

Abstract
Carbon monoxide (CO) is an endogenous paracrine and autocrine gaseous messenger that regulates physiological functions in a wide variety of tissues. CO induces vasodilation by activating arterial smooth muscle large–conductance Ca2+-activated potassium (BKCa) channels. However, the mechanism by which CO activates BKCa channels remains unclear. Here, we tested the hypothesis that CO activates BKCa channels by binding to channel-bound heme, a BKCa channel inhibitor, and altering the interaction between heme and the conserved heme-binding domain (HBD) of the channel α subunit C terminus. Data obtained using thin-layer chromatography, spectrophotometry, mass spectrometry (MS), and MS-MS indicate that CO modifies the binding of reduced heme to the α subunit HBD. In contrast, CO does not alter the interaction between the HBD and oxidized heme (hemin), to which CO cannot bind. Consistent with these findings, electrophysiological measurements of native and cloned (cbv) cerebral artery smooth muscle BKCa channels show that CO reverses BKCa channel inhibition by heme but not by hemin. Site-directed mutagenesis of the cbv HBD from CKACH to CKASR abolished both heme-induced channel inhibition and CO-induced activation. Furthermore, on binding CO, heme switches from being a channel inhibitor to an activator. These findings indicate that reduced heme is a functional CO receptor for BKCa channels, introduce a unique mechanism by which CO regulates the activity of a target protein, and reveal a novel process by which a gaseous messenger regulates ion channel activity.
Keywords: vascular smooth muscle, vasodilation, potassium channels, signal transduction
Large-conductance Ca2+-activated potassium (BKCa) channels regulate the physiological functions of many tissues, including smooth muscle, neuronal and endocrine cells.1 BKCa channels are typically composed of pore-forming α subunits that are encoded by the Slo1 (or KCNMA1) gene, and accessory β subunits that modulate channel gating.2 In smooth muscle cells, BKCa channels regulate cellular membrane potential and, thus, Ca2+ entry through voltage-gated Ca2+ channels, providing a mechanism to control contractility.3
BKCa channel activity is regulated by a variety of signaling molecules, including intracellular Ca2+ ([Ca2+]i), protein kinases,4-6 tyrosine kinases,7 cytochrome P-450 metabolites of arachidonic acid,8 and heme.9 BKCa channels are also activated by physiologically relevant gases, including O2, CO, and NO. Although these gases can use cellular signaling pathways, O2, CO, and NO also activate BKCa channels in cell-free membrane patches isolated from the intracellular milieu.10-12
Carbon monoxide is a physiological paracrine and autocrine messenger and neurotransmitter produced by heme oxygenase (HO) catalyzed metabolism of heme.13-15 Heme is found in virtually all cell types, and many cell types contain HO-2, including arterial smooth muscle cells, endothelial cells, and neurons. CO regulates a variety of physiological processes, including growth, smooth muscle contractility, neuronal excitability, and apoptosis.12,13,16 CO generated in both endothelial and smooth muscle cells induces vasodilation by activating BKCa channels.12,16,17 Recently, O2 has been reported to stimulate BKCa channels by acting as a substrate for HO-2, leading to the generation of CO, the downstream channel activator.11 Because CO activates arterial smooth muscle and carotid body BKCa channels in excised membrane patches,11,12,18,19 CO may bind directly to the channel protein itself or to a tightly associated regulatory molecule. However, how relatively inert CO interacts with and activates BKCa channels is unclear.
Free heme regulates human BKCa channel activity by binding with high affinity to a conserved amino acid sequence (CXXCH, where X is any amino acid) that is located between the 2 regulator of conductance for K+ (RCK) domains present in the α subunit.9,20 A recognized property of reduced heme, particularly when contained within heme proteins, is its ability to bind diatomic gases, including CO. Thus, we tested the hypothesis that CO activates BKCa channels by binding to heme and modifying its interaction with the α subunit heme-binding domain.
Materials and Methods
Thin-Layer Chromatography
Native heme-binding pocket peptide (DAKEVKRAFFYCKACHDDITDPK, BKPP) and double mutant (C to S and H to R) peptide (DAKEVKRAFFYCKASRDDITDPK; BKMP) were dissolved in water (stock solution: 1 mmol/L). Peptides (10 μL) were applied 2 cm from the bottom edge of 5×20 cm silica gel 60 plates and allowed to dry. Deoxygenated Krebs (dKrebs) was prepared by vigorously diffusing N2 through Krebs solution in a parafilm-sealed Erlenmeyer flask. Fe2+-heme solution was prepared by dissolving Fe3+-hemin in basic (pH 8) dKrebs and reducing it with sodium dithionate (SDT) (2 mmol/L). Exposure of porphyrins to light and air were minimized. Heme (10 μL, 1 mmol/L) was applied 3 cm from the bottom edge (1 cm above the peptide), and the plate was immediately placed in a gas controlled, environmental chamber development tank. The chromatography solvent was ethanol/isopropanol/PBS (15/15/70). Solvent migration time was 2 hours in atmospheres of either 100% N2, 100% CO, or 10% CO and 90% N2 (data for 100% and 10% CO were the same and, thus, combined as CO). Under these conditions, heme does not move, but the peptide migrates into the heme spot. If the peptide binds to the heme, its migration will be either stopped or slowed. Plates were dried in room air and peptides stained with naphthol blue black (500 mg dissolved in methanol:acetic acid:water [45:10:45], 3 minutes), rinsed with water (3 minutes), and destained with methanol:acetic acid:water (90:2:8). Following digital imaging, line scan intensity was quantified with NIH Image.
