenous opioids such as β-endorphin, methionine-enkephalin, and dynorphin-A are released from immune cells in inflamed tissue (44, 45). Neurotrophic factors such as BDNF and nerve growth factor are up-regulated under these conditions as well (8, 46). Further, in studies involving mice with mutant Kir3 channels (weaver), analgesia after opioid treatment is decreased, suggesting a role for Kir3 itself in opioid-mediated analgesia (41). Thus, inflammatory pain represents a scenario in which tyrosine kinase cascades coexist with G protein-coupled receptor stimulation in anatomical regions associated with pain transmission such as the dorsal horn of the spinal cord, potentially modulating neuronal excitability by a Kir3-dependent mechanism. Further, in the heart GPCRs such as angiotensin II type 1 receptor mobilize the tyrosine kinase Src, which plays a role in modulating excitability by increasing cellular calcium (32). A concomitant increase in GTPase activity following Src phosphorylation of Kir3 might likewise contribute to the increase in cardiac excitability. Thus, regulation of Kir3 functioning by tyrosine kinase cascades may provide an important means for physiologically regulating cardiac and neuronal excitability during processes such as development, inflammatory responses, synaptic plasticity, and cardiac excitability.
Acknowledgments
We thank Dr. Stephen Van Dien for advice on kinetic analyses. We also thank AMGEN for generously supplying BDNF.
Footnotes
This work was supported by United States Public Health Service Grants DA11672 from the National Institute on Drug Abuse and GM07270 from NIGMS, National Institutes of Health.
The abbreviations used are: Kir3 or GIRK, G protein-coupled inwardly rectifying potassium channel; GPCR, G protein-coupled receptor; BDNF, brain-derived neurotrophic factor; DAMGO, [d-Ala2, N-MePhe4-Gly-Ol5]enkephalin; MOR, mu opioid receptor; ND96, low potassium vehicle; HK+, high potassium buffer; RGS4, regulator of G protein signaling, subtype 4; GAP, GTPase-activating protein; trkB, tyrosine kinase receptor for BDNF.
References
- 1.Dascal N. Cell Signal. 1997;9:551–573. doi: 10.1016/s0898-6568(97)00095-8. [DOI] [PubMed] [Google Scholar]
- 2.Yi BA, Minor DL, Jr, Lin YF, Jan YN, Jan LY. Proc Natl Acad Sci U S A. 2001;98:11016–11023. doi: 10.1073/pnas.191351798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ishii M, Inanobe A, Kurachi Y. Proc Natl Acad Sci U S A. 2002;99:4325–4330. doi: 10.1073/pnas.072073399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kobrinsky E, Mirshahi T, Zhang H, Jin T, Logothetis DE. Nat Cell Biol. 2000;2:507–514. doi: 10.1038/35019544. [DOI] [PubMed] [Google Scholar]
- 5.Sui JL, Petit-Jacques J, Logothetis DE. Proc Natl Acad Sci U S A. 1998;95:1307–1312. doi: 10.1073/pnas.95.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lesage F, Guillemare E, Fink M, Duprat F, Heurteaux C, Fosset M, Romey G, Barhanin J, Lazdunski M. J Biol Chem. 1995;270:28660–28667. doi: 10.1074/jbc.270.48.28660. [DOI] [PubMed] [Google Scholar]
- 7.Rogalski S, Appleyard S, Pattillo A, Terman G, Chavkin C. J Biol Chem. 2000;275:25082–25088. doi: 10.1074/jbc.M000183200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woolf CJ, Costigan M. Proc Natl Acad Sci U S A. 1999;96:7723–7730. doi: 10.1073/pnas.96.14.7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peleg S, Varon D, Ivanina T, Dessauer CW, Dascal N. Neuron. 2002;33:87–99. doi: 10.1016/s0896-6273(01)00567-0. [DOI] [PubMed] [Google Scholar]
- 10.Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, Wittinghofer A. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 11.Rittinger K, Walker PA, Eccleston JF, Smerdon SJ, Gamblin SJ. Nature. 1997;389:758–762. doi: 10.1038/39651. [DOI] [PubMed] [Google Scholar]
- 12.Nassar N, Hoffman GR, Manor D, Clardy JC, Cerione RA. Nat Struct Biol. 1998;5:1047–1052. doi: 10.1038/4156. [DOI] [PubMed] [Google Scholar]
- 13.Kovoor A, Lester HA. Neuron. 2002;33:6–8. doi: 10.1016/s0896-6273(01)00572-4. [DOI] [PubMed] [Google Scholar]
- 14.Sprang SR. Science. 1997;277:329–330. doi: 10.1126/science.277.5324.329. [DOI] [PubMed] [Google Scholar]
- 15.Vetter IR, Wittinghofer A. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 16.Gutfreund, H. (1995) Kinetics for the Life Sciences: Receptors, Transmitters, and Catalysts, pp. 19–34, Cambridge University Press, Cambridge
- 17.Sakmann, B., and Neher, E. (1983) Single-channel Recording, pp. 135–175, Plenum Press, New York