Spectrophotometry
CO was prepared as a saturated solution (1 mmol/L) by vigorously bubbling distilled water with 100% CO for 2 hours under a headspace gas of 100% CO. CO dilutions were produced by injecting CO (1 mmol/L) into aqueous solutions using syringes without air space. CO concentrations in solutions were measured by gas chromatography–mass spectrometry. Under reducing conditions (0.5 mmol/L SDT and anoxic Krebs), CO (1 mmol/L) was added to heme to make final concentrations of 20, 40, and 100 μmol/L. The stock solution of BKPP peptide (1 mmol/L) was prepared in water and stored at −20°C. For experimentation, BKPP peptide was dissolved in dKrebs to final concentrations of 20 to 100 μmol/L. All solutions were protected from air and light, and the spectra were taken immediately after mixing in a parafilm-sealed optical cuvette with N2-purged headspace. To investigate interactions between heme and CO under reducing conditions, the absorption spectrum of heme solution was recorded first, then CO was added to make a final concentration of 20 to 100 μmol/L in the cuvette, and spectra were recorded again. To investigate the effects of BKPP on heme-CO interactions under reducing conditions, BKPP was dissolved in dKrebs to make final concentrations of 20 to 40 μmol/L and was added to the heme solution: (1) in the absence of CO; (2) immediately before CO; and (3) immediately after CO; and the absorption spectra of heme solutions were recorded. To investigate interactions among hemin, CO, and BKPP under nonreducing conditions, the above experimental protocol was repeated in the absence of SDT. Scans were performed using an Ultraspec 2100 UV/visible spectrophotometer (Amersham, Piscataway, NJ) and analyzed using Biochrom Data Capture spreadsheet interface software. Each experiment was repeated at least 5 times.
Mass Spectrometry
A Micromass Qtof2 mass spectrometer with a nanoelectrospray ionization probe was used for mass spectrometry (MS), spraying 20 to 150 μmol/L peptide or protein solutions. For MS, collision energies of 5 or 8 V were used. The voltages were kept very low to enable peak visualization of weakly bound ion complexes. Spray needle voltage was 1.8 kV, and cone voltage was 5V. Solutions were sprayed in 25 mmol/L ammonium bicarbonate, 12% to 25% acetonitrile. In all cases, stable native protein or peptide charge states were formed. For MS-MS, progressively increasing voltages were used while fixing on 1 precursor m/z and scanning the fragments. MS-MS plots demonstrated affinity of the heme for peptide or protein.
Hemin solutions were made in small amounts of 0.2 N ammonium hydroxide, with final dilution to 1 mg/mL and 70% acetonitrile, and added to 1 mmol/L BKPP. Oxygen was kept from solutions by N2 purging. Dithiothreitol was used at 2 to 5 times hemin on a molar basis to reduce the hemin to heme. Myoglobin was dissolved in 50 mmol/L ammonium bicarbonate (500 μmol/L). SDT at a 1.3-fold higher concentration than myoglobin was used to reduce the integral heme iron, followed by desalting on a Sephadex G-25 column.
Electrophysiology
Smooth muscle cells were isolated from resistance-size (≈150 μm) rat cerebral arteries using an enzyme procedure that was similar to a procedure that has been previously described.21 Cos-1 cells were transfected with cloned rat cerebral artery smooth muscle cell slo (cbv1) or cbv1, in which CKACH was mutated to CKASR between amino acids 612 and 616. Channel currents were measured from inside-out membrane patches using an Axopatch 200B patch-clamp amplifier and a Digidata 1322A. Bath solution contained (in mmol/L): 140 KCl, 5.2 CaCl2, 1 MgCl2, 5 EGTA, 1.6 HEDTA, and 10 HEPES (pH 7.2); and pipette solution contained (in mmol/L): 140 KCl, 2 MgCl2, 2 CaCl2, and 10 HEPES (pH 7.4). The free [Ca2+] bathing the intracellular membrane surface was 10 μmol/L, as confirmed with a Ca2+-sensitive and reference electrode (Corning). Following preparation, bath solutions were placed in a gas impermeant container and continually perfused through the patch-clamp chamber at a rate of 4 mL min−1. O2 pressure in the patch-clamp chamber was monitored using a PO2 electrode (Extech Instruments). Dilution of heme to a 100 nmol/L concentration in bath solution was accompanied by addition of 2 μmol/L dithionite. Dithionite (2 μmol/L) did not alter BKCa channel activity (data not shown). BKCa channel open probability (Po) was calculated from the following equation: Po=NPo/N, where N is the total number of channels in the patch (determined by application of 100 μmol/L free Ca2+ at +40 mV). Where appropriate, data were fit with a Hill equation: , where Po is the open probability, Kd is the dissociation constant, n is the Hill coefficient, and Pmax is maximal Po.
cDNA Synthesis From Cerebral Artery Smooth Muscle Cells
Following isolation from rat basilar and middle-cerebral arteries,22 smooth muscle cells were identified under an inverted microscope using Hoffmann optics and individually aspirated into Eppendorf tubes containing ≈50 μL of PicoPure extraction buffer (Arcturus). Total RNA from ≈100 smooth muscle cells was isolated using the PicoPure Kit (Arcturus), and first-strand cDNAs were reverse transcribed by SuperscriptII Reverse transcriptase with oligo(dT) primer (Invitrogen).
Polymerase Chain Reaction Amplification of slo1-Conserved Fragment and 3′-RACE
Amplification of rat cerebral artery smooth muscle cell slo1 (KCNMA1) fragments was performed using the following polymerase chain reaction (PCR) primers: forward, 5′-gca agt gat gcc aaa gaa gtt a-3′; reverse, 5′-atc tgt cca ttc cag gag gt-3′. PCR amplification was performed on an Amplitron II-Thermocycler (Barnstead Thermolyne) using 2 μL of the first strand cDNA in a final volume of 50 μL of system with 2 U of Platinum TaqDNA polymerase (Invitrogen). Based on the amplified rat slo1 fragment sequences, we designed 2 primers: GSP1, cca gtc tgt ctc att cct ccc ac; GSP2, ctg tcc act cca tcc cgt cca to conduct 3′RACE. Target 3′ cDNA ends were amplified by 2 rounds of PCR under optimal conditions using the Invitrogen 3′-RACE System.
Full-Length slo1 Amplification and Sequence Analysis
After comparing the sequences of slo1 3′ cDNA ends of rat cerebral artery smooth muscle cells, another 2 primers for first-round PCR were designed: 5 out, tcc tcc tct tcc tcc tcg tcc tcg g; 3 out, acg tca cca ttt atg cag ttt gtc ag. For the second round of PCR, primers were as follows: 5 in, gtc cac gag ccc aag atg gat gcg c; 3 in, cct ggg aat caa cat tca tct tca act tc. Target cDNAs were amplified by nested PCR in optimal conditions; first-round PCR was conducted in a total volume of 50 μL containing the following: 20 mmol/L Tris-HCl (pH 8.4); 50 mmol/L KCl; 2 mmol/L MgCl2; 300 nmol/L primer 5 out; 300 nmol/L primer 3 out; 200 μmol/L of each dNTP; 2 μL cDNA; and 1.5 U of DNA polymerase (Expand High Fidelity PCR system; Roche).
A band of ≈3.6 kb was amplified, rescued, and ligated to the pCR-XL-Topo vector (Invitrogen). Using restriction enzyme mapping and sequence analysis, we screened 2 slo1 isoforms (from ≈18 clones) that contained the 3.6-kb insert. After full sequencing, information corresponding to the 2 isoforms of rat cerebral artery myocyte slo1 (termed cbv1 and cbv2) was deposited into GenBank (AY330293 and AY330294).
Cbv1 Insertion for Mammalian Expression and Mutagenesis
PCR primers were designed starting from the second putative Met: forward, cac caa gat gga tgc gct cat cat ccc; reverse, tct gta aac cat ttc ttt ttt ctg ttt gtc gcg. A Kozak sequence was introduced at the 5′ end to improve expression in mammalian cells. Amplified PCR fragments were then ligated directly to the pcDNA3/V5/His-TOPO vector (Invitrogen).
A double mutation was introduced in cbv1 to eliminate the heme binding motif (CKACH to CKASR), using Quickchange (Stratagene). The oligo sequences used were as follows: forward, gca ttt ttt tac tgc aag gcc tct cgt gat gac gtc ac; reverse, gtg acg tca tca cga gag gcc ttg cag taa aaa aat gc. Fidelity of desired mutations and absence of unwanted mutations was verified by sequencing.
Cell Culture and Transfection
Cos1 cells were maintained in DMEM (Cellgro) supplemented with 10% FBS (Gibco) and 1% penicillin/streptomycin. pCDNA3 encoding cbv1 and pEGFP-C3 were cotransfected into Cos1 cells using FuGENE 6 (Roche). Cells were cultured at 37°C in 95% air/5% CO2 and used between 24 and 96 hours following transfection. Transfected cells were positively identified by their green fluorescence; enhanced green fluorescent protein was excited at 450 to 490 nm and light visualized at 500 to 550 nm.
Statistics
Summary data are expressed as mean±SEM. ANOVA, paired or unpaired Students t, Student–Newman–Keuls, and/or Wilcoxon matched-pairs signed-rank test were performed according to experimental design and data distribution (Kolmogorov and Smirnov distance). P<0.05 was considered significant.
An expanded Materials and Methods section is provided in the online data supplement available at http://circres.ahajournals.org.
Results
To investigate whether CO can interfere with binding of heme to the BKCa channel heme-binding domain, a novel thin-layer chromatography (TLC) approach was used. A 23 amino acid peptide corresponding to the hslo sequence23 that contained the conserved heme-binding domain (CKACH) was constructed (BK>Ca channel pocket peptide, BKPP). BKPP was spotted on silica gel TLC plates and solvent progression carried the peptide to immobile heme where it either bound or migrated through. Sample TLC chromatograms illustrating passage of BKPP in a N2atmosphere or a CO containing atmosphere are shown in Figure 1A. Relative BKPP migration through reduced (ferrous, Fe2+) heme (hereafter termed heme) or oxidized (ferric, Fe3+) heme (termed hemin) were compared with freely migrating peptide (no heme or hemin) on the same plate (Figure 1B and 1C). In a N2 atmosphere, only ≈12% of BKPP passed through heme, whereas in a CO-containing atmosphere, BKPP passage increased to ≈63%. In contrast, CO did not alter the block of BKPP migration by hemin, to which CO cannot bind. To investigate whether heme blocked BKPP migration by binding to the heme-binding domain, a peptide containing a double mutation in the heme-binding domain (CKACH to CKASR, termed BKMP) was constructed. A similar mutation has been shown to abolish heme inhibition of BKCa channels.9 Even in a purely N2 atmosphere, heme did not prevent migration of BKMP.
Figure 1.
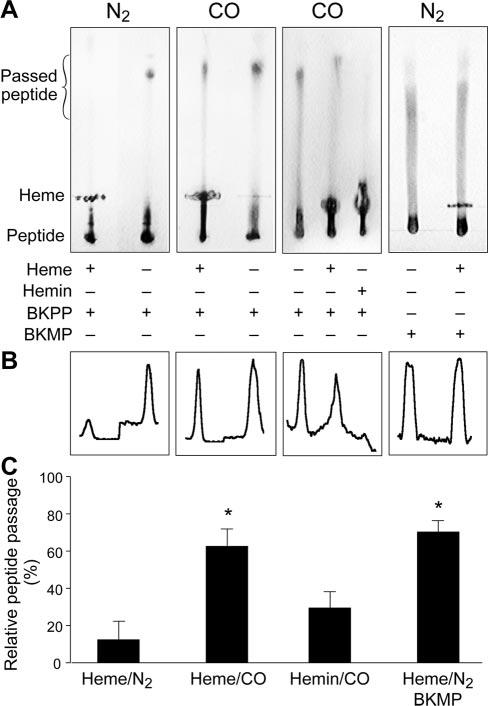
Binding of 23 amino acid heme-binding domain-containing peptides (BKPP or BKMP) to heme or hemin, detected by TLC. Peptides migrate, but heme and hemin are immobile. A, In an N2 environment, heme blocked migration of the BKPP, indicating binding (first plate). In a CO-containing atmosphere, BKPP passed through heme, indicating binding was attenuated (second and third plate). However, hemin blocked BKPP migration even in a CO atmosphere (third plate). Heme did not prevent BKMP migration even in a N2 atmosphere (fourth plate). The area marked passed peptide illustrates where line scans were obtained to collect data for B and C. B, Density plots for plates illustrated in A. C, Summary data of all trials; n=8, 8, 6, 14, respectively. *P<0.05 compared with BKPP/heme/N2.
Spectrophotometry was used as a complimentary approach to investigate heme to peptide molecular interactions. Heme produced an absorbance peak at ≈390 nm (the Soret band). Addition of CO (20 to 100 μmol/L) to heme produced a concentration-dependent increase in a peak at ≈410 nm, indicating CO binding to heme (Figure 2A through 2D). In contrast, addition of CO (20 to 100 μmol/L) to hemin did not alter the spectra, suggesting no interaction (data shown for 20 μmol/L CO in Figure 2A). Addition of BKPP (20 μmol/L) to a heme–CO mixture, or addition of CO to a heme–BKPP mixture, reduced the 410-nm absorption peak (Figure 2C and 2D). In contrast, BKPP (20 to 100 μmol/L) alone did not alter heme spectra (data not shown).