- 18.Doupnik C, Davidson N, Lester H, Kofuji P. Proc Natl Acad Sci U S A. 1997;94:10461–10466. doi: 10.1073/pnas.94.19.10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dascal N, Ifune C, Hopkins R, Snutch TP, Lubbert H, Davidson N, Simon MI, Lester HA. Brain Res. 1986;387:201–209. doi: 10.1016/0169-328x(86)90026-4. [DOI] [PubMed] [Google Scholar]
- 20.Vivaudou M, Chan KW, Sui JL, Jan LY, Reuveny E, Logothetis DE. J Biol Chem. 1997;272:31553–31560. doi: 10.1074/jbc.272.50.31553. [DOI] [PubMed] [Google Scholar]
- 21.Mosser VA, Amana IJ, Schimerlik MI. J Biol Chem. 2002;277:922–931. doi: 10.1074/jbc.M104210200. [DOI] [PubMed] [Google Scholar]
- 22.Srinivasa SP, Watson N, Overton MC, Blumer KJ. J Biol Chem. 1998;273:1529–1533. doi: 10.1074/jbc.273.3.1529. [DOI] [PubMed] [Google Scholar]
- 23.Sprang SR. Curr Opin Struct Biol. 1997;7:849–856. doi: 10.1016/s0959-440x(97)80157-1. [DOI] [PubMed] [Google Scholar]
- 24.Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Cell. 1997;89:251–261. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- 25.Nissen P, Kjeldgaard M, Thirup S, Polekhina G, Reshetnikova L, Clark BF, Nyborg J. Science. 1995;270:1464–1472. doi: 10.1126/science.270.5241.1464. [DOI] [PubMed] [Google Scholar]
- 26.Holmes TC, Fadool DA, Ren R, Levitan IB. Science. 1996;274:2089–2091. doi: 10.1126/science.274.5295.2089. [DOI] [PubMed] [Google Scholar]
- 27.Holmes TC, Fadool DA, Levitan IB. J Neurosci. 1996;16:1581–1590. doi: 10.1523/JNEUROSCI.16-05-01581.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ling S, Woronuk G, Sy L, Lev S, Braun AP. J Biol Chem. 2000;275:30683–30689. doi: 10.1074/jbc.M004292200. [DOI] [PubMed] [Google Scholar]
- 29.Bowlby MR, Fadool DA, Holmes TC, Levitan IB. J Gen Physiol. 1997;110:601–610. doi: 10.1085/jgp.110.5.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Felsch JS, Cachero TG, Peralta EG. Proc Natl Acad Sci U S A. 1998;95:5051–5056. doi: 10.1073/pnas.95.9.5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu XM, Askalan R, Keil GJ, Salter MW. Science. 1997;275:674–678. doi: 10.1126/science.275.5300.674. [DOI] [PubMed] [Google Scholar]
- 32.Haendeler J, Berk BC. Regul Pept. 2000;95:1–7. doi: 10.1016/s0167-0115(00)00133-6. [DOI] [PubMed] [Google Scholar]
- 33.Alleva E, Santucci D. Physiol Behav. 2001;73:313–320. doi: 10.1016/s0031-9384(01)00498-x. [DOI] [PubMed] [Google Scholar]
- 34.Huang X, Morielli A, Peralta E. Cell. 1993;75:1145–1156. doi: 10.1016/0092-8674(93)90324-j. [DOI] [PubMed] [Google Scholar]
- 35.Huang CL, Feng S, Hilgemann DW. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- 36.Huang C, Slesinger P, Casey P, Jan Y, Jan L. Neuron. 1995;15:1133–1143. doi: 10.1016/0896-6273(95)90101-9. [DOI] [PubMed] [Google Scholar]
- 37.Huang C, Jan Y, Jan L. FEBS Lett. 1997;405:291–298. doi: 10.1016/s0014-5793(97)00197-x. [DOI] [PubMed] [Google Scholar]
- 38.He C, Zhang H, Mirshahi M, Logothetis D. J Biol Chem. 1999;274:12517–12524. doi: 10.1074/jbc.274.18.12517. [DOI] [PubMed] [Google Scholar]
- 39.Rak A, Fedorov R, Alexandrov K, Albert S, Goody RS, Gallwitz D, Scheidig AJ. EMBO J. 2000;19:5105–5113. doi: 10.1093/emboj/19.19.5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seewald MJ, Korner C, Wittinghofer A, Vetter IR. Nature. 2002;415:662–666. doi: 10.1038/415662a. [DOI] [PubMed] [Google Scholar]
- 41.Ikeda K, Kobayashi T, Kumanishi T, Niki H, Yano R. Neurosci Res. 2000;38:113–116. doi: 10.1016/s0168-0102(00)00144-9. [DOI] [PubMed] [Google Scholar]
- 42.Kovoor A, Chen CK, He W, Wensel TG, Simon MI, Lester HA. J Biol Chem. 2000;275:3397–3402. doi: 10.1074/jbc.275.5.3397. [DOI] [PubMed] [Google Scholar]
- 43.Keren-Raifman T, Bera AK, Zveig D, Peleg S, Witherow DS, Slepak VZ, Dascal N. FEBS Lett. 2001;492:20 –28. doi: 10.1016/s0014-5793(01)02220-7. [DOI] [PubMed] [Google Scholar]
- 44.Cabot PJ, Carter L, Schafer M, Stein C. Pain. 2001;93:207–212. doi: 10.1016/S0304-3959(01)00322-0. [DOI] [PubMed] [Google Scholar]
- 45.Cabot PJ, Carter L, Gaiddon C, Zhang Q, Schafer M, Loeffler JP, Stein C. J Clin Invest. 1997;100:142–148. doi: 10.1172/JCI119506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mannion RJ, Costigan M, Decosterd I, Amaya F, Ma QP, Holstege JC, Ji RR, Acheson A, Lindsay RM, Wilkinson GA, Woolf CJ. Proc Natl Acad Sci U S A. 1999;96:9385–9390. doi: 10.1073/pnas.96.16.9385. [DOI] [PMC free article] [PubMed] [Google Scholar]