Figure 2.
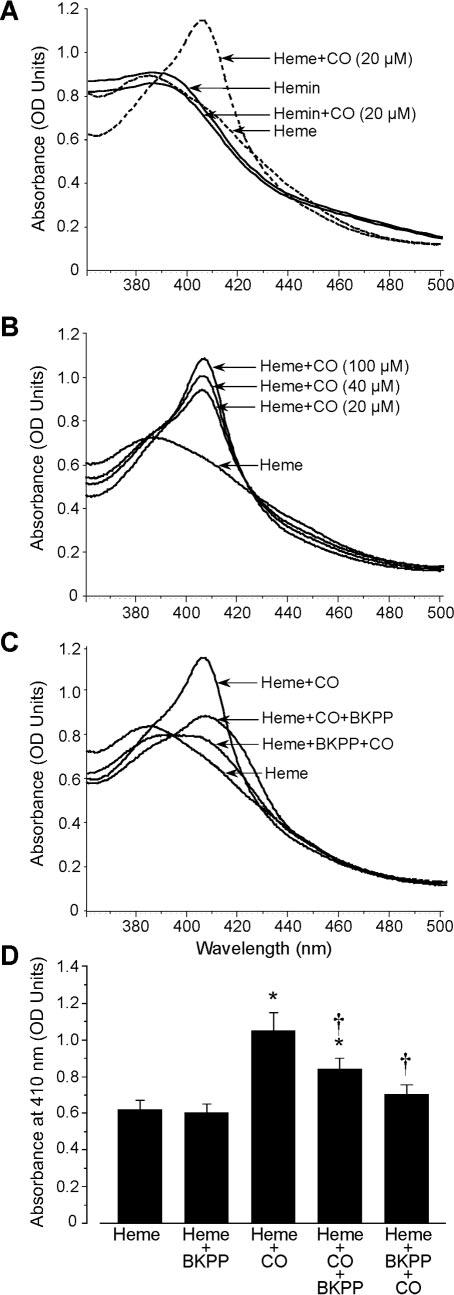
Absorbance spectra of heme and BKPP in the absence and presence of CO. A, CO (20 μmol/L) right shifted the major absorbance peak of heme, suggesting interaction. In contrast, addition of CO (20 μmol/L) to hemin did not alter spectra, suggesting no interaction. B, CO increased heme absorbance at ≈410 nm in a concentration-dependent manner. C, Effect of CO (20 μmol/L) and BKPP on the heme spectrum. Either CO was added to heme and then the mixture pooled with peptide or peptide was combined with the heme first, as indicated. D, Absorbance at 410 nm for combinations in C (n=5 for each). In A through D, heme and hemin concentrations were 20 μmol/L. *P<0.05 compared with control heme absorbance, †P<0.05 compared with heme-CO absorbance.
To specifically identify heme bound to BKPP, electrospray ionization mass spectrometry was used. Figure 3A unequivocally identifies a molecule comprised of heme bound to BKPP, the theoretical mass matching the experimental. To determine relative binding of heme to BKPP and to BKMP, we measured the proportion of peptide with adherent heme. Heme was added to peptide in a 2-to-1 ratio. As expected, many more BKPP molecules bound heme (28±6%) than did BKMP molecules (7±2%), consistent with the TLC results. Next, MS-MS techniques were used to investigate the effect of CO on the binding of heme within myoglobin, a native heme protein, and the effect of CO on the binding of heme to BKPP. For myoglobin, collision energy curves show that CO causes release of heme from the protein at lower energies than when CO is absent (Figure 3B). Heme and BKPP produce a multicomponent curve (Figure 3C). Some heme binds loosely with only low collision energies required to separate it from BKPP. The strength of this low-affinity binding is increased by CO. However, some heme binding is much stronger, and this high affinity binding is abolished by CO (Figure 3C, inset).
Figure 3.
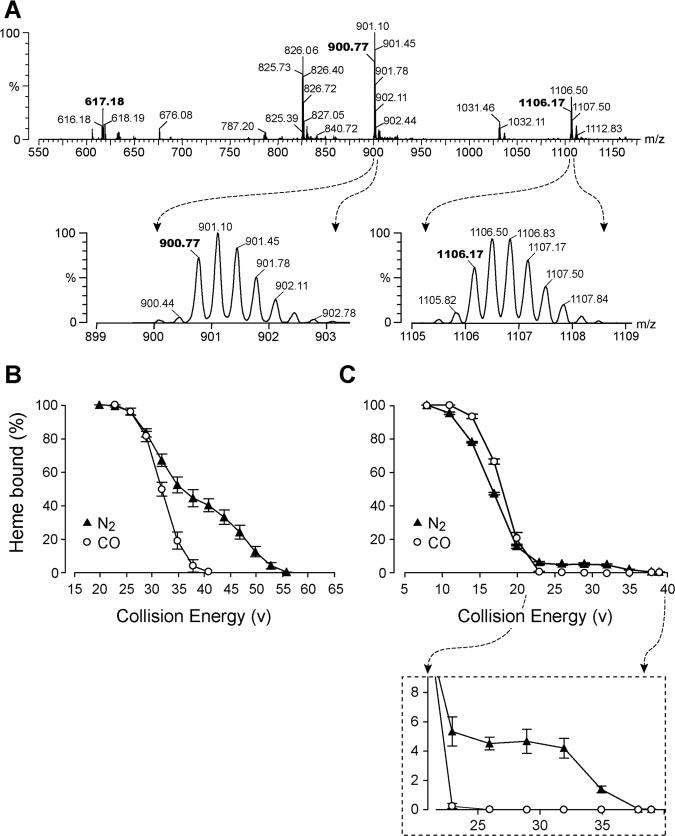
A, MS spectra of heme bound to BKPP. The upper trace is abundance from 550 to 1200 m/z. The molecular weight of the reduced heme 1+ ion is 617.18. The reduced peptide molecular weight is 2699.28, shown here as a 900.77m/z, 3+ ion. Addition of heme to the peptide yields 1106.17 m/z, 3+; 826 is a 3+ ion fragment in which PK (pro-lys) at the C terminal of BKPP is removed. The 2 magnified peaks below the spectral scan show the detailed isotopes of BKPP and BKPP with bound heme, proving the 3+ charge state of each. That is, the inverse of the difference between any 2 isotopic peaks is the charge state (1 /0.33 mz=3+). The 2 very small isotopic peaks preceding both the 900.77 and the 1106.17 reduced isotopic peaks are oxidized forms, indicating the nearly completely reduced condition of the compounds. B, CO alters the affinity of heme for myoglobin. The affinity of reduced heme for myoglobin and the effect of CO on that affinity were determined by elevating collision energy of the mass spectrometer. Heme was released once the collision energy of the mass spectrometer exceeded the affinity of heme for myoglobin. CO lowered the affinity of heme for myoglobin (n=4). C, CO alters the affinity of heme for BKPP (n=4). The plot shows an initial increase in heme/BKPP affinity in the presence of CO, then significantly less binding at higher collision energy levels (expanded as an inset below Figure 3C).
Because CO binds to heme, and heme binds to BKCa channels, CO may activate BKCa channels by binding to channel associated heme. Therefore, the activity of single cerebral artery smooth muscle cell BKCa channels were recorded using the inside-out patch-clamp configuration with 10 μmol/L free Ca2+ at the intracellular membrane surface. At +40 mV, hemin reduced BKCa channel open probability (Po) with a half-maximal inhibitory concentration of 5.1 nmol/L and a Hill coefficient of 1.03 (Figure 4A and 4B). BKCa channel inhibition by hemin (100 nmol/L) was similar at −40 (to 3.9±1.2% of control; n=7, P<0.05) and +40 mV (Figure 4A through 4C).
Figure 4.
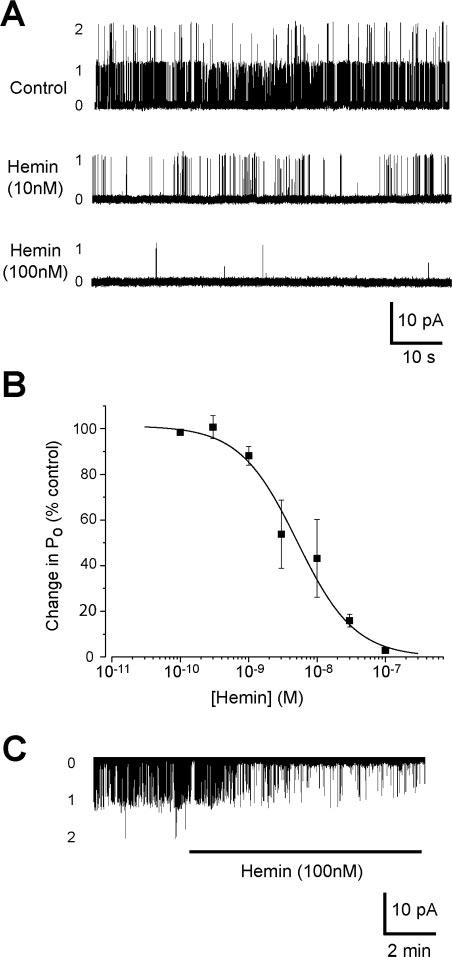
Heme and hemin inhibit BKCa channels in inside-out membrane patches from cerebral artery smooth muscle cells. Bath (ie, intracellular) free [Ca2+] was 10 μmol/L. A, Original recordings at a membrane potential of +40 mV illustrating concentration-dependent BKCa channel inhibition by hemin. B, Concentration-dependent BKCa channel inhibition by hemin at +40 mV. Solid line illustrates Boltzmann fit to the data; n=1, 3, 6, 4, 5, 3, and 11 for 100 pmol/L, 300 pmol/L, 1 nmol/L, 3 nmol/L, 10 nmol/L, 30 nmol/L, and 100 nmol/L, respectively. C, Hemin inhibits BKCa channels at −40 mV. *P<0.05 compared with control.
Subsequent electrophysiological experiments were performed with a physiological intracellular PO2 of 20 mm Hg that would lessen oxidation of reduced heme to hemin. Reducing O2 from 150 mm Hg (ambient air) to 20 mm Hg decreased mean BKCa channel Po from 0.32±0.08 to 0.12±0.04, or to 45.7±6.8% of control (n=17, P<0.05), consistent with previous reports.11 With a PO2 of 20 mm Hg, hemin and heme (100 nmol/L each) reduced mean channel Po similarly (Figure 5A and 5C). More importantly, and consistent with the spectra and TLC data, CO activated BKCa channels that were inhibited by heme, but CO did not alter the activity of channels inhibited by hemin (Figure 5A and 5C). Moreover, following CO-induced BKCa channel activation, addition of heme further activated BKCa channels, suggesting CO–heme is an activator (Figure 5B and 5D). In contrast, hemin blocked BKCa channels when applied in the continued presence of CO (Figure 5B and 5D).
Figure 5.
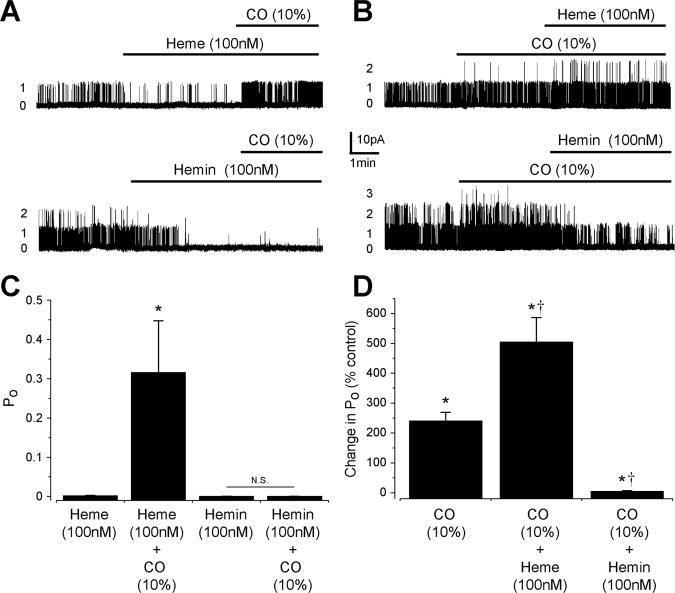
CO reverses BKCa channel inhibition by heme but not by hemin. A, Original recordings of arterial smooth muscle BKCa channels at +40 mV. Heme blocked BKCa channels, and subsequent addition of CO (10%) induced channel activation. Hemin also blocked BKCa channels, but subsequent addition of CO did not alter channel activity. B, Original recordings illustrating BKCa channel activation by CO and subsequent effects of heme or hemin addition. C, Summary data indicating CO reverses inhibition of BKCa channels by heme (n=6) but does not alter the activity of channels inhibited by hemin (n=4). *P<0.05 compared with heme. D, Mean effects of CO (10%, n=11) and subsequent heme (n=6) or hemin (n=5) addition. *P<0.05 compared with 100%, †P<0.05 compared with CO.
To investigate whether CO activates BKCa channels by binding to α subunit–associated, endogenous heme, BKCa channels were cloned from smooth muscle cells of rat cerebral arteries and termed cbv1 and cbv2. Sequence analysis of full-length cbv1 (GenBank accession no. AAP82453) and cbv2 (GenBank accession no. AAP82454) indicated that the conserved heme-binding domain (CKACH) is located between amino acids 612 and 616. Cbv1 and cvb2 channels expressed in cos-1 cells exhibited properties similar to native cerebral artery smooth muscle cell BKCa channels, including slope conductance, voltage-dependence, Ca2+ sensitivity, and pharmacology (online data supplement). As illustrated in Figure 6, wild-type cbv1 channels were inhibited by heme (500 nmol/L and 5 μmol/L) and activated by CO, consistent with regulation of native arterial smooth muscle cell BKCa channels (Figure 5). In addition, CO applied in the continued presence of heme was a more effective cbv1 channel activator than CO alone. In contrast, heme-binding domain mutated cbv1 channels were insensitive to heme (500 nmol/L and 5 μmol/L), CO (10%), and the combination of CO+heme (Figure 6B and 6D).
Figure 6.
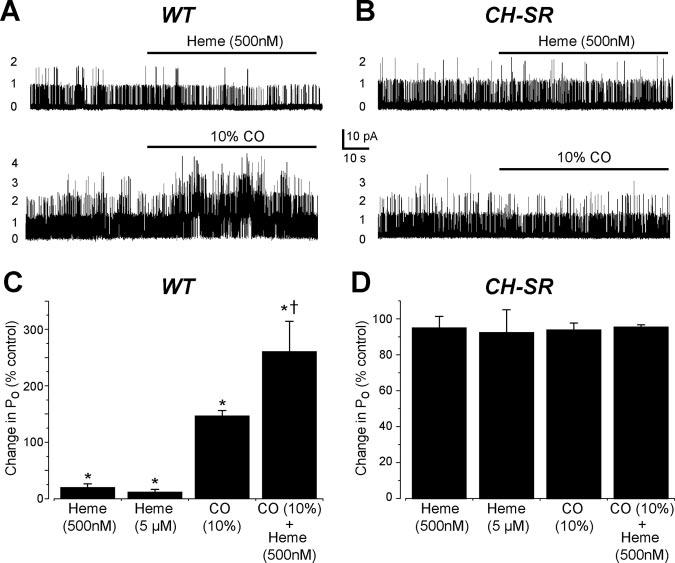
Mutation of the BKCa channel heme-binding domain abolishes heme inhibition and CO activation. A, Original recordings of cbv1 BKCa channels measured in inside-out patches illustrating inhibition by heme (500 nmol/L) and activation by CO (10%). WT indicates wild type. B, A 2 amino acid mutation of the heme-binding domain (CH to SR [CH-SR]) abolishes heme-induced cbv1 channel inhibition and CO activation. C, Summary data illustrating effects of heme (500 nmol/L, n=6; 5 μmol/L, n=6), CO (n=4), and CO+heme (n=10) on cbv1 channels. D, Mean data illustrating effects of heme (500 nmol/L, n=8; 5 μmol/L, n=8), CO (n=8), and CO+heme (n=13) on mutant cbv1 channels. *P<0.05 compared with 100%, †P<0.05 compared with CO.
Discussion
The current report provides direct experimental evidence for a mechanism by which CO interacts with and activates BKCa channels. Data indicate CO activates BKCa channels by binding to endogenous channel-bound heme and altering the interaction between heme and the heme-binding domain.
CO activates arterial smooth muscle and carotid body BKCa channels in excised membrane patches and activates cslo and mslo channels when expressed in the absence of auxiliary β subunits.11,18,19,24 Thus, β subunits are not required for CO-induced BKCa channel activation, indicating that CO acts on the pore-forming α subunit itself or another tightly associated regulatory molecule. The hypothesis that CO may influence activity by binding directly to BKCa channels has been proposed.25,26 However, under physiological conditions CO should not bind directly to proteins, and no direct experimental evidence or chemical mechanism to support such binding and activation has been obtained.12
Cellular heme is produced in the reduced state and, being hydrophobic, incorporates readily into lipid bilayers.27 Data indicate that native ferrous heme bound to amino acids 612 to 616 of the BKCa channel provides a receptor for CO. Within the heme-binding domain is an integral histidine that binds the iron center of heme. Mutation of the histidine within the heme-binding domain prevented hemin-induced BKCa channel inhibition.9 In other studies, chemical modification of histidines abolished CO-induced BKCa channel activation.26,28 Thus, to investigate the importance of the heme-binding domain in CO-induced BKCa channel activation, cbv1 channels were constructed that contained a 2 amino acid mutation in the heme-binding domain, similarly to the mutation present in the BKMP (ie, from CKACH to CKASR). Mutation of the histidine and the proximal cysteine that provides disulphide bridge binding with the heme porphyrin markedly attenuated heme–BKPP binding, the ability of heme to block BKCa channels, and the ability of CO to activate BKCa channels. In addition, BKCa channels with native heme-binding domains are insensitive to CO if the heme-binding domain is occupied with hemin that does not bind CO. Binding of CO to heme reduces the inhibitory interaction and may generate an activator configuration, leading to the previously reported increase in BKCa channel Ca2+ sensitivity.19 In smooth muscle cells, an increase in BKCa channel Ca2+ sensitivity would enhance coupling to Ca2+ sparks and increase transient BKCa current frequency and amplitude, leading to membrane hyperpolarization and, ultimately, vasodilation.17,19 CO-to-heme binding is a readily reversible carbon monoxygenation that is dependent on CO partial pressure. Therefore, activation of cellular CO production would increase PCO, leading to an increase in CO to heme binding and BKCa channel activation, whereas reducing PCO would lead to the removal of CO from channel-bound heme and a decrease in BKCa channel activity.
CO changed the binding curve of heme to BKPP; therefore, its shape is similar to that of myoglobin with CO. As a short sequence, BKPP does not have the binding strength of native myoglobin; therefore, the curve is shifted left. Although the shape of the N2 (non-CO) curves are different for myoglobin and BKPP, they both have higher affinity binding than their CO counterparts. The multicomponent curve of BKPP and heme strongly suggests different kinds of heme to BKPP binding can occur under the nonphysiological conditions necessitated by MS. Some heme binds relatively loosely, perhaps nonspecifically, and low collision energies separate it from the peptide. This low affinity binding is actually strengthened by CO. Beyond this, ≈10% of the heme adheres much more strongly to the peptide, and this adherence is virtually eliminated by CO.
Hemin has been demonstrated to regulate heterologously expressed hslo1 channels,9,20 but whether hemin or heme regulate native BKCa channels was unknown. Thus, we investigated hemin, heme, and CO regulation of BKCa channels in smooth muscle cells of small cerebral arteries that regulate blood pressure and flow. In smooth muscle cells, BKCa channels are activated by Ca2+ sparks that elevate intracellular Ca2+ within the micromolar concentration range.3,29,30 Therefore, regulation was studied with 10 μmol/L Ca2+ present at the BKCa channel inner-membrane surface. Data show that the half-maximal inhibitory concentration of heme for arterial smooth muscle BKCa channels was approximately 1-order of magnitude lower than that previously described for hslo channels, suggesting higher heme sensitivity.9 One explanation for different heme sensitivity may be a valine for isoleucine substitution 3 amino acids distal to the histidine in the heme-binding domain of the rat BKCa channel (GenBank accession nos. AAA92290 and AY330293).23 Although just outside the conserved heme-binding domain, this substitution may improve heme accessibility to critical amino acids. However, native rat cerebral artery smooth muscle BKCa channels were also noticeably more sensitive to hemin and heme than cbv1 channels when expressed in cos cells. Another potential explanation for the difference in heme sensitivity is that cerebral artery smooth muscle cells are enriched in β1 subunits.31 Although β subunits are not obligatory for heme inhibition, the presence of these accessory channel subunits may increase heme sensitivity. The fact that both heme and β1 subunits alter BKCa channel Ca2+ sensitivity1,2,20,31 is consistent with this hypothesis.
HO and BKCa channels are membrane colocalized.11 Conceivably, HO activation may not only generate CO, a BKCa channel activator, but may also reduce membrane associated heme, a BKCa channel blocker, to generate the CO. Both of these effects would elevate BKCa channel activity. Thus, it is possible that compartmentalization of the CO generator (HO), CO receptor (heme), and downstream target (BKCa channel) may regulate cellular excitability through more than 1 local signaling pathway.
O2 regulates BKCa channel activity by acting as a reactant for HO catabolism of heme.11 However, O2 also regulates BKCa channel activity when NADPH, another obligatory reactant, is absent, and thus HO cannot metabolize heme.11 Thus, additional HO-independent O2 sensors appear to exist for BKCa channels. Conceivably, heme may also act as a binding site for other gaseous messengers, including O2. As such, heme may act as a receptor that regulates BKCa channel activity and thus cellular excitability in response to diverse physiological and pathological stimuli. In summary, data here indicate that heme is a functional CO receptor for BKCa channels.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health and the American Heart Association. We thank Robert Appling, Jane Dickerson, and Alex Fedinec for technical assistance with TLC and Dominic Desiderio for mass spectrometric equipment.
References
- 1.Orio P, Rojas P, Ferreira G, Latorre R. New disguises for an old channel: MaxiK channel beta-subunits. News Physiol Sci. 2002;17:156–161. doi: 10.1152/nips.01387.2002. [DOI] [PubMed] [Google Scholar]
- 2.Tanaka Y, Koike K, Alioua A, Shigenobu K, Stefani E, Toro L. Beta1-subunit of MaxiK channel in smooth muscle: a key molecule which tunes muscle mechanical activity. J Pharmacol Sci. 2004;94:339–347. doi: 10.1254/jphs.94.339. [DOI] [PubMed] [Google Scholar]
- 3.Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. Am J Physiol. 2000;278:C235–C256. doi: 10.1152/ajpcell.2000.278.2.C235. [DOI] [PubMed] [Google Scholar]
- 4.Kume H, Takai A, Tokuno H, Tomita T. Regulation of Ca2+-dependent K+ channel activity in tracheal myocytes by phosphorylation. Nature. 1989;341:152–154. doi: 10.1038/341152a0. [DOI] [PubMed] [Google Scholar]
- 5.Robertson BE, Schubert R, Hescheler J, Nelson MT. cGMP-dependent protein kinase activates Ca-activated K channels in cerebral artery smooth muscle cells. Am J Physiol. 1993;265:C299–C303. doi: 10.1152/ajpcell.1993.265.1.C299. [DOI] [PubMed] [Google Scholar]
- 6.Bonev AD, Jaggar JH, Rubart M, Nelson MT. Activators of protein kinase C decrease Ca2+ spark frequency in smooth muscle cells from cerebral arteries. Am J Physiol. 1997;273:C2090–C2095. doi: 10.1152/ajpcell.1997.273.6.C2090. [DOI] [PubMed] [Google Scholar]
- 7.Alioua A, Mahajan A, Nishimaru K, Zarei MM, Stefani E, Toro L. Coupling of c-Src to large conductance voltage- and Ca2+-activated K+ channels as a new mechanism of agonist-induced vasoconstriction. Proc Natl Acad Sci U S A. 2002;99:14560–14565. doi: 10.1073/pnas.222348099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zou AP, Fleming JT, Falck JR, Jacobs ER, Gebremedhin D, Harder DR, Roman RJ. 20-HETE is an endogenous inhibitor of the large-conductance Ca2+- activated K+ channel in renal arterioles. Am J Physiol. 1996;270:R228–R237. doi: 10.1152/ajpregu.1996.270.1.R228. [DOI] [PubMed] [Google Scholar]
- 9.Tang XD, Xu R, Reynolds MF, Garcia ML, Heinemann SH, Hoshi T. Haem can bind to and inhibit mammalian calcium-dependent Slo1 BK channels. Nature. 2003;425:531–535. doi: 10.1038/nature02003. [DOI] [PubMed] [Google Scholar]
- 10.Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- 11.Williams SE, Wootton P, Mason HS, Bould J, Iles DE, Riccardi D, Peers C, Kemp PJ. Hemoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science. 2004;306:2093–2097. doi: 10.1126/science.1105010. [DOI] [PubMed] [Google Scholar]
- 12.Wang R. Resurgence of carbon monoxide: an endogenous gaseous vasorelaxing factor. Can J Physiol Pharmacol. 1998;76:1–15. doi: 10.1139/cjpp-76-1-1. [DOI] [PubMed] [Google Scholar]
- 13.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 14.Boehning D, Moon C, Sharma S, Hurt KJ, Hester LD, Ronnett GV, Shugar D, Snyder SH. Carbon monoxide neurotransmission activated by CK2 phosphorylation of heme oxygenase-2. Neuron. 2003;40:129–137. doi: 10.1016/s0896-6273(03)00596-8. [DOI] [PubMed] [Google Scholar]
- 15.Xue L, Farrugia G, Miller SM, Ferris CD, Snyder SH, Szurszewski JH. Carbon monoxide and nitric oxide as coneurotransmitters in the enteric nervous system: evidence from genomic deletion of biosynthetic enzymes. Proc Natl Acad Sci U S A. 2000;97:1851–1855. doi: 10.1073/pnas.97.4.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryter SW, Morse D, Choi AM. Carbon monoxide: to boldly go where NO has gone before. Sci STKE. 2004;2004:RE6. doi: 10.1126/stke.2302004re6. [DOI] [PubMed] [Google Scholar]
- 17.Jaggar JH, Leffler CW, Cheranov SY, Tcheranova D, Shuyu S, Cheng X. Carbon monoxide dilates cerebral arterioles by enhancing the coupling of Ca2+ sparks to Ca2+-activated K+ channels. Circ Res. 2002;91:610–617. doi: 10.1161/01.res.0000036900.76780.95. [DOI] [PubMed] [Google Scholar]
- 18.Riesco-Fagundo AM, Perez-Garcia MT, Gonzalez C, Lopez-Lopez JR. O2 modulates large-conductance Ca2+-dependent K+ channels of rat chemoreceptor cells by a membrane-restricted and CO-sensitive mechanism. Circ Res. 2001;89:430–436. doi: 10.1161/hh1701.095632. [DOI] [PubMed] [Google Scholar]
- 19.Xi Q, Tcheranova D, Parfenova H, Horowitz B, Leffler CW, Jaggar JH. Carbon monoxide activates KCa channels in newborn cerebral arteriole smooth muscle cells by increasing the apparent Ca2+-sensitivity of a-subunits. Am J Physiol. 2004;286:H610–H618. doi: 10.1152/ajpheart.00782.2003. [DOI] [PubMed] [Google Scholar]
- 20.Horrigan FT, Heinemann SH, Hoshi T. Heme regulates allosteric activation of the Slo1 BK channel. J Gen Physiol. 2005;126:7–21. doi: 10.1085/jgp.200509262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaggar JH. Intravascular pressure regulates local and global Ca2+ signaling in cerebral artery smooth muscle cells. Am J Physiol. 2001;281:C439–C448. doi: 10.1152/ajpcell.2001.281.2.C439. [DOI] [PubMed] [Google Scholar]
- 22.Liu P, Xi Q, Ahmed A, Jaggar JH, Dopico AM. Essential role for smooth muscle BK channels in alcohol-induced cerebrovascular constriction. Proc Natl Acad Sci U S A. 2004;101:18217–18222. doi: 10.1073/pnas.0406096102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCobb DP, Fowler NL, Featherstone T, Lingle CJ, Saito M, Krause JE, Salkoff L. A human calcium-activated potassium channel gene expressed in vascular smooth muscle. Am J Physiol. 1995;269:H767–H777. doi: 10.1152/ajpheart.1995.269.3.H767. [DOI] [PubMed] [Google Scholar]
- 24.Wu L, Cao K, Lu Y, Wang R. Different mechanisms underlying the stimulation of KCa channels by nitric oxide and carbon monoxide. J Clin Invest. 2002;110:691–700. doi: 10.1172/JCI15316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang R, Wu L, Wang Z. The direct effect of carbon monoxide on KCa channels in vascular smooth muscle cells. Pflugers Arch. 1997;434:285–291. doi: 10.1007/s004240050398. [DOI] [PubMed] [Google Scholar]
- 26.Wang R, Wu L. Interaction of selective amino acid residues of KCa channels with carbon monoxide. Exp Biol Med (Maywood) 2003;228:474–480. doi: 10.1177/15353702-0322805-09. [DOI] [PubMed] [Google Scholar]
- 27.Balla G, Vercellotti GM, Muller-Eberhard U, Eaton J, Jacob HS. Exposure of endothelial cells to free heme potentiates damage mediated by granulocytes and toxic oxygen species. Lab Invest. 1991;64:648–655. [PubMed] [Google Scholar]
- 28.Wang R, Wu L. The chemical modification of KCa channels by carbon monoxide in vascular smooth muscle cells. J Biol Chem. 1997;272:8222–8226. doi: 10.1074/jbc.272.13.8222. [DOI] [PubMed] [Google Scholar]
- 29.Perez GJ, Bonev AD, Nelson MT. Micromolar Ca2+ from sparks activates Ca2+-sensitive K+ channels in rat cerebral artery smooth muscle. Am J Physiol. 2001;281:C1769–C1775. doi: 10.1152/ajpcell.2001.281.6.C1769. [DOI] [PubMed] [Google Scholar]
- 30.ZhuGe R, Fogarty KE, Tuft RA, Walsh JV., Jr Spontaneous transient outward currents arise from microdomains where BK channels are exposed to a mean Ca2+ concentration on the order of 10 microM during a Ca2+ spark. J Gen Physiol. 2002;120:15–27. doi: 10.1085/jgp.20028571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature. 2000;407:870–876. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